
The Thermal Behavior of Lyocell Fibers Containing Bis(trimethylsilyl)acetylene

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Fiber Spinning
2.3. Structure Characterization
2.4. Mechanical Properties
2.5. Scanning Electron Microscopy (SEM)
2.6. Thermal Properties
3. Results
3.1. Structure of Cellulose and Composite Fibers
3.2. Morphology of Cellulose and Composite Fibers
3.3. Mechanical Properties
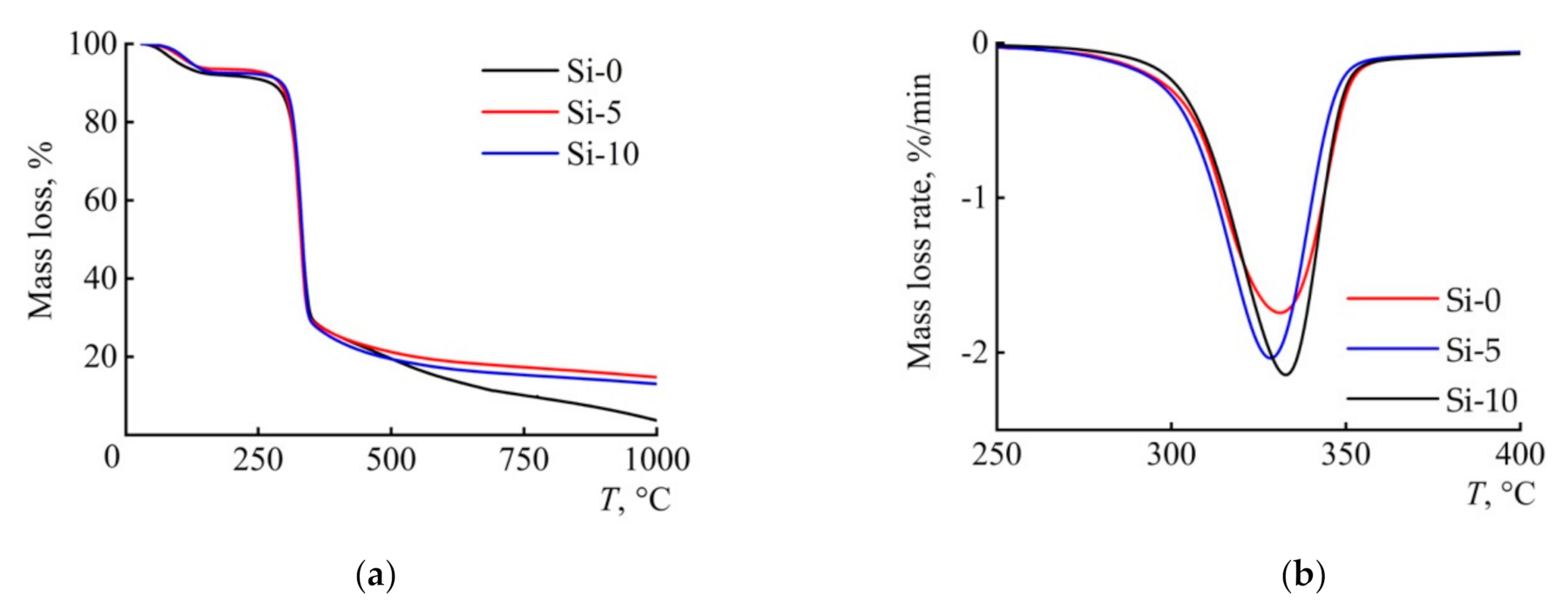
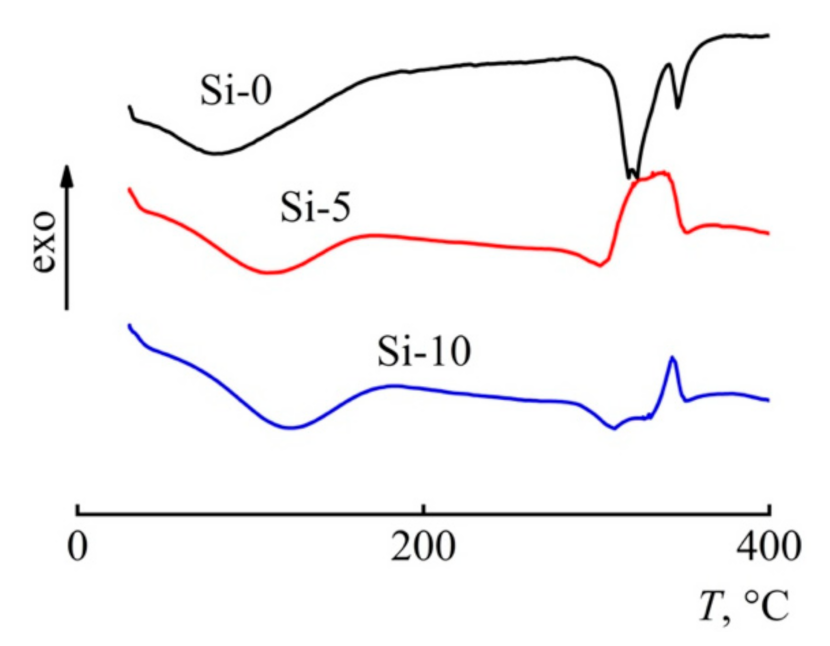
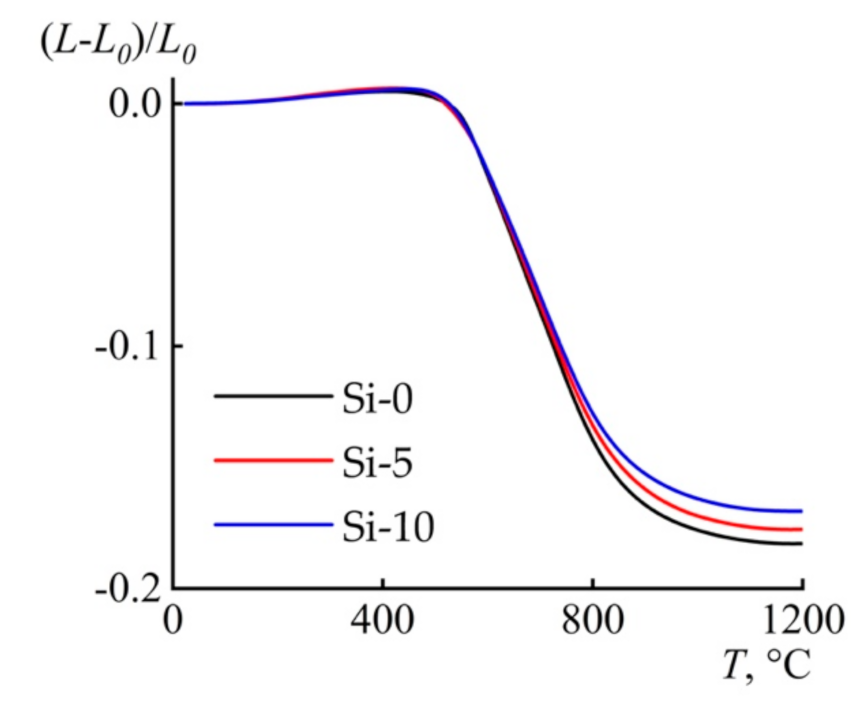
3.4. Thermal Behavior
3.5. Morphology and Structure of Carbonized Fibers
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Konkin, A.A. Carbon and Other Heat-Resistant Fibrous Materials; Khimiya: Moscow, Russia, 1974. [Google Scholar]
- Nashchokin, A.V.; Malakho, A.P.; Galiguzov, A.A.; Kulakov, V.V.; Seleznev, A.N.; Avdeev, V.V. Dependence of the mechanical properties, morphology, and structural characteristics of different types of carbon fibers on treatment temperature. Fibre Chem. 2012, 44, 180–185. [Google Scholar] [CrossRef]
- Lian, F.; Liu, J.; Xue, Y.; Ma, Z.; Liang, J. The demonstration of a dense and stable circumferential layer in the subsurface of polyacrylonitrile fibers for carbon fibers. Fibers Polym. 2013, 14, 243–249. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Wu, G.-P.; Li, R.-M.; Li, X.-L.; Lu, C.-X. Fracture mechanisms of polyacrylonitrile-based high-strength type carbon fibers. Fibers Polym. 2014, 15, 2541–2543. [Google Scholar] [CrossRef]
- Varshavsky, V.Y. Carbon Fibers, 2nd ed.; FGUP PIK VINITI: Moscow, Russia, 2007. [Google Scholar]
- Karacan, I.; Erdogan, G. The influence of thermal stabilization stage on the molecular structure of polyacrylonitrile fibers prior to the carbonization stage. Fibers Polym. 2012, 13, 295–302. [Google Scholar] [CrossRef]
- Saklakova, E.V.; Astashkina, O.V.; Medvedeva, N.G.; Kuzikova, I.L.; Lysenko, A.A. Antibacterial Properties of Carbon Fibers Containing Metal Nano- and Microparticles. Fibre Chem. 2015, 47, 324–328. [Google Scholar] [CrossRef]
- Hu, X.C.; Zhang, H.H.; Shao, H.; Zhang, T.; Guo, L. Process for preparing precursor fiber of cellulose base carbon fiber. Patent CN 1587457A, 3 February 2004. [Google Scholar]
- Makarov, I.S.; Golova, L.K.; Bondarenko, G.N.; Skvortsov, I.Y.; Berkovich, A.K.; Bermeshev, M.V.; Mironova, M.V. Carbon—Silicon-Carbide Fibers Prepared from Solid Solutions of Cellulose in N-Methylmorpholine-N-Oxide with Added Tetraethoxysilane. Fibre Chem. 2017, 49, 231–236. [Google Scholar] [CrossRef]
- Kulkarni, R.; Ochoa, O. Transverse and Longitudinal CTE Measurements of Carbon Fibers and their Impact on Interfacial Residual Stresses in Composites. J. Compos. Mater. 2006, 40, 733–754. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Wu, G.-H. Interface and thermal expansion of carbon fiber reinforced aluminum matrix composites. Trans. Nonferrous Met. Soc. China 2010, 20, 2148–2151. [Google Scholar] [CrossRef]
- Pradere, C.; Sauder, C. Transverse and longitudinal coefficient of thermal expansion of carbon fibers at high temperatures (300–2500K). Carbon 2008, 46, 1874–1884. [Google Scholar] [CrossRef]
- Belenkov, E.A. Formation of graphite structure in carbon fiber. Chelyabinsk Phys. Math. J. 1998, 6, 42–53. [Google Scholar]
- Bochek, A.M. Effect of hydrogen bonding on cellulose solubility in aqueous and nonaqueous solvents. Russ. J. Appl. Chem. 2003, 76, 1711–1719. [Google Scholar] [CrossRef]
- Golova, L.K.; Borodina, O.E.; Kuznetsova, L.K.; Lyubova, T.A.; Krylova, T.B. The solid-phase MMO process. Fibre Chem. 2000, 32, 243–251. [Google Scholar]
- Ciechanska, D.; Wesolowska, E.; Wawro, D. An introduction to cellulosic fibres. In Handbook of Textile Fibre Structure; Eichhorn, S.J., Hearle, J.W.S., Jaffe, M., Kikutani, T., Eds.; Woodhead Publishing: Cambridge, UK, 2009; Volume 2, pp. 3–61. [Google Scholar]
- Burger, C.; Maron, R.; Michels, C.; Mick, K.P. New types of cellulose materials obtained by an alternative spinning method. Fibre Chem. 1996, 28, 22–27. [Google Scholar] [CrossRef]
- Golova, L.K.; Borodina, O.E.; Rudinskaya, G.Y.; Papkov, S.P. Optical properties and structure of highly concentrated solutions of cellulose in N-methylmorpholine N-oxide. Fibre Chem. 2001, 33, 140–144. [Google Scholar] [CrossRef]
- Golova, L.K.; Kulichikhin, V.G.; Papkov, S.P. Mechanism of dissolution of cellulose in non-aqueous dissolving systems. Rev. Polym. Sci. U.S.S.R. 1986, 28, 1995–2011. [Google Scholar]
- Golova, L.K. Processing of cellulose via highly concentrated “solid solutions”. Fibre Chem. 1996, 28, 5–16. [Google Scholar] [CrossRef]
- Golova, L.K. New cellulose fiber lyocell. Russ. J. Gen. Chem. 2002, XLVI, 49–57. [Google Scholar]
- Sporl, J.M.; Beyer, R.; Abels, F.; Cwik, T.; Muller, A.; Hermanutz, F.; Buchmeister, M.R. Cellulose-derived carbon fibers with improved carbon yield and mechanical properties. Macromol. Mater. Eng. 2017, 302, 1–10. [Google Scholar] [CrossRef]
- Huang, X. Fabrication and properties of carbon fibers. Materials 2009, 2, 2369–2403. [Google Scholar] [CrossRef]
- Chernenko, D.N. Development and research of technological process of obtaining carbon fabrics from hydrated cellulose fibers. Bachelor’s Thesis, Research Institute of Structural Materials based on graphite “NIIgraphit”, Moscow, Russia, 2015. [Google Scholar]
- Sporl, J.M.; Ota, A.; Son, S.; Massonne, K.; Hermanutz, F.; Buchmeiser, M.R. Carbon fibers prepared from ionic liquid-derived cellulose precursors. Mater. Today Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef]
- Kazakov, M.E.; Trushnikov, A.M.; Yunitskaya, M.L. The Method of Obtaining Carbon Fiber Material. Patent RF 2045472, 10 October 1995. [Google Scholar]
- Olri, P.; Plezantene, E.; Louison, S.; Paye, R. Carbonization of Cellulosic Fibrous Materials in the Presence of an Organosilicon Compound. Patent RF 2256013, 10 July 2005. [Google Scholar]
- Trushnikov, A.M.; Kazakov, M.E.; Gridina, Y.F.; Vazheva, L.D.; Borisova, L.K. The Method of Obtaining Carbon Fiber Material. Patent RF 2047674, 10 November 1995. [Google Scholar]
- Olri, P.; Louison, S.; Kazakov, M.E.; Trushnikov, A.M. The Method of Obtaining Carbon Fiber Material. Patent RF 2258773, 20 August 2005. [Google Scholar]
- Wizon, I.; Robertson, J.A. Continuous filament ceramic fibers via the viscose process. J. Polym. Sci. Part. C 1967, 19, 267–281. [Google Scholar] [CrossRef]
- Makarov, I.S.; Golova, L.K.; Kuznetsova, L.K.; Bondarenko, G.N.; Skvortsov, I.Y.; Mironova, M.V.; Bermeshev, M.V. Composite fibers based on cellulose and tetraethoxysilane: Preparation, structure and properties. Fibre Chem. 2017, 49, 101–107. [Google Scholar] [CrossRef]
- Golova, L.K.; Makarov, I.S.; Bondarenko, G.N.; Mironova, M.V.; Berkovich, A.K.; Shandryuk, G.A.; Vinogradov, M.I.; Bermeshev, M.V.; Kulichikhin, V.G. Composite Fibers Based on Cellulose and Vinyltriethoxysilane as Precursors of Carbon Materials. Polym. Sci. Ser. B. 2020, 62, 152–162. [Google Scholar] [CrossRef]
- Makarov, I.S.; Golova, L.K.; Mironova, M.V.; Vinogradov, M.I.; Kulichikhin, V.G. Composite fibres based on cellulose and vinyltriethoxysilane: Preparation, properties and carbonization. Mat. Sci. Eng. 2018, 347, 012032. [Google Scholar] [CrossRef]
- Makarov, I.S.; Golova, L.K.; Mironova, M.V.; Vinogradov, M.I.; Bermeshev, M.V.; Berkovich, A.K.; Kulichikhin, V.G. Structural and morphological features of carbon-silicon-carbide fibers based on cellulose and triethoxyvinylsilane. Fibre Chem. 2018, 50, 79–84. [Google Scholar]
- Golova, L.K.; Romanov, V.V.; Lunina, O.B.; Platonov, V.A.; Papkov, S.P.; Khorozova, O.D.; Yakshin, V.V.; Belasheva, T.P.; Sokira, A.N. Method for Producing a Solution for Spinning Fibers. Patent RF 1645308, 30 April 1991. [Google Scholar]
- Blackburn, R. Biodegradable and Sustainable Fibres; Woodhead Publishing: Cambridge, UK, 2005. [Google Scholar]
- Abu-Rous, M.; Ingolic, E.; Schuster, K.C. Visualisation of the nano-structure of Tencel® (Lyocell) and other cellulosics as an approach to explaining functional and wellness properties in textiles. Lenzing. Ber. 2006, 85, 31–37. [Google Scholar]
- Hideno, A. Comparison of the Thermal degradation properties of crystalline and amorphous cellulose, as well as treated lignocellulosic biomass. BioResources 2016, 11, 6309–6319. [Google Scholar] [CrossRef]
- Kulichikhin, V.G.; Skvortsov, I.Y.; Mironova, M.V.; Ozerin, A.N.; Kurkin, T.S.; Berkovich, A.K.; Frenkin, E.I.; Malkin, A.Y. From polyacrylonitrile, its solutions, and filaments to carbon fibers II. Spinning PAN-precursors and their thermal treatment. Adv. Polym. Technol. 2018, 37, 1099–1113. [Google Scholar] [CrossRef]
- Niekraszewicz, B.; Czarnecki, P. Modified cellulose fibers prepared by the N-Methylmorpholine-N-oxide (NMMO) process. J. Appl. Polym. Sci. 2002, 86, 907–916. [Google Scholar] [CrossRef]
- Makarov, I.S.; Golova, L.K.; Vinogradov, M.I.; Levin, I.S.; Shandryuk, G.A.; Arkharova, N.A.; Golubev, Y.V.; Berkovich, A.K.; Eremin, T.V.; Obraztsova, E.D. The effect of alcohol precipitants on structural and morphological features and thermal properties of lyocell fibers. Fibers 2020, 8, 43. [Google Scholar] [CrossRef]
- Broido, A. A simple, sensitive graphical method of treating thermogravimetric analysis data. J. Polym. Sci. Part. A-2 1969, 7, 1761–1773. [Google Scholar] [CrossRef]
- Brown, M.E.; Maciejewski, M.; Vyazovkin, S.; Nomen, R.; Sempere, J.; Burnham, A.; Opfermann, J.; Strey, R.; Anderson, H.L.; Kemmler, A.; et al. Computational aspects of kinetic analysis: Part A: The ICTAC kinetics project-data, methods and results. Thermochim. Acta 2000, 355, 125–143. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Burnham, A.K.; Criado, J.M.; Pérez-Maqueda, L.A.; Popescu, C.; Sbirrazzuoli, N. ICTAC kinetics committee recommendations for performing kinetic computations on thermal analysis data. Thermochim. Acta 2011, 520, 1–19. [Google Scholar]
- Tanaka, H. The theory and practice of thermoanalytical kinetics of solid-state reactions. J. Therm. Anal. Calorim. 2005, 80, 795–797. [Google Scholar] [CrossRef]
- Bacon, R. Carbon fibers from rayon precursors. In Chemistry and Physics of Carbon; Walker, P.L., Jr., Thrower, P.A., Eds.; Marcel Dekker: New York, NY, USA, 1974; Volume 9, p. 1. [Google Scholar]
- Belenkov, E.A.; Iakovlev, D.V. Peculiarities of the form analysis of profiles X-Ray diffraction lines for carbon materials. Part II. Relation of the profiles form and crystals distribution on the sizes. Bull. Chelyabinsk Sci. Cent. Ural Branch RAS 2001, 2, 37–45. [Google Scholar]
- Cho, S.Y.; Yun, Y.S.; Jin, H.J. Carbon nanofibers prepared by the carbonization of self-assembled cellulose nanocrystals. Macromol. Res. 2014, 22, 753–756. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Resonant raman spectroscopy of disordered, amorphous, and diamondlike carbon. Phys. Rev. B Condens. Matter. 2001, 64, 075414. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B Condens. Matter. 2000, 61, 14095–14107. [Google Scholar]
- Dychalska, A.; Popielarski, P.; Franków, W.; Fabisiak, K.; Paprocki, K.; Szybowicz, M. Study of CVD diamond layers with amorphous carbon admixture by Raman scattering spectroscopy. Mater. Sci. 2015, 33, 799–805. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Diameter (μm) | Tensile Strength (MPa) | Elongation (%) | Modulus (GPa) |
|---|---|---|---|---|
| Si-0 | 17.0 ± 2.8 | 590 ± 22 | 11.0 ± 0.8 | 11.0 ± 0.9 |
| Si-5 | 12.0 ± 2.3 | 565 ± 34 | 7.0 ± 2.1 | 14.8 ± 2.1 |
| Si-10 | 12.5 ± 1.5 | 515 ± 43 | 7.0 ± 1.2 | 13.4 ± 2.3 |
| Sample | Tmax (°C) | DTGmax (% min−1) | Tonset (°C) | Ea1 (kJ mol−1) |
|---|---|---|---|---|
| Si-0 | 331 | −1.74 | 281 | 234 |
| Si-5 | 328 | −2.03 | 279 | 162 |
| Si-10 | 332 | −2.14 | 279 | 172 |
| Sample | (L−L0) 1 × 103/L0, 25–400 °C (step I) | CTE × 106, (average from 30 °C to 400 °C) | (L−L0) × 103/L0, 25–1200 °C (step II) |
|---|---|---|---|
| Si-0 | 5.02 | 13.50 | −181.45 |
| Si-5 | 6.31 | 16.99 | −175.61 |
| Si-10 | 5.58 | 14.98 | −168.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makarov, I.; Vinogradov, M.; Mironova, M.; Shandryuk, G.; Golubev, Y.; Berkovich, A. The Thermal Behavior of Lyocell Fibers Containing Bis(trimethylsilyl)acetylene. Polymers 2021, 13, 537. https://doi.org/10.3390/polym13040537
Makarov I, Vinogradov M, Mironova M, Shandryuk G, Golubev Y, Berkovich A. The Thermal Behavior of Lyocell Fibers Containing Bis(trimethylsilyl)acetylene. Polymers. 2021; 13(4):537. https://doi.org/10.3390/polym13040537
Chicago/Turabian StyleMakarov, Igor, Markel Vinogradov, Maria Mironova, Georgy Shandryuk, Yaroslav Golubev, and Anna Berkovich. 2021. "The Thermal Behavior of Lyocell Fibers Containing Bis(trimethylsilyl)acetylene" Polymers 13, no. 4: 537. https://doi.org/10.3390/polym13040537
APA StyleMakarov, I., Vinogradov, M., Mironova, M., Shandryuk, G., Golubev, Y., & Berkovich, A. (2021). The Thermal Behavior of Lyocell Fibers Containing Bis(trimethylsilyl)acetylene. Polymers, 13(4), 537. https://doi.org/10.3390/polym13040537