
Influence of Oligopeptide Length and Distribution on Polyisoprene Properties


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Samples
2.2.1. B-2A-PIP
2.2.2. Synthesis of B-3A-PIP
2.2.3. Synthesis of R-2A-PIP
2.2.4. Synthesis of R-3A-PIP
2.3. Sample Molding Process
2.4. Characterization
3. Results and Discussion
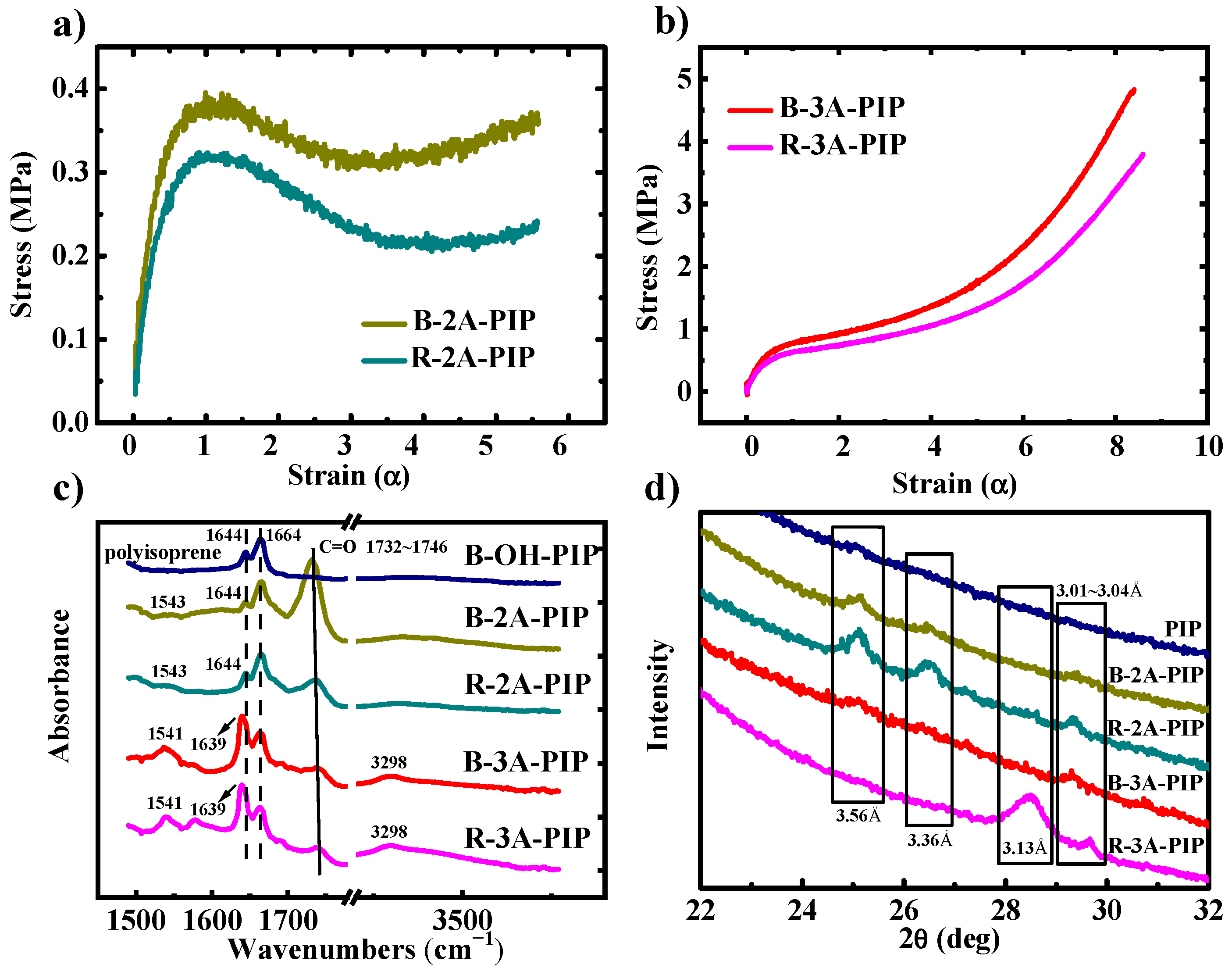
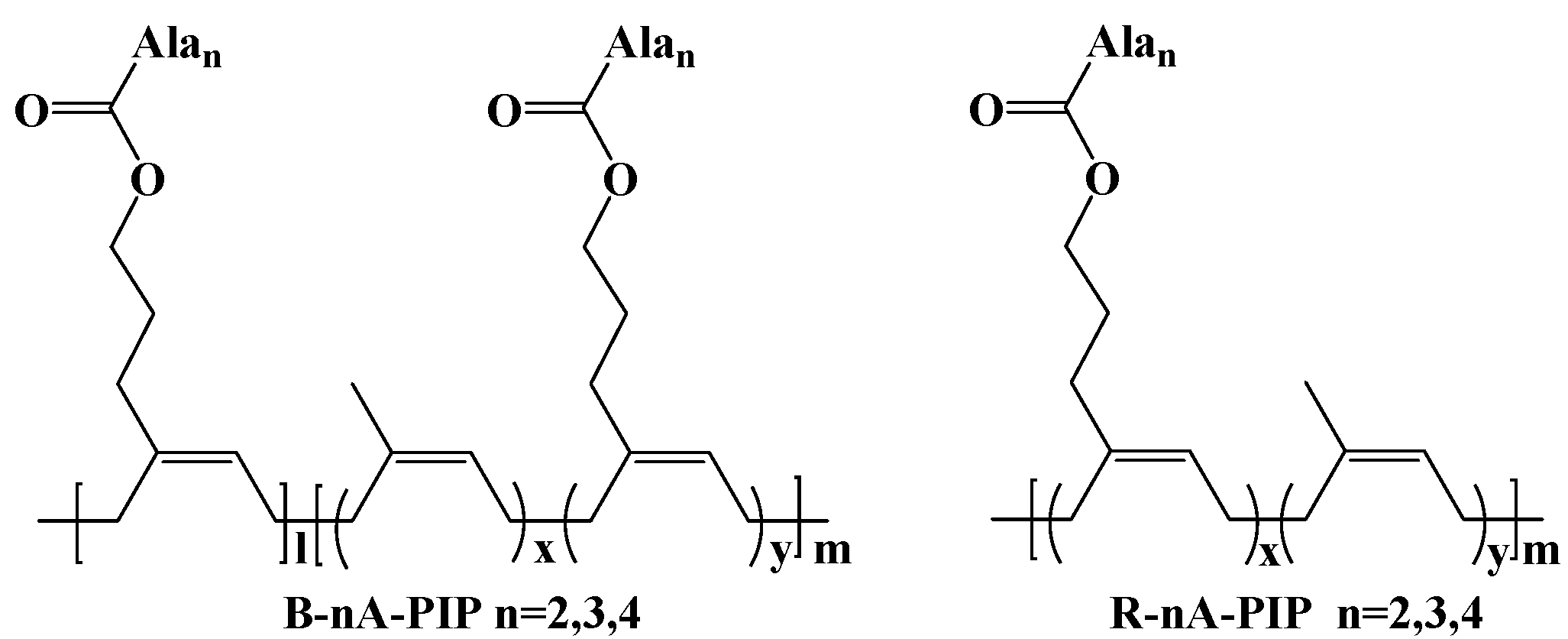
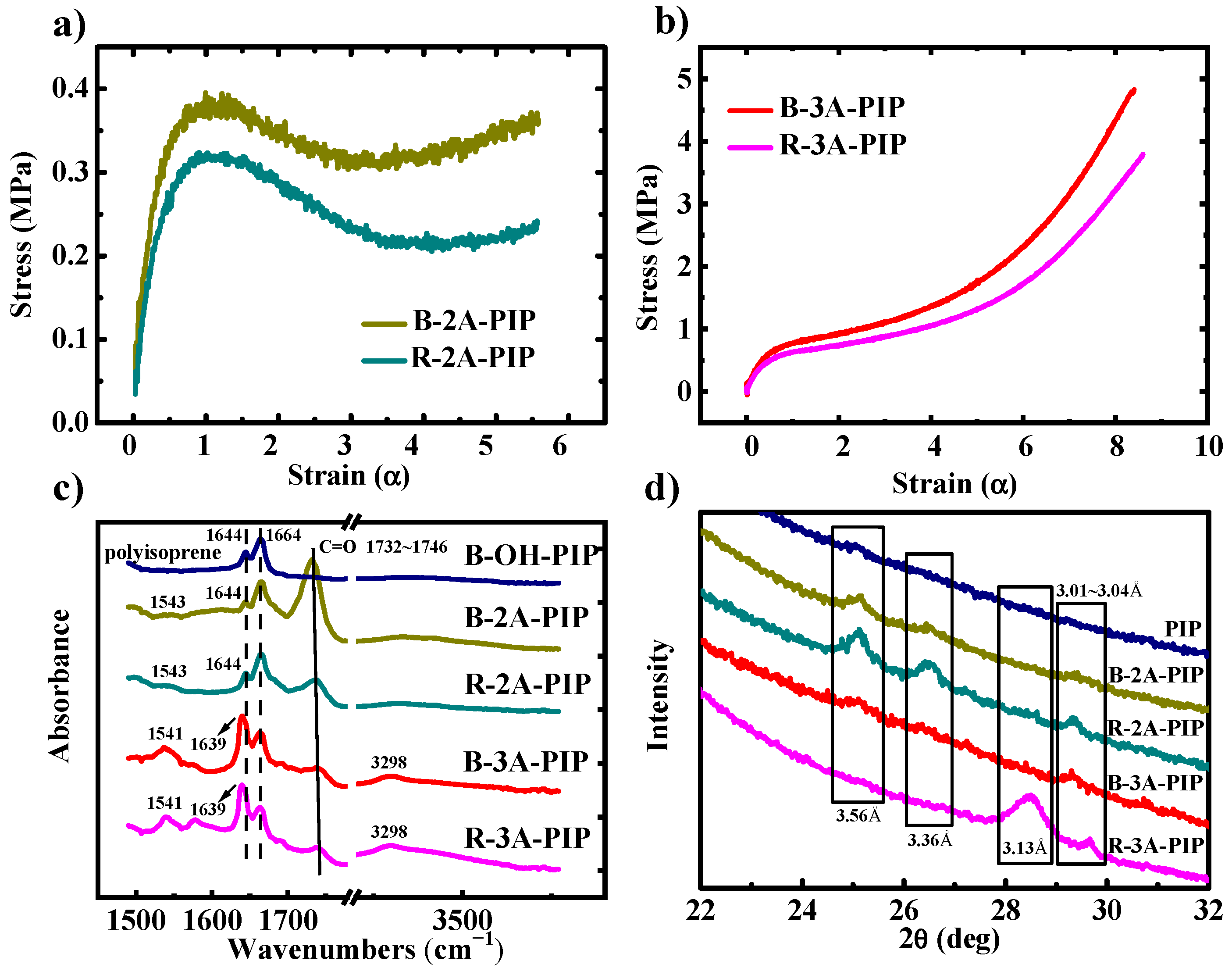
3.1. Mechanical Properties and Secondary Structure Analysis
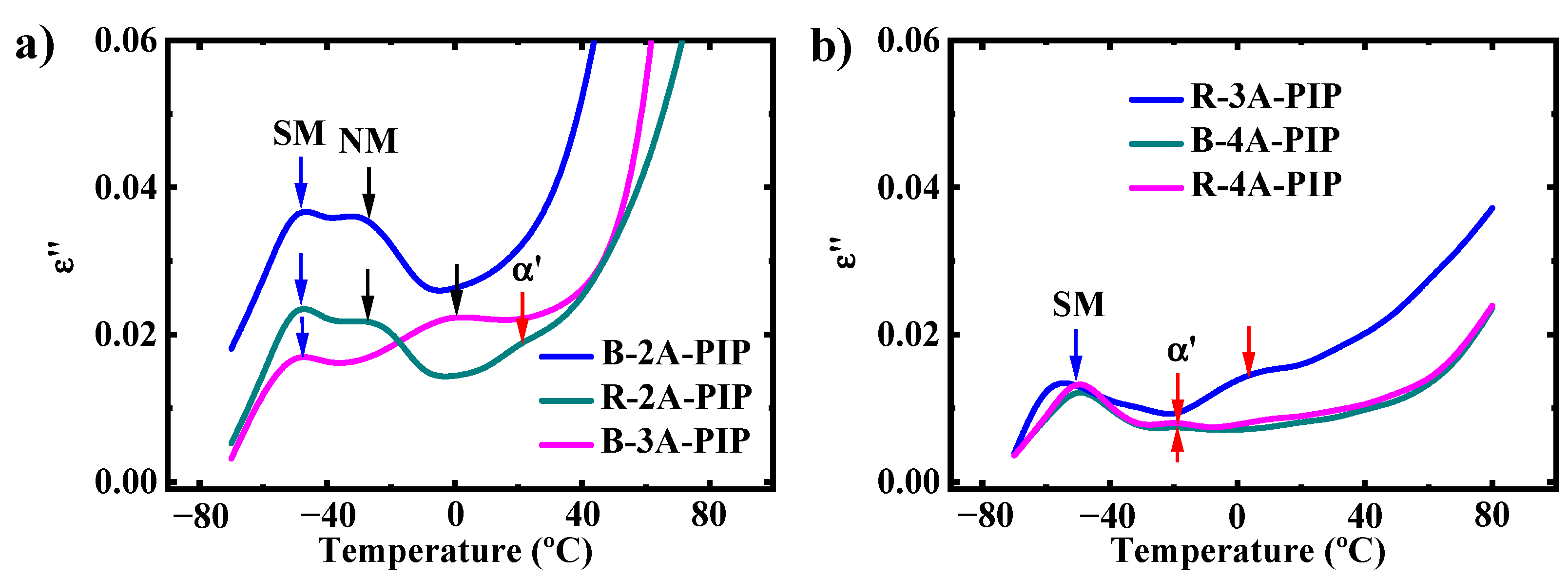
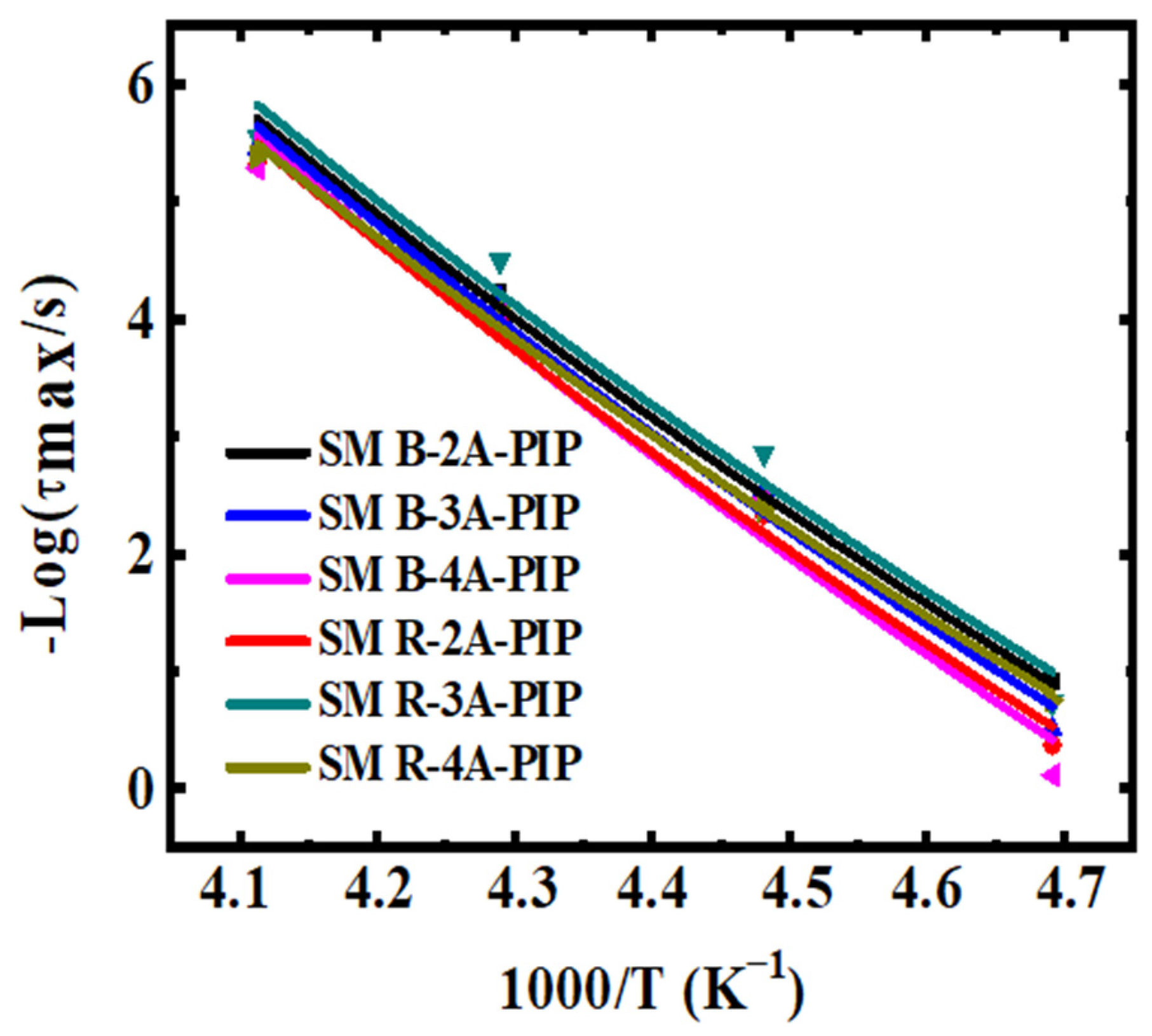
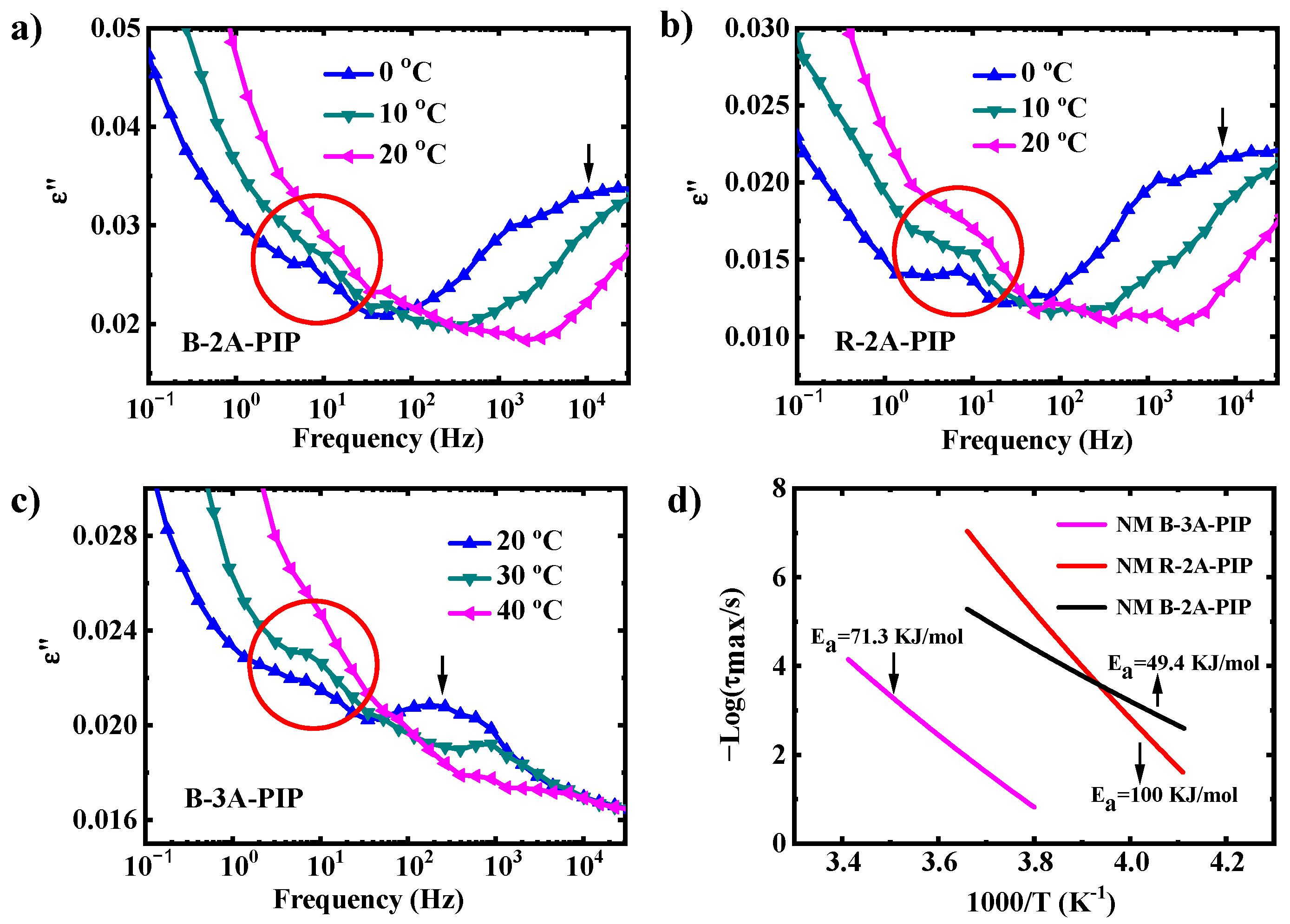
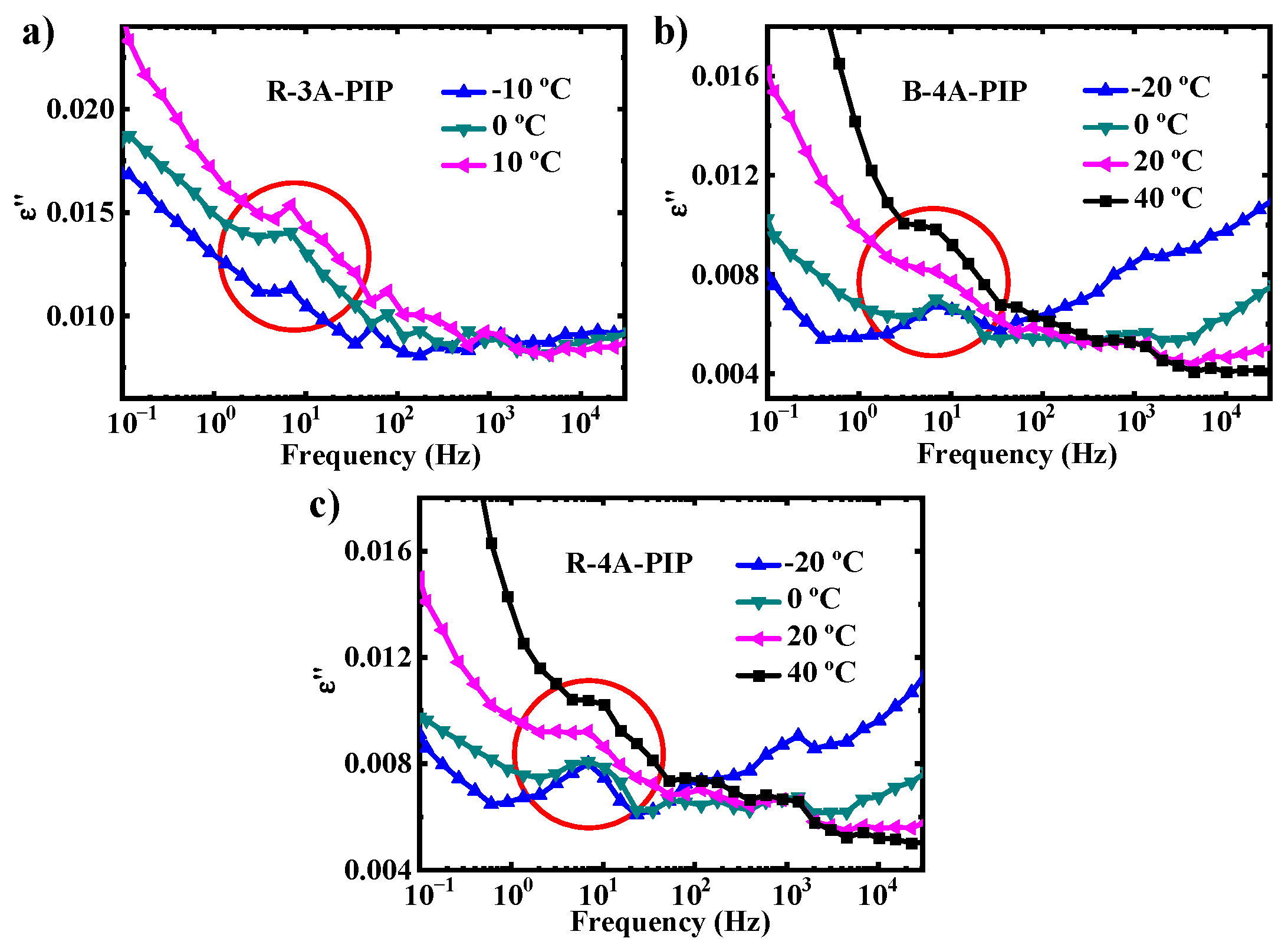
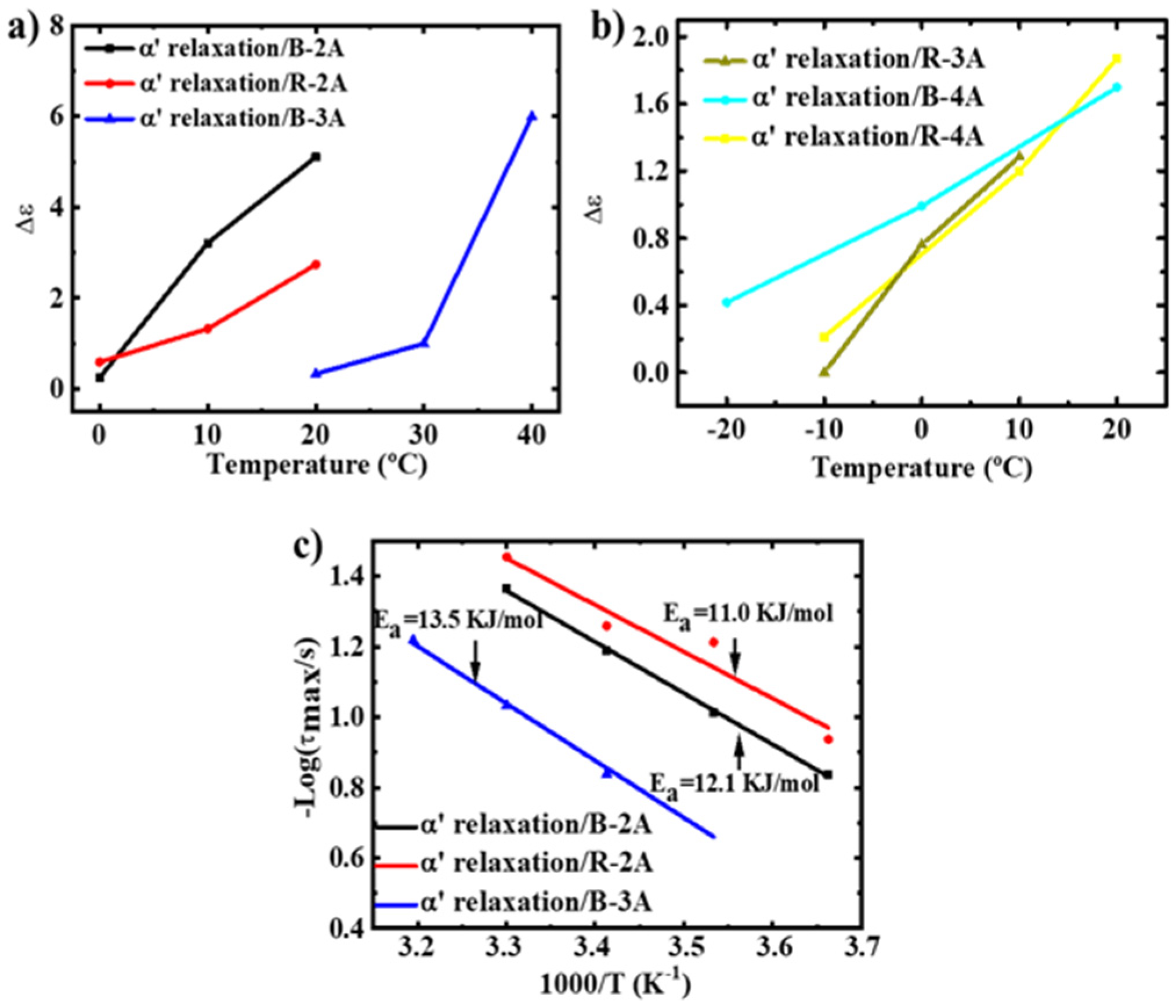
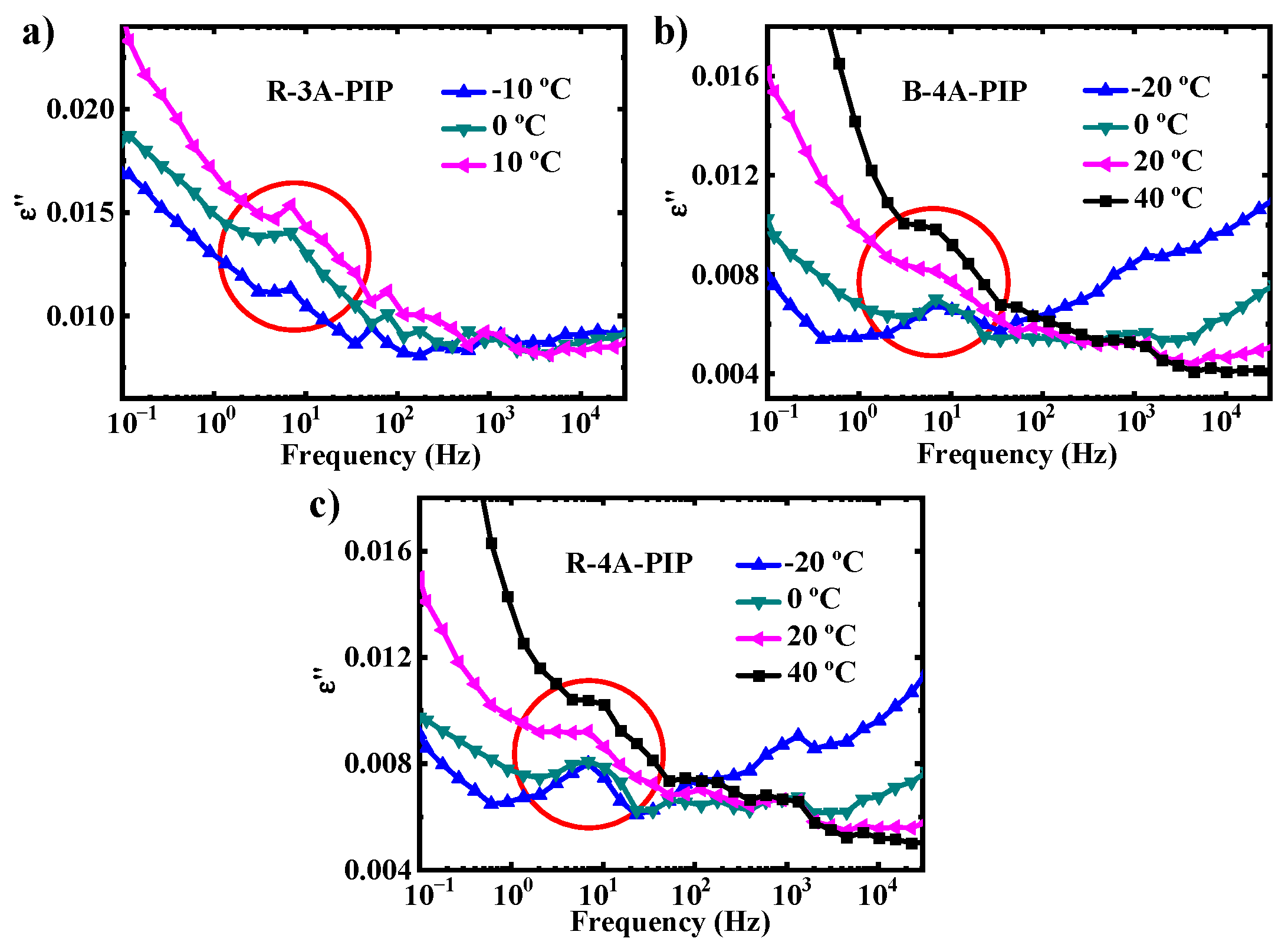
3.2. Molecular Dynamics-SM Relaxation, NM Relaxation and α′ Relaxation
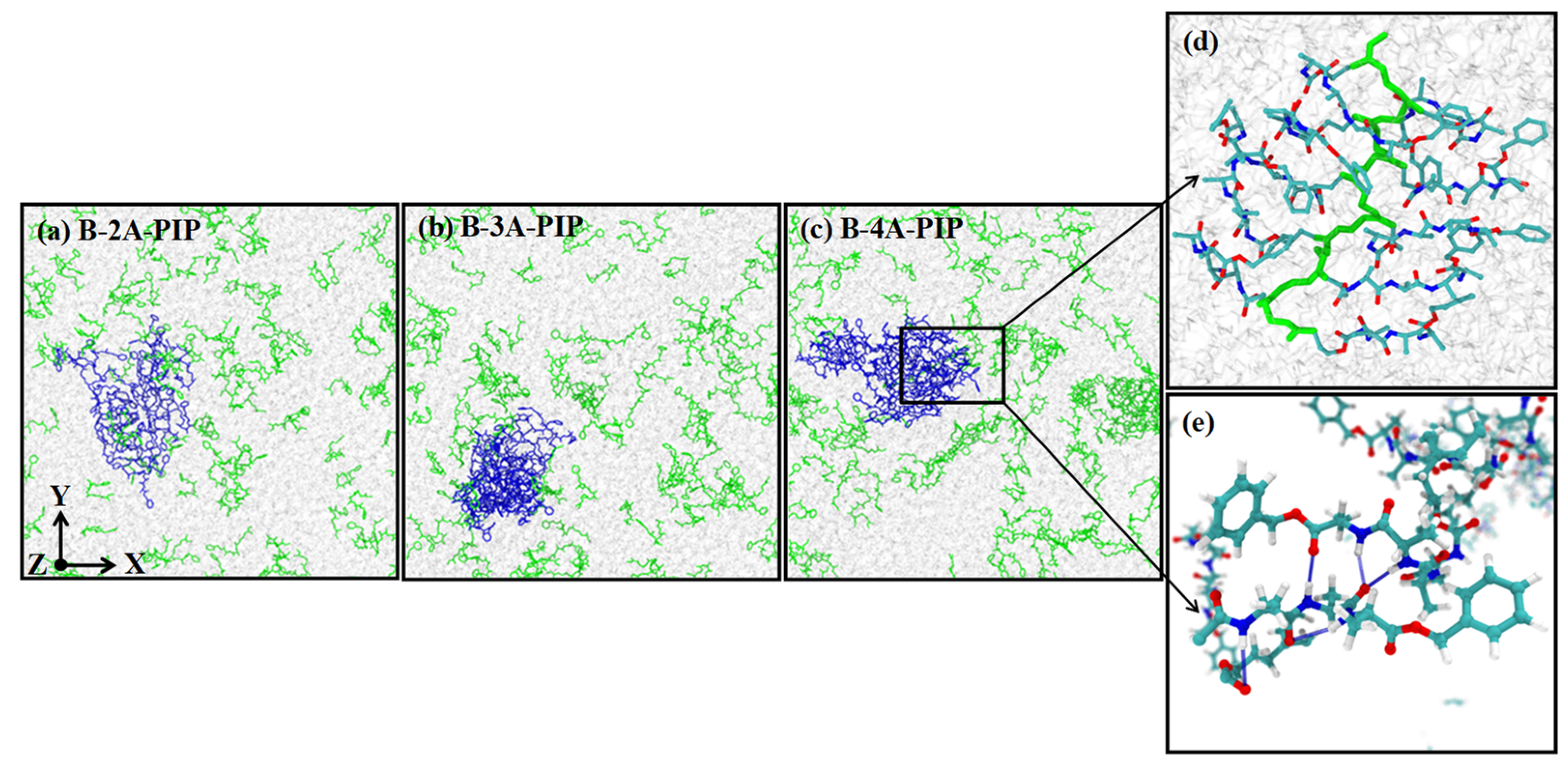
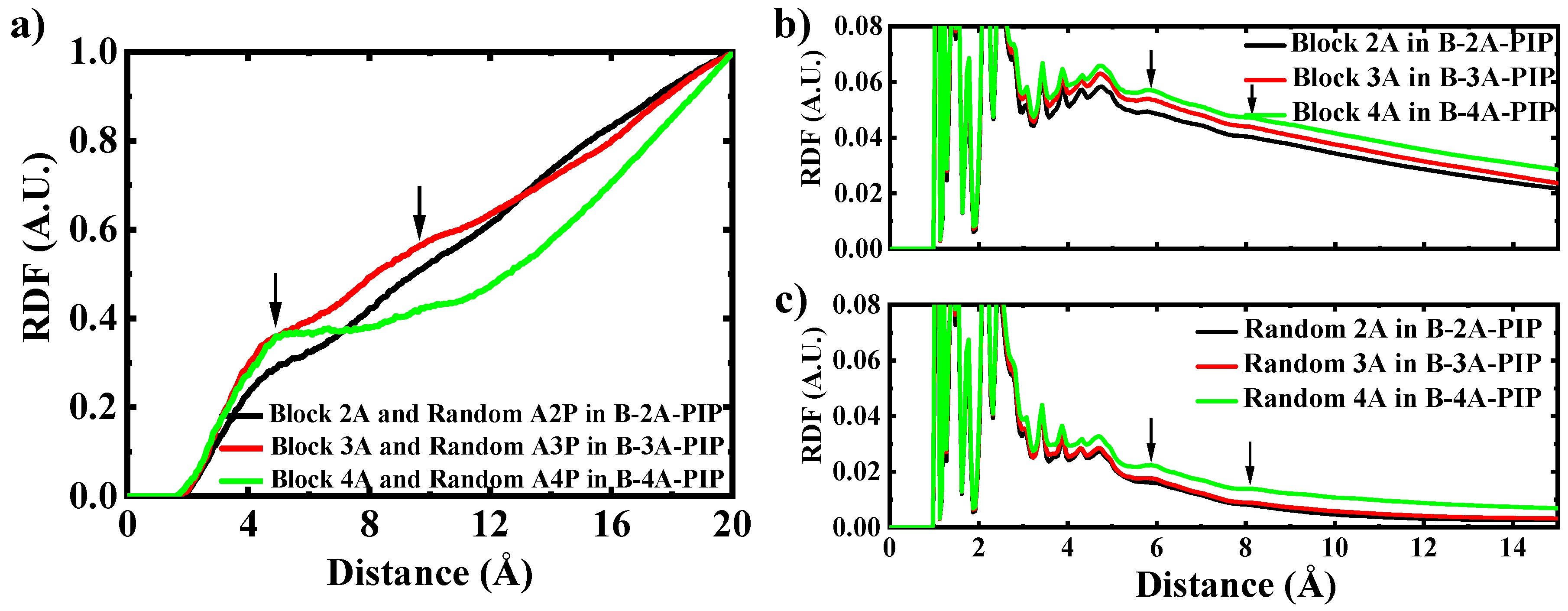
3.3. Classical Molecular Dynamics Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deng, F.; Ito, M.; Noguchi, T.; Wang, L.; Ueki, H.; Niihara, K.; Kim, Y.A.; Endo, M.; Zheng, Q.S. Elucidation of the Reinforcing Mechanism in Carbon Nanotube/Rubber Nanocomposites. ACS Nano 2011, 5, 3858–3866. [Google Scholar] [CrossRef] [PubMed]
- Amnuaypornsri, S.; Sakdapipanich, J.; Tanaka, Y. Green Strength of Natural Rubber: The Origin of the Stress-Strain Behavior of Natural Rubber. J. Appl. Polym. Sci. 2010, 111, 2127–2133. [Google Scholar] [CrossRef]
- Zhu, L.; Tian, X.; Pan, Y.; Chang, T.; Wang, K.; Niu, G.; Zhang, L.; Wang, C.; Han, W. Optimization of Serial Modular Continuous Mixing Process Parameters for Natural Rubber Composites Reinforced by Silica/Carbon Black. Polymers 2020, 12, 416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toki, S.; Che, J.; Rong, L.; Hsiao, B.S.; Amnuaypornsri, S.; Nimpaiboon, A.; Sakdapipanich, J. Entanglements and Networks to Strain-Induced Crystallization and Stress–Strain Relations in Natural Rubber and Synthetic Polyisoprene at Various Temperatures. Macromolecules 2013, 46, 5238–5248. [Google Scholar] [CrossRef]
- Ehabe, E.; Bonfils, F.; Aymard, C.; Akinlabi, A.K.; Sainte, B.J. Modelling of Mooney Viscosity Relaxation in Natural Rubber. Polym. Test. 2005, 24, 620–627. [Google Scholar] [CrossRef]
- Nimpaiboon, A.; Amnuaypornsri, S.; Sakdapipanich, J. Influence of Gel Content on the Physical Properties of Unfilled and Carbon Black Filled Natural Rubber Vulcanizates. Polym. Test. 2013, 32, 1135–1144. [Google Scholar] [CrossRef]
- Toki, S.; Hsiao, B.S.; Amnuaypornsri, S.; Sakdapipanich, J. New Insights into the Relationship between Network Structure and Strain-Induced Crystallization in Un-Vulcanized and Vulcanized Natural Rubber by Synchrotron X-ray Diffraction. Polymer 2009, 50, 2142–2148. [Google Scholar] [CrossRef]
- Tanaka, Y. Structural Characterization of Natural Polyisoprenes: Solve the Mystery of Natural Rubber Based on Structural Study. Rubber Chem. Technol. 2001, 74, 355–375. [Google Scholar] [CrossRef]
- Karino, T.; Ikeda, Y.; Yasuda, Y.; Kohjiya, S.; Shibayama, M.J.B. Nonuniformity in Natural Rubber as Revealed by Small-Angle Neutron Scattering, Small-Angle X-ray Scattering, and Atomic Force Microscopy. Biomacromolecules 2007, 8, 693–699. [Google Scholar] [CrossRef]
- Tanaka, Y.; Tarachiwin, L. Recent Advances in Structural Characterization of Natural Rubber. Rubber Chem. Technol. 2009, 82, 283–314. [Google Scholar] [CrossRef]
- Qin, X.; Wang, J.; Zhang, Y.; Wang, Z.; Li, S.; Zhao, S.; Tan, T.; Liu, J.; Zhang, L. Self-Assembly Strategy for Double Network Elastomer Nanocomposites with Ultralow Energy Consumption and Ultrahigh Wear Resistance. Adv. Funct. Mater. 2020, 30, 2003429. [Google Scholar] [CrossRef]
- Ilyas, M.; Haque, M.A.; Yue, Y.; Kurokawa, T.; Nakajima, T.; Nonoyama, T.; Gong, J.P. Water-Triggered Ductile–Brittle Transition of Anisotropic Lamellar Hydrogels and Effect of Confinement on Polymer Dynamics. Macromolecules 2017, 50, 8169–8177. [Google Scholar] [CrossRef]
- Zhang, Q.; Niu, S.; Wang, L.; Lopez, J.; Chen, S.; Cai, Y.; Du, R.; Liu, Y.; Lai, J.-C.; Liu, L.; et al. An Elastic Autonomous Self-Healing Capacitive Sensor Based on a Dynamic Dual Crosslinked Chemical System. Adv. Mater. 2018, 30, 1801435. [Google Scholar] [CrossRef]
- Lessard, J.J.; Scheutz, G.M.; Sung, S.H.; Lantz, K.A.; Epps, T.H., 3rd; Sumerlin, B.S. Block Copolymer Vitrimers. J. Am. Chem. Soc. 2020, 142, 283–289. [Google Scholar] [CrossRef]
- Ji, F.; Li, J.; Weng, Y.; Ren, J. Synthesis of Pla-Based Thermoplastic Elastomer and Study on Preparation and Properties of Pla-Based Shape Memory Polymers. Mater. Res. Express 2019, 7, 015315. [Google Scholar] [CrossRef]
- Moyassari, A.; Gkourmpis, T.; Hedenqvist, M.S.; Gedde, U.W. Molecular Dynamics Simulations of Short-Chain Branched Bimodal Polyethylene: Topological Characteristics and Mechanical Behavior. Macromolecules 2019, 52, 807–818. [Google Scholar] [CrossRef] [Green Version]
- Scavuzzo, J.; Tomita, S.; Cheng, S.; Liu, H.; Gao, M.; Kennedy, J.P.; Sakurai, S.; Cheng, S.Z.D.; Jia, L. Supramolecular Elastomers: Self-Assembling Star-Blocks of Soft Polyisobutylene and Hard Oligo(Β-Alanine) Segments. Macromolecules 2015, 48, 1077–1086. [Google Scholar] [CrossRef]
- Müller, M.; Fischer, E.W.; Kremer, F.; Seidel, U.; Stadler, R. The Molecular Dynamics of Thermoreversible Networks as Studied by Broadband Dielectric Spectroscopy. Colloid. Polym. Sci. 1995, 273, 38–46. [Google Scholar] [CrossRef]
- Nawamawat, K.; Sakdapipanich, J.T.; Ho, C.C. Effect of Deproteinized Methods on the Proteins and Properties of Natural Rubber Latex During Storage. Macromol. Symp. 2010, 288, 95–103. [Google Scholar] [CrossRef]
- Cerveny, S.; Zinck, P.; Terrier, M.; Arrese-Igor, S.; Alegría, A.; Colmenero, J. Dynamics of Amorphous and Semicrystalline 1,4-Trans-Poly(Isoprene) by Dielectric Spectroscopy. Macromolecules 2008, 41, 8669–8676. [Google Scholar] [CrossRef]
- Tang, M.; Zhang, R.; Li, S.; Zeng, J.; Luo, M.; Xu, Y.X.; Huang, G. Towards a Supertough Thermoplastic Polyisoprene Elastomer Based on a Biomimic Strategy. Angew. Chem. Int. Ed. 2018, 57, 15836–15840. [Google Scholar] [CrossRef]
- Loiseau, N.; Gomis, J.-M.; Santolini, J.; Delaforge, M.; André, F. Predicting the Conformational States of Cyclic Tetrapeptides. Biopolymers 2003, 69, 363–385. [Google Scholar] [CrossRef]
- Havriliak, S.; Negami, S. A Complex Plane Representation of Dielectric and Mechanical Relaxation Processes in Some Polymers. Polymer 1967, 8, 161–210. [Google Scholar] [CrossRef]
- Díaz-Calleja, R. Comment on the Maximum in the Loss Permittivity for the Havriliak−Negami Equation. Macromolecules 2000, 33, 8924. [Google Scholar] [CrossRef]
- Liu, J.; Wu, S.; Tang, Z.; Lin, T.; Guo, B.; Huang, G. New Evidence Disclosed for Networking in Natural Rubber by Dielectric Relaxation Spectroscopy. Soft Matter 2015, 11, 2290–2299. [Google Scholar] [CrossRef]
- Ortizserna, P.; Díazcalleja, R.; Sanchis, M.J.; Floudas, G.; Nunes, R.C.; Martins, A.F.; Visconte, L.L. Dynamics of Natural Rubber as a Function of Frequency, Temperature, and Pressure. A Dielectric Spectroscopy Investigation. Macromolecules 2010, 43, 5094–5102. [Google Scholar] [CrossRef]
- Tang, Z.; Zhang, L.; Feng, W.; Guo, B.; Liu, F.; Jia, D. Rational Design of Graphene Surface Chemistry for High-Performance Rubber/Graphene Composites. Macromolecules 2015, 47, 8663–8673. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. Gromacs: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van, d.S.D.; Lindahl, E.; Hess, B.; Kutzner, C.; van, d.S.D.; Lindahl, E.; Hess, B.; Kutzner, C.; et al. Gromacs 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. Gromacs 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. Gromacs: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; van der Spoel, D. Gromacs 3.0: A Package for Molecular Simulation and Trajectory Analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the Opls All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Rizzo, R.C.; Jorgensen, W.L. Opls All-Atom Model for Amines: Resolution of the Amine Hydration Problem. J. Am. Chem. Soc. 1999, 121, 4827–4836. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the Opls-Aa Force Field for Proteins Via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B. 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Price, M.L.P.; Ostrovsky, D.; Jorgensen, W.L. Gas-Phase and Liquid-State Properties of Esters, Nitriles, and Nitro Compounds with the Opls-Aa Force Field. J. Comput. Chem. 2001, 22, 1340–1352. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Chen, D.; Shao, H.; Yao, W.; Huang, B. Fourier Transform Infrared Spectral Analysis of Polyisoprene of a Different Microstructure. Int. J. Polymer. Sci. 2013, 2013, 7866–7873. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Krishnaji, S.; Tokareva, O.R.; Kaplan, D.; Cebe, P. Influence of Water on Protein Transitions: Morphology and Secondary Structure. Macromolecules 2014, 47, 8107–8114. [Google Scholar] [CrossRef]
- Fawcett, J.K.; Camerman, N.; Camerman, A. The Structure of the Tripeptide L -Alanyl- L -Alanyl- L -Alanine. Acta Crystallogr. Sect. B: Struct. Sci. 2010, 31, 658–665. [Google Scholar] [CrossRef]
- Asakura, T.; Yazawa, K.; Horiguchi, K.; Suzuki, F.; Nishiyama, Y.; Nishimura, K.; Kaji, H. Difference in the Structures of Alanine Tri- and Tetra-Peptides with Antiparallel Β-Sheet Assessed by X-ray Diffraction, Solid-State Nmr and Chemical Shift Calculations by Gipaw. Biopolymers 2014, 101, 13–20. [Google Scholar] [CrossRef]
- Takusagawa, F.; Fumagalli, A.; Koetzle, T.F.; Shore, S.G.; Schmitkons, T.; Fratini, A.V.; Morse, K.W.; Wei, C.-Y.; Bau, R. Neutron and X-ray Diffraction Studies of Tris(Methyldiphenylphosphine)[Tetrahydroborato(1-)]Copper, Cu[P(C6h5)2ch3]3(Bh4). The First Accurate Characterization of an Unsupported Metal-Hydrogen-Boron Bridge Bond. J. Am. Chem. Soc. 1981, 103, 5165–5171. [Google Scholar] [CrossRef]
- Ratajczak-Sitarz, M.; Katrusiak, A. Coupling of Molecular Orientation with the Hydrogen-Bond Dimensions and H-Sites in Carboxylic Acids. J. Mol. Struct. 2011, 995, 29–34. [Google Scholar] [CrossRef]
- Kona, F.R.; Buac, D.; A, M.B. Molecular Dynamics in Linear and Multi-Armed Star Polymers of Cis-Polyisoprene as Studied by Broadband Dielectric Spectroscopy. Macromolecules 1991, 23, 1826–1830. [Google Scholar]
- Boese, D.; Kremer, F.; Fetters, L.J. Further Investigation on the Molecular Dynamics in Linear and Multiarmed Star Polymers of Cis -Polyisoprene Studied by Dielectric Spectroscopy. Polymer 1990, 31, 1831–1837. [Google Scholar] [CrossRef]
- Adachi, K.; Kotaka, T. Influence of Entanglement on the Dielectric Normal Mode Process of Cis-Polyisoprene. Macromolecules 1984, 17, 120–122. [Google Scholar] [CrossRef]
- Adachi, K.; Kotaka, T. Dielectric Normal Mode Process in Undiluted Cis-Polyisoprene. Macromolecules 1985, 18, 466–472. [Google Scholar] [CrossRef]
- Lewis, C.L.; Stewart, K.; Anthamatten, M. The Influence of Hydrogen Bonding Side-Groups on Viscoelastic Behavior of Linear and Network Polymers. Macromolecules 2014, 47, 729–740. [Google Scholar] [CrossRef]
- Mijović, J.; Ristić, S.; Kenny, J. Dynamics of Six Generations of Pamam Dendrimers as Studied by Dielectric Relaxation Spectroscopy. Macromolecules 2007, 40, 5212–5221. [Google Scholar] [CrossRef]
- Boese, D.; Kremer, F. Molecular Dynamics in Bulk Cis-Polyisoprene as Studied by Dielectric Spectroscopy. Macromolecules 1990, 23, 829–835. [Google Scholar] [CrossRef]
- Schönhals, A.; Kremer, F.; Schlosser, E. Scaling of the A Relaxation in Low-Molecular Weight Glass-Forming Liquids and Polymers. Phys. Rev. Lett. 1991, 67, 999–1002. [Google Scholar] [CrossRef]
- Asakura, T.; Nishimura, A.; Aoki, A.; Naito, A. Packing Structure of Antiparallel Β-Sheet Polyalanine Region in a Sequential Model Peptide of Nephila Clavipes Dragline Silk Studied Using 13c Solid-State Nmr and Md Simulation. Biomacromolecules 2019, 20, 3884–3894. [Google Scholar] [CrossRef]
- Fawcett, J.K.; Camerman, N.; Camerman, A. The Structure of the Tripeptide L-Alanyl-L-Alanyl-L-Alanine. Acta Cryst. B 1975, 31, 658–665. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-C.; Zhang, R.; Li, S.; Huang, G.; Tang, M.; Xu, Y.-X. Influence of Oligopeptide Length and Distribution on Polyisoprene Properties. Polymers 2021, 13, 4408. https://doi.org/10.3390/polym13244408
Wang C-C, Zhang R, Li S, Huang G, Tang M, Xu Y-X. Influence of Oligopeptide Length and Distribution on Polyisoprene Properties. Polymers. 2021; 13(24):4408. https://doi.org/10.3390/polym13244408
Chicago/Turabian StyleWang, Chang-Cheng, Rong Zhang, Shiqi Li, Guangsu Huang, Maozhu Tang, and Yun-Xiang Xu. 2021. "Influence of Oligopeptide Length and Distribution on Polyisoprene Properties" Polymers 13, no. 24: 4408. https://doi.org/10.3390/polym13244408
APA StyleWang, C.-C., Zhang, R., Li, S., Huang, G., Tang, M., & Xu, Y.-X. (2021). Influence of Oligopeptide Length and Distribution on Polyisoprene Properties. Polymers, 13(24), 4408. https://doi.org/10.3390/polym13244408