
Effect of Ultra-High-Molecular-Weight Molecular Chains on the Morphology, Crystallization, and Mechanical Properties of Polypropylene

 , ,
, ,
Abstract
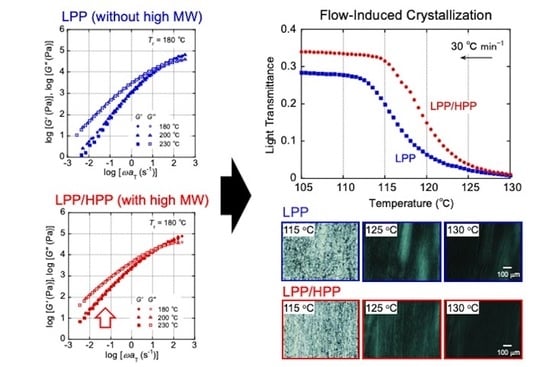
:
1. Introduction
2. Experimental
2.1. Materials
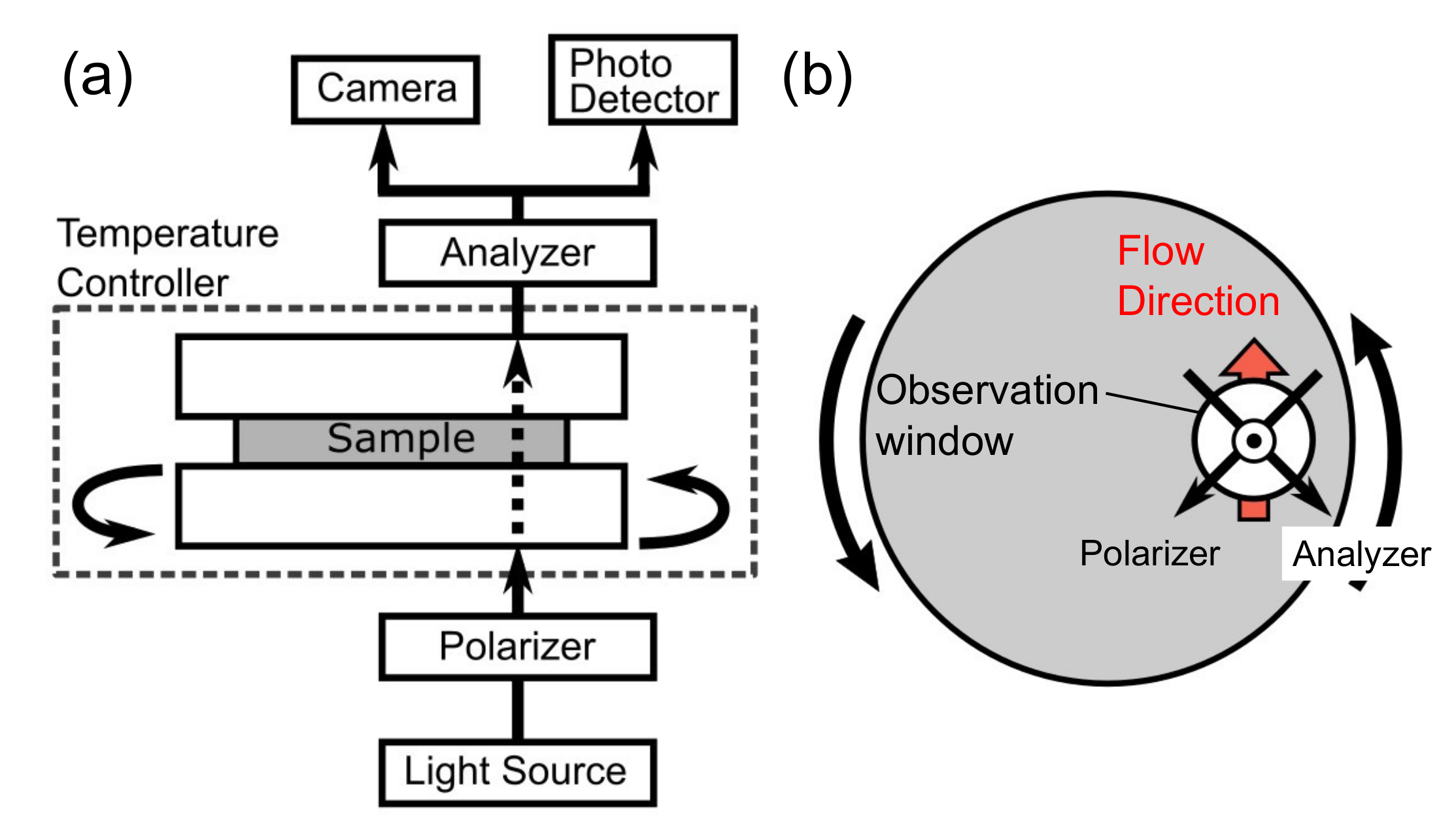
2.2. Measurements
3. Results and Discussion
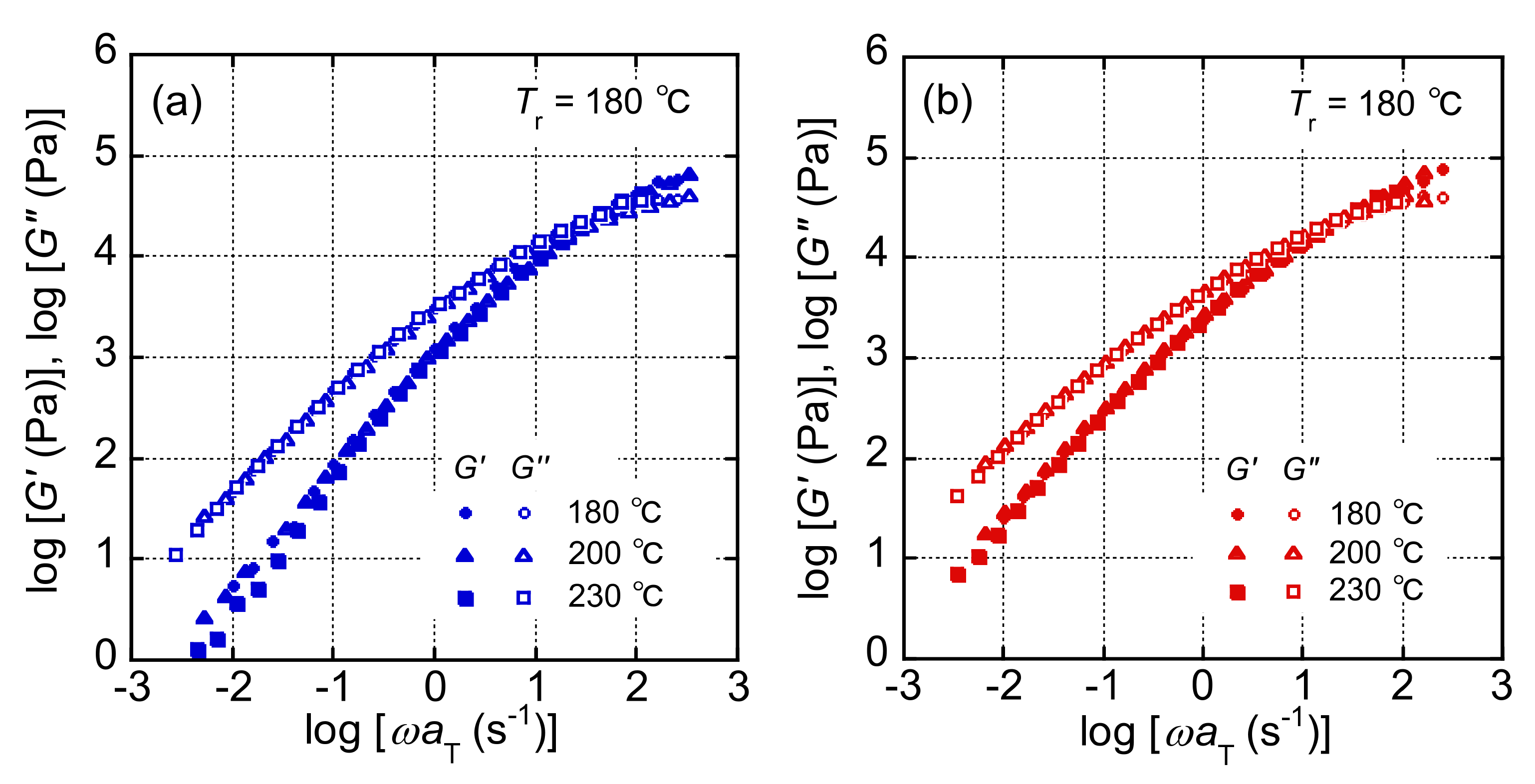
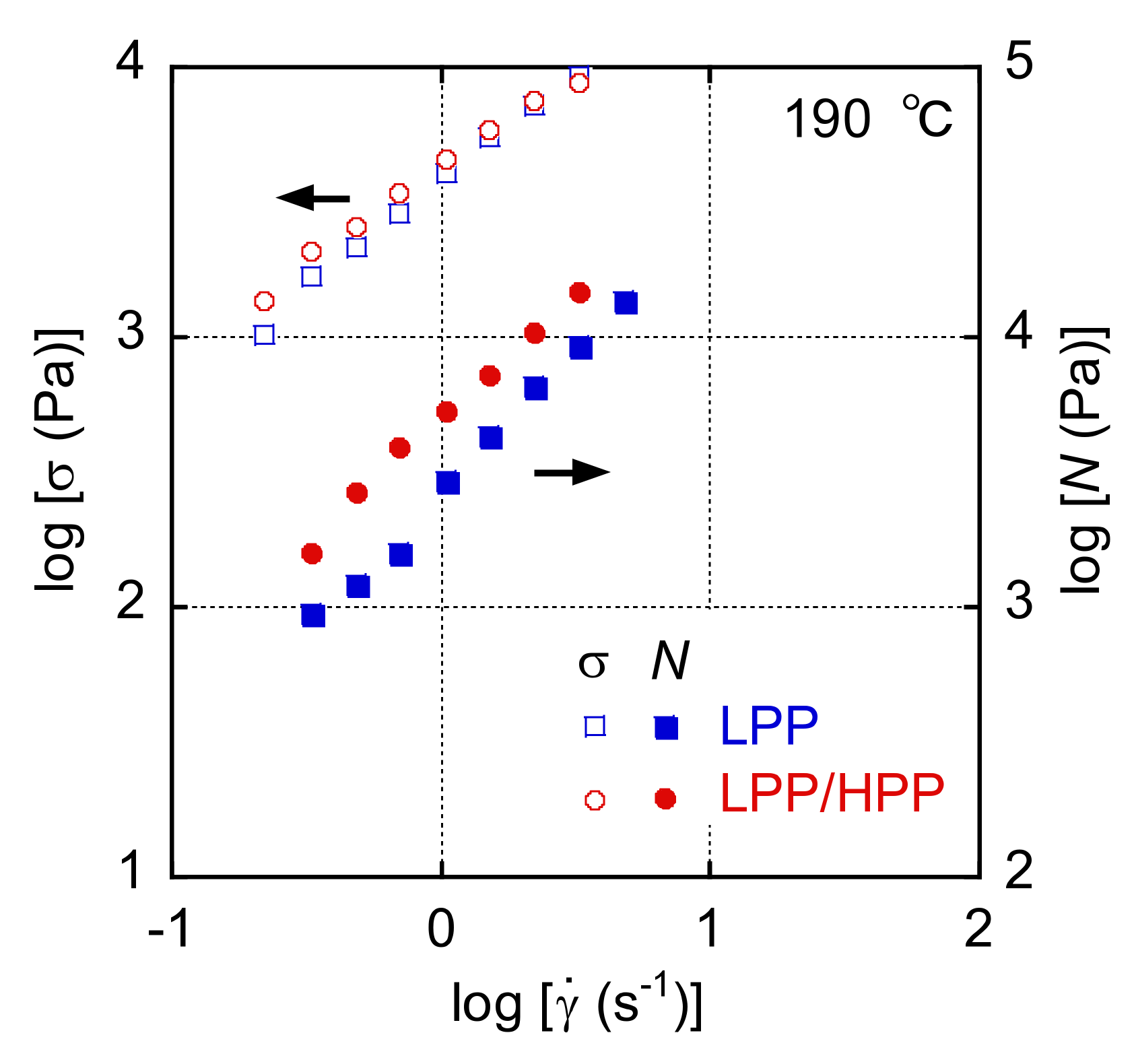
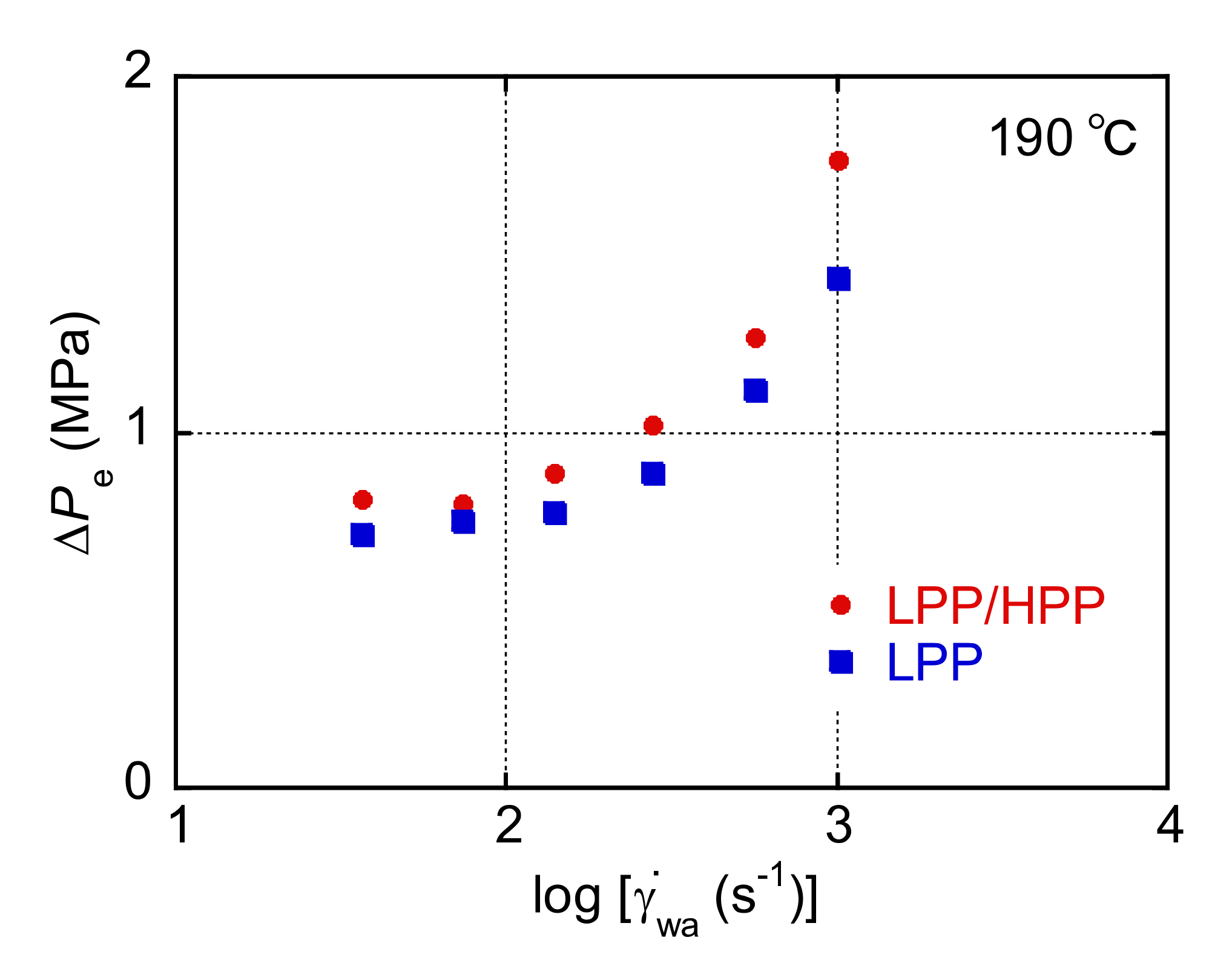
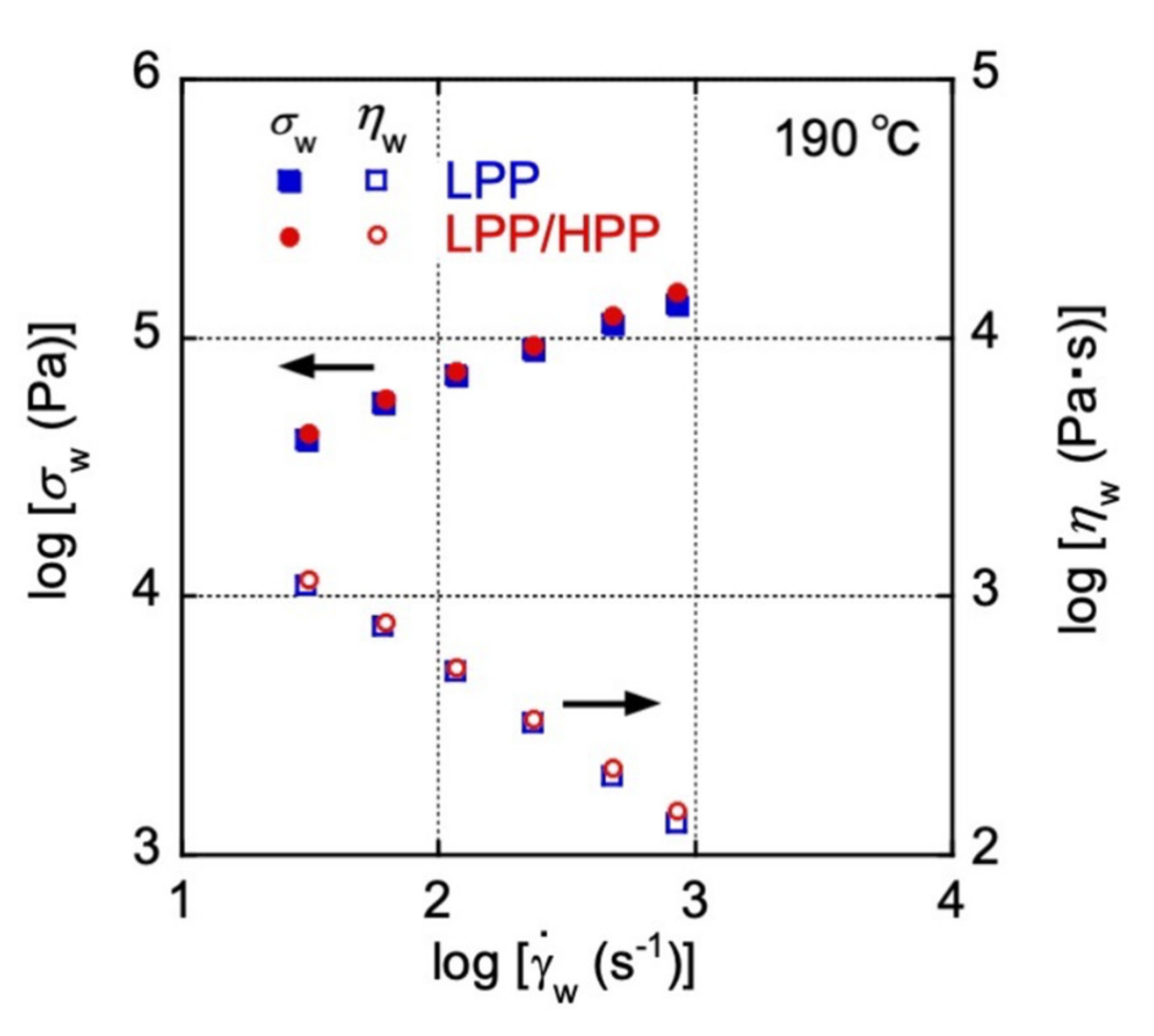
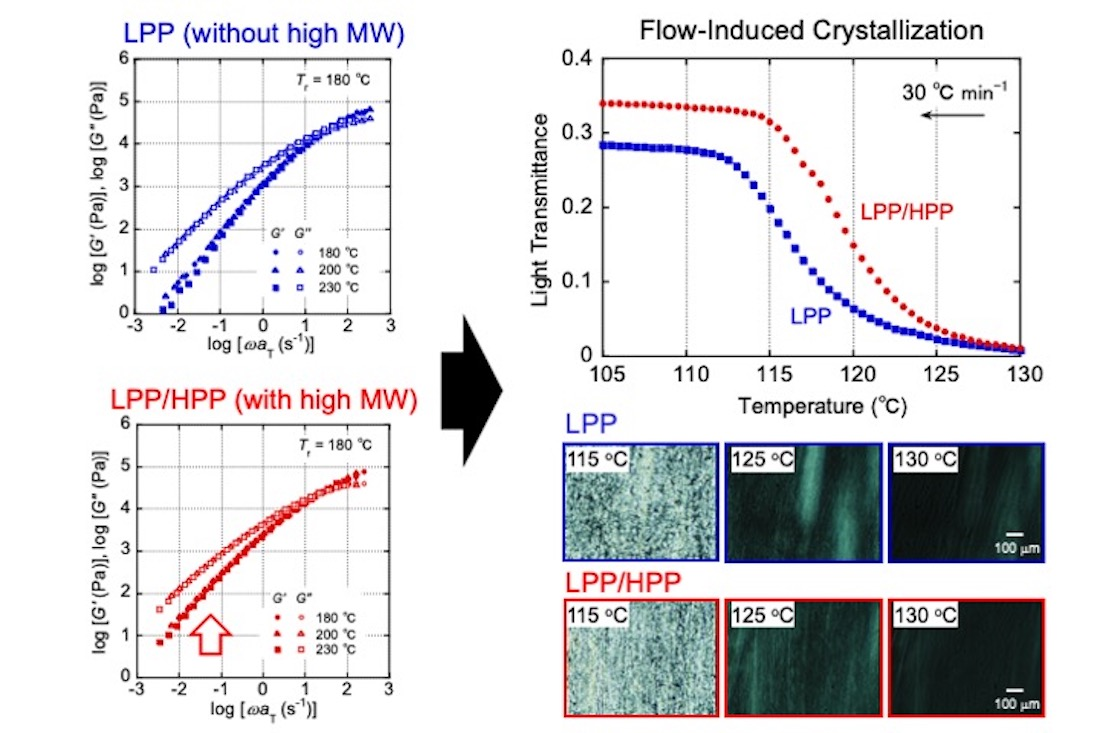
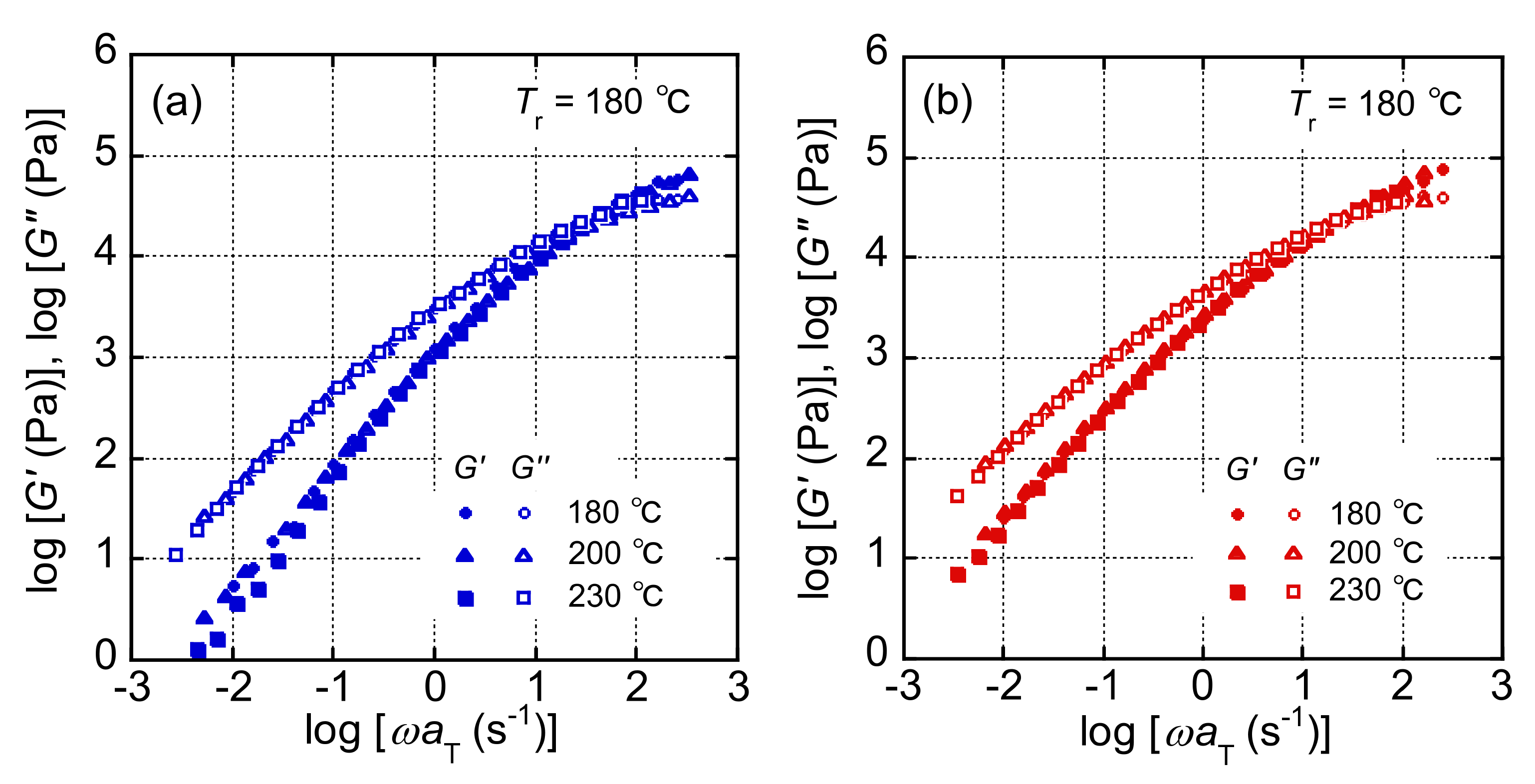
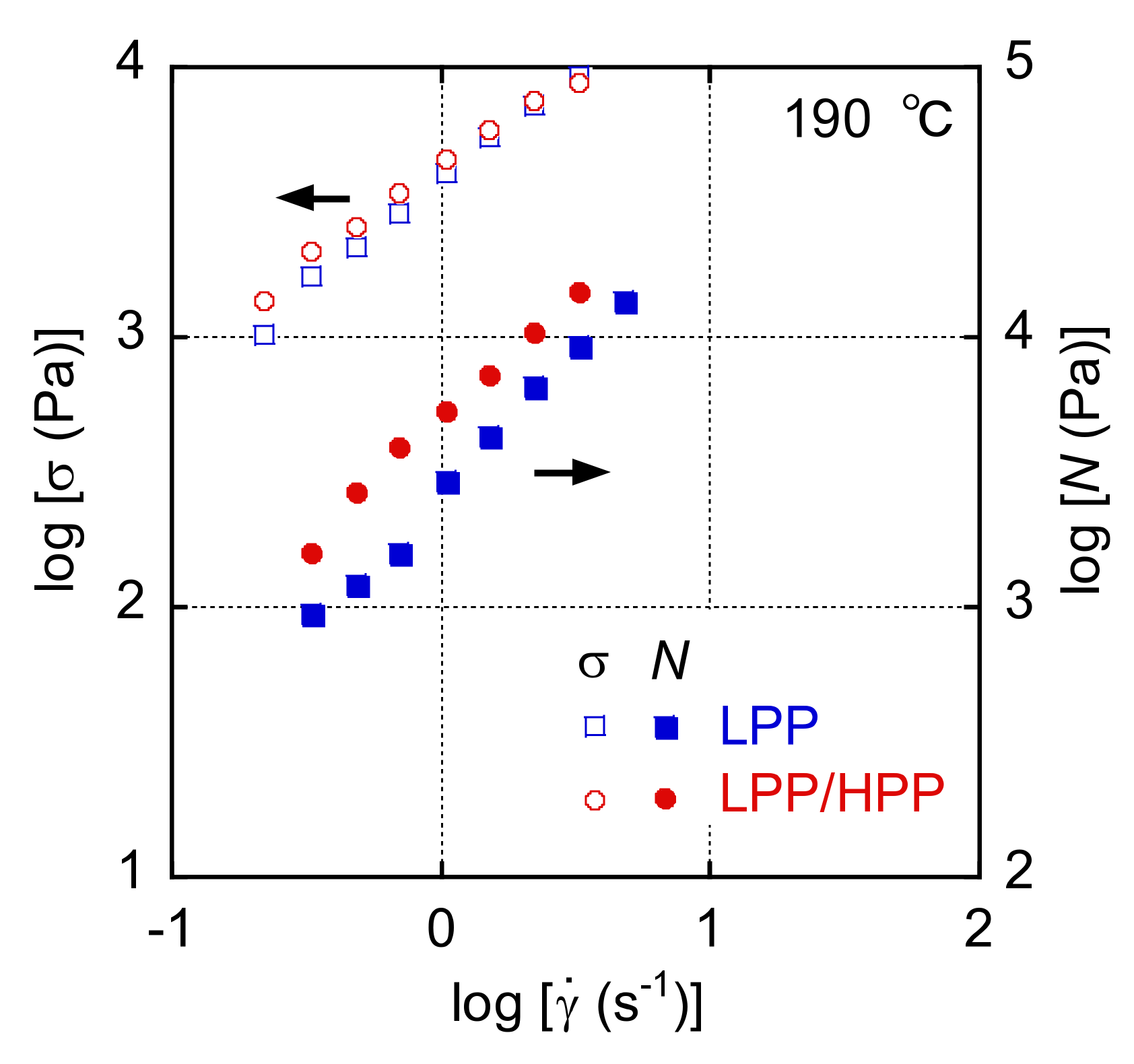
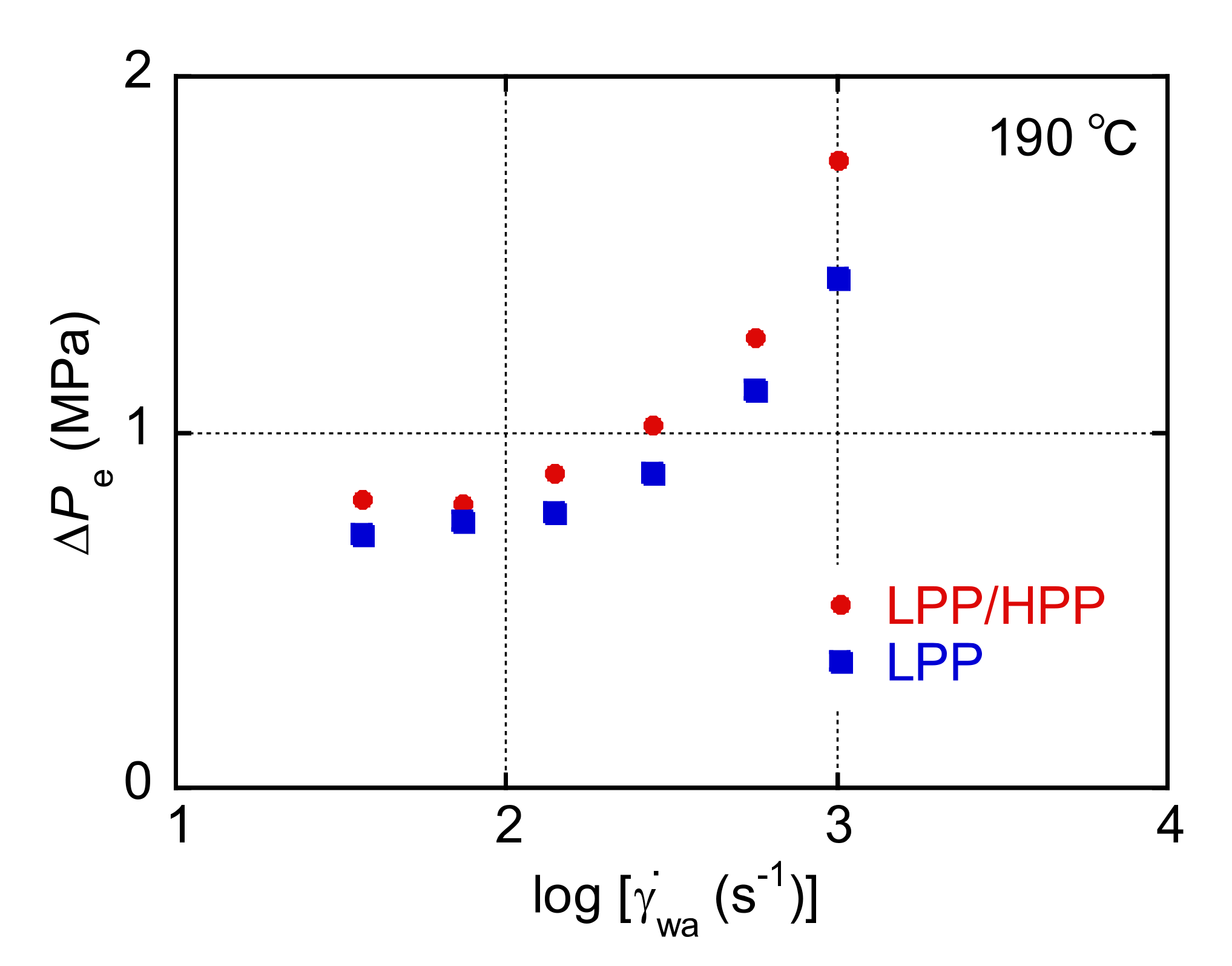
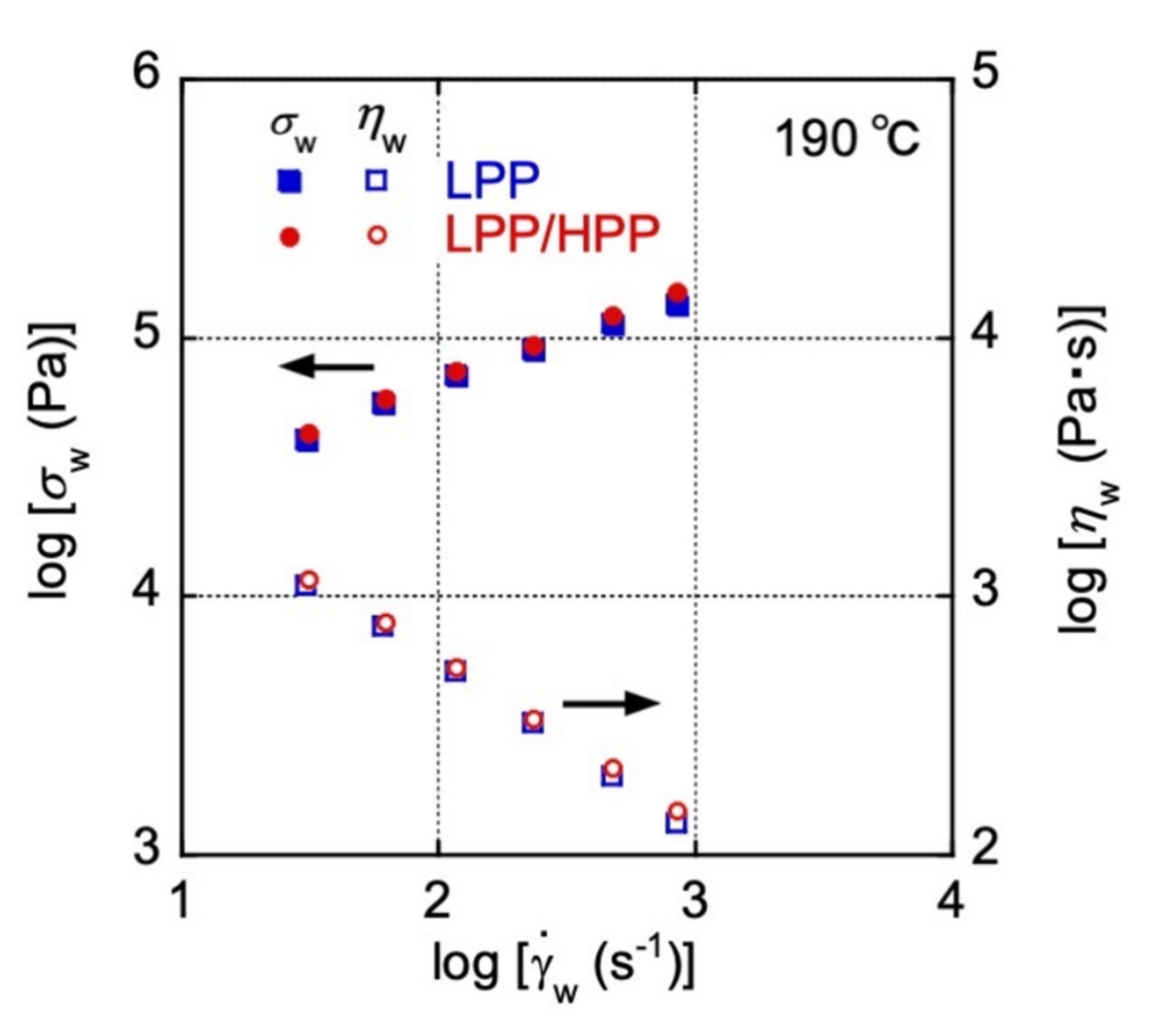
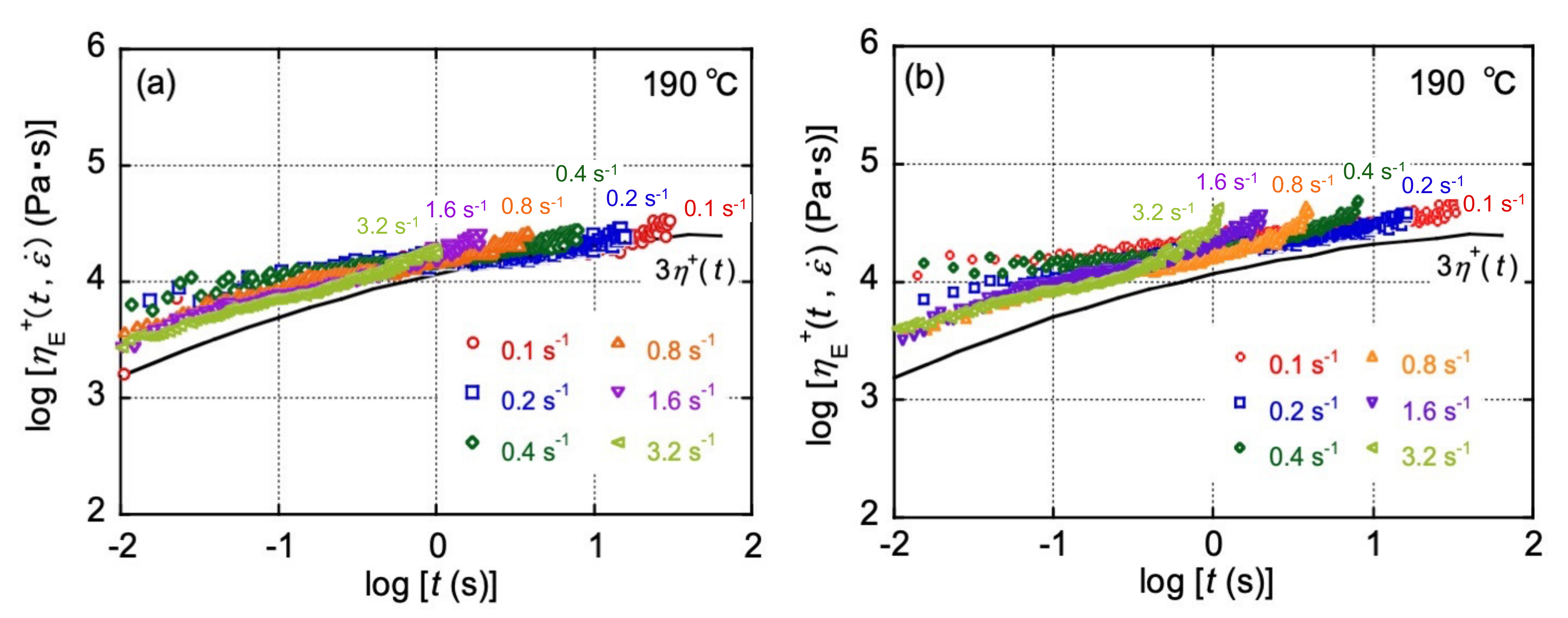
3.1. Rheological Properties
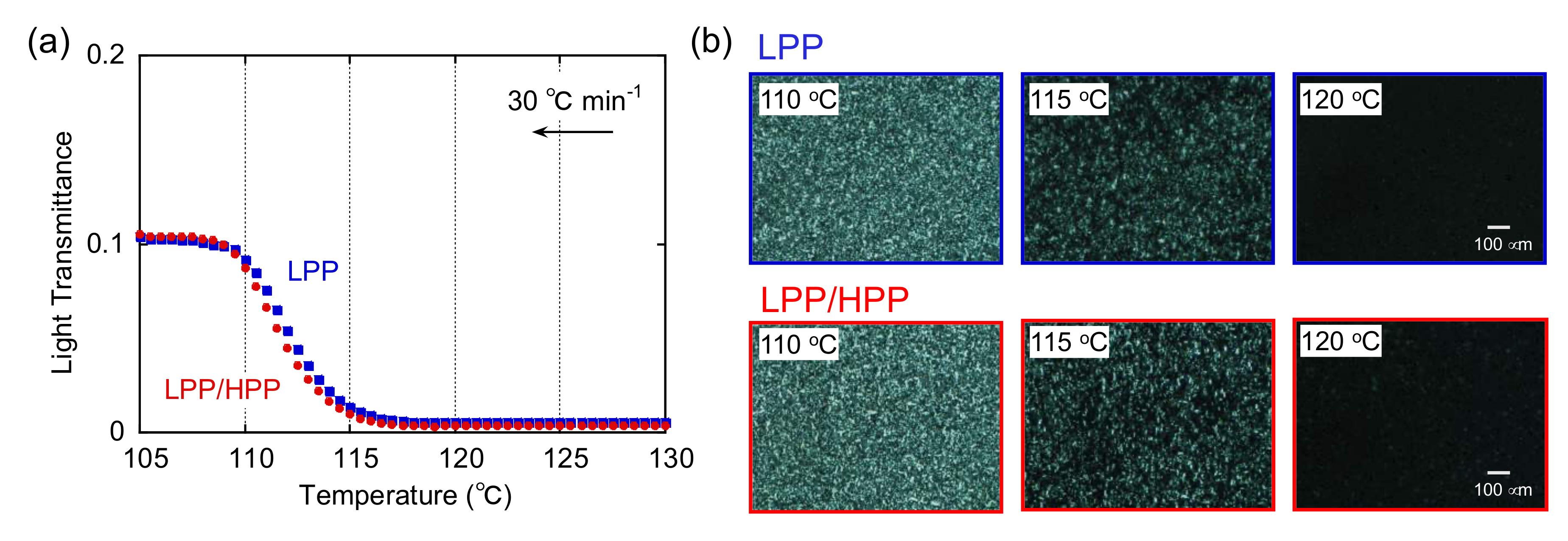
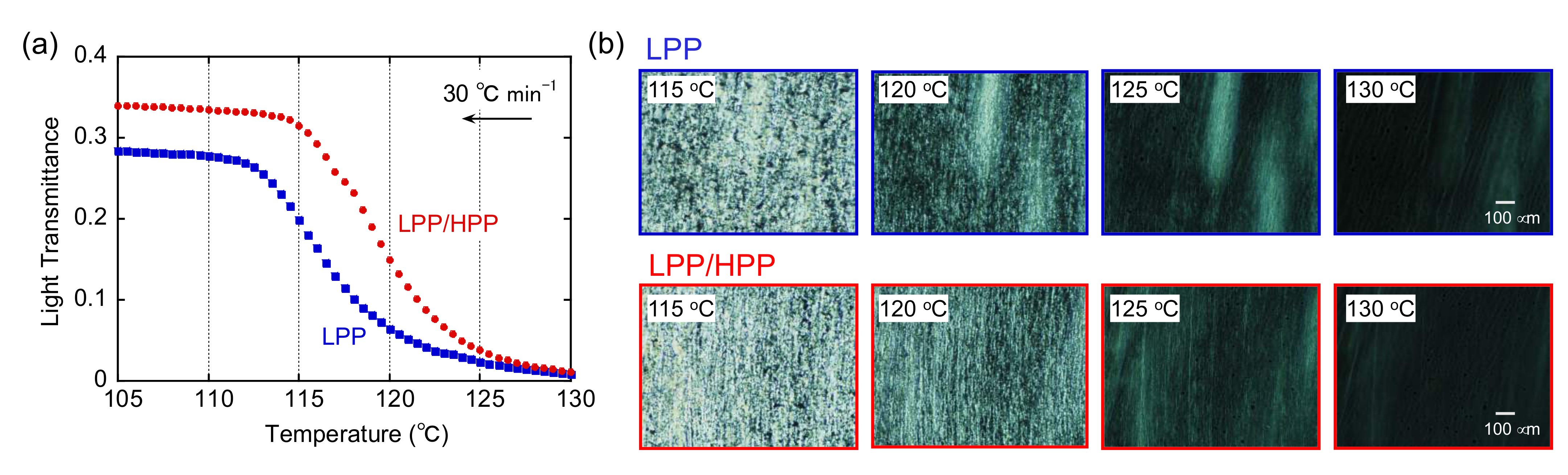
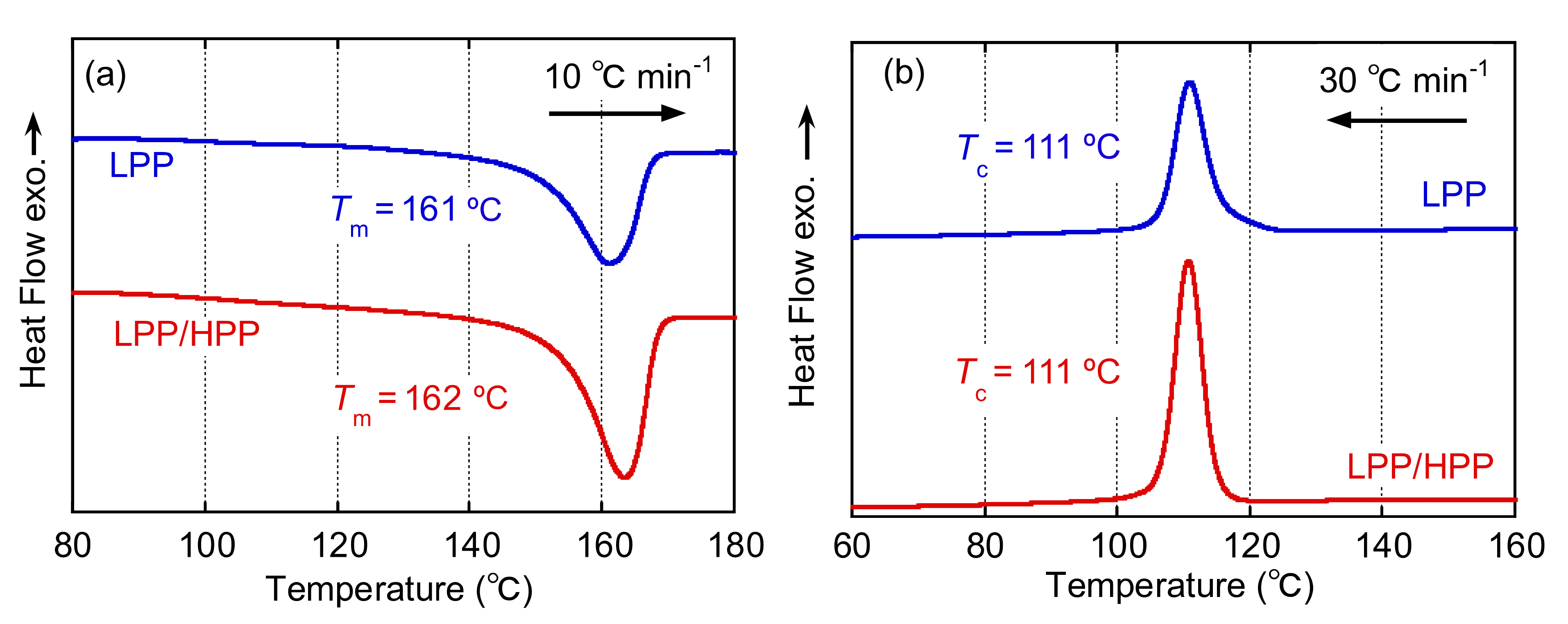
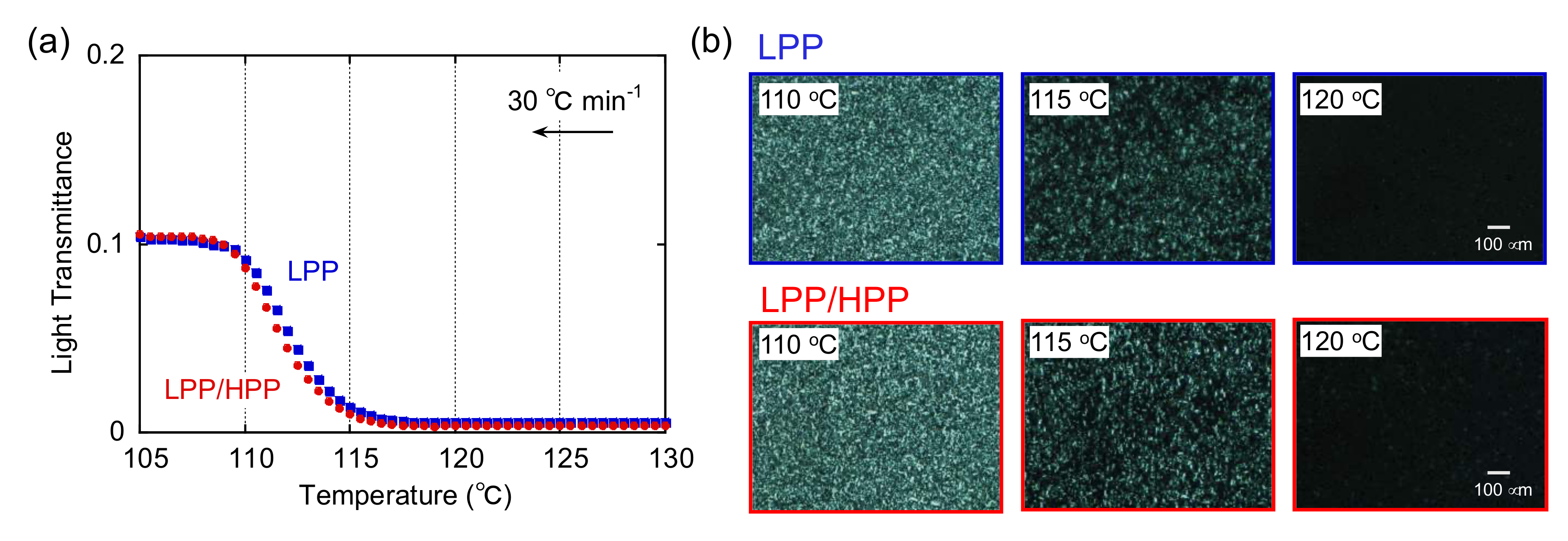
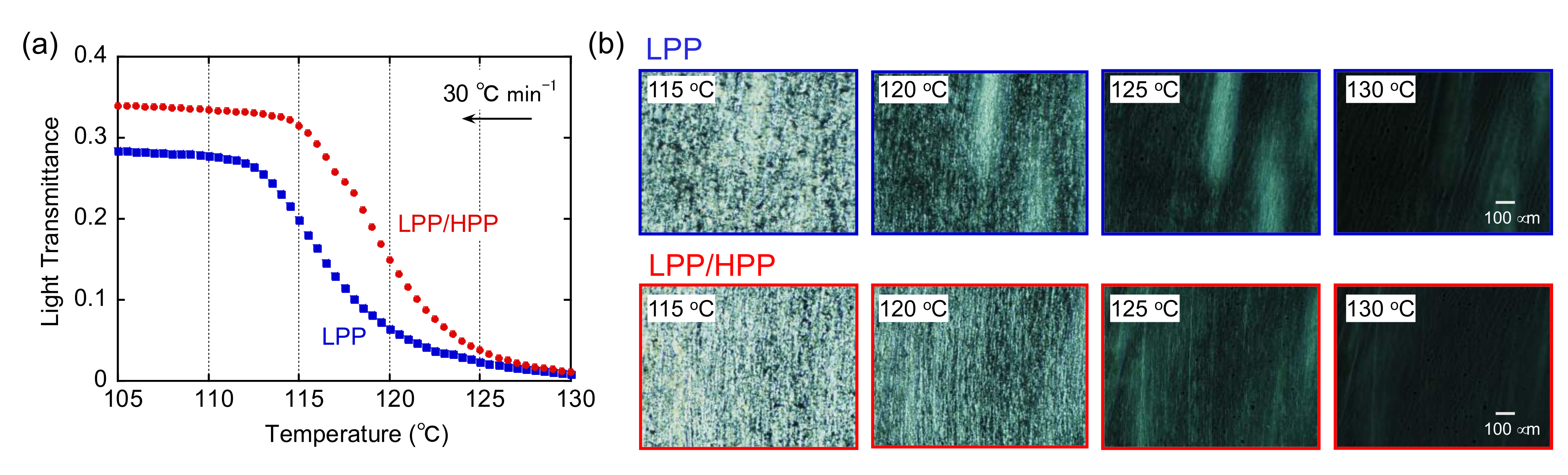
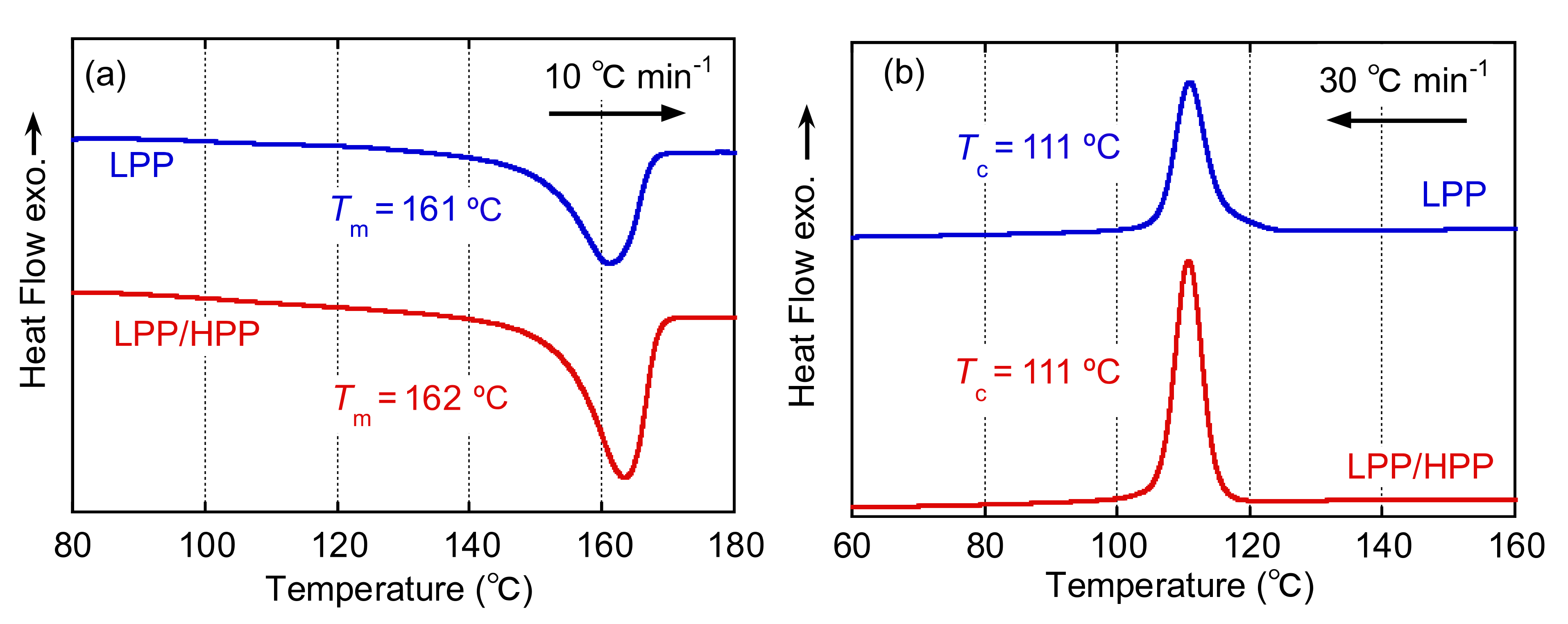
3.2. Crystallization Behavior
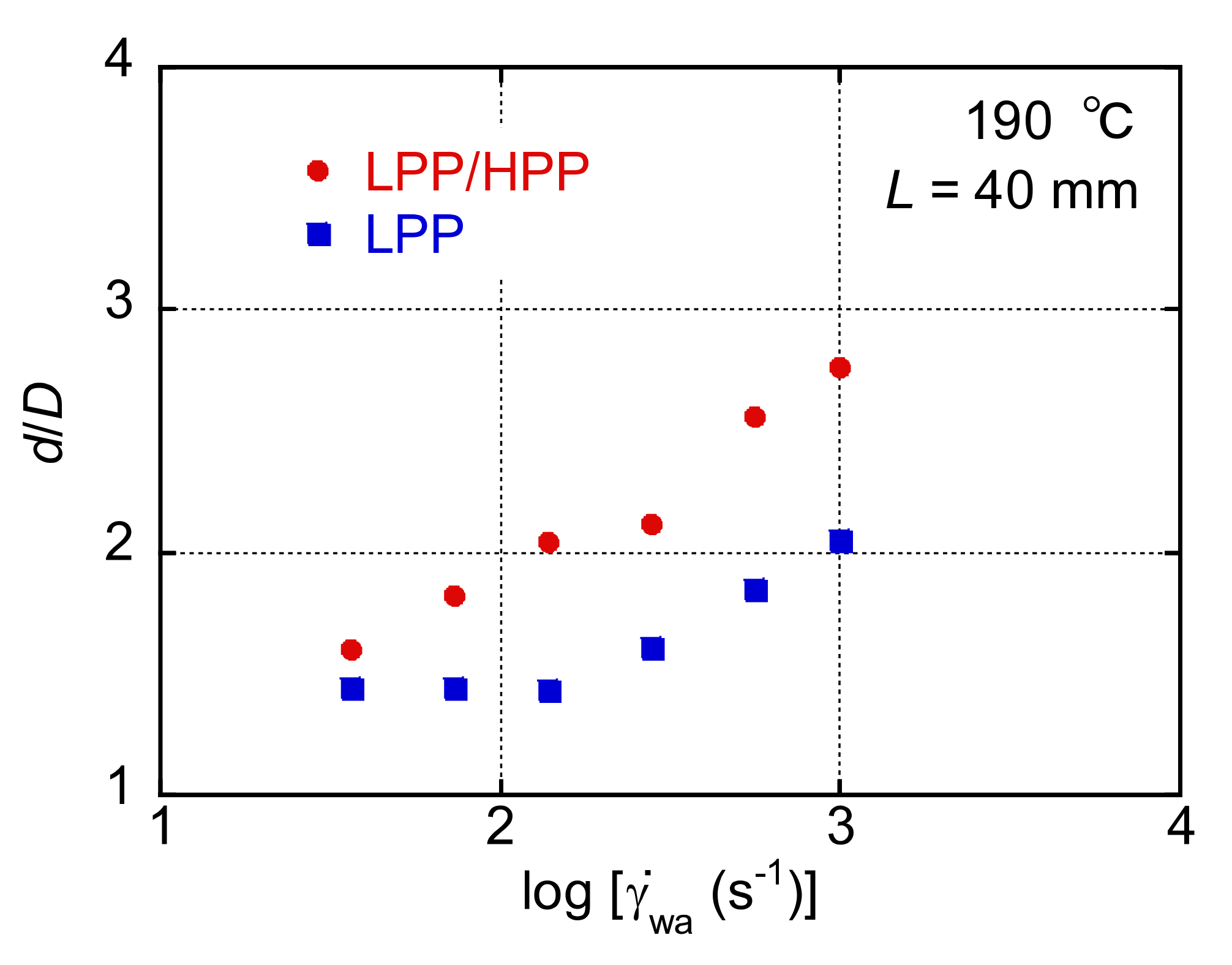
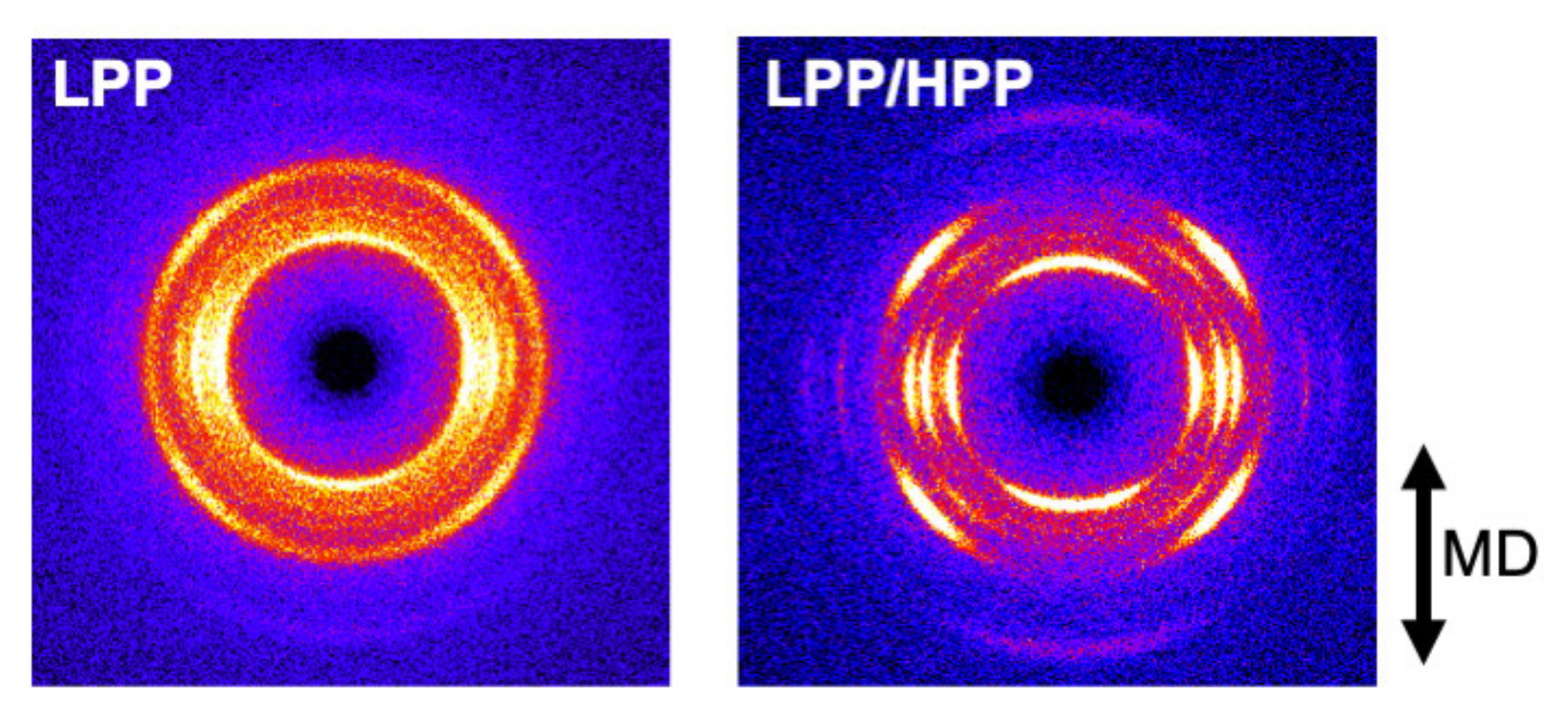
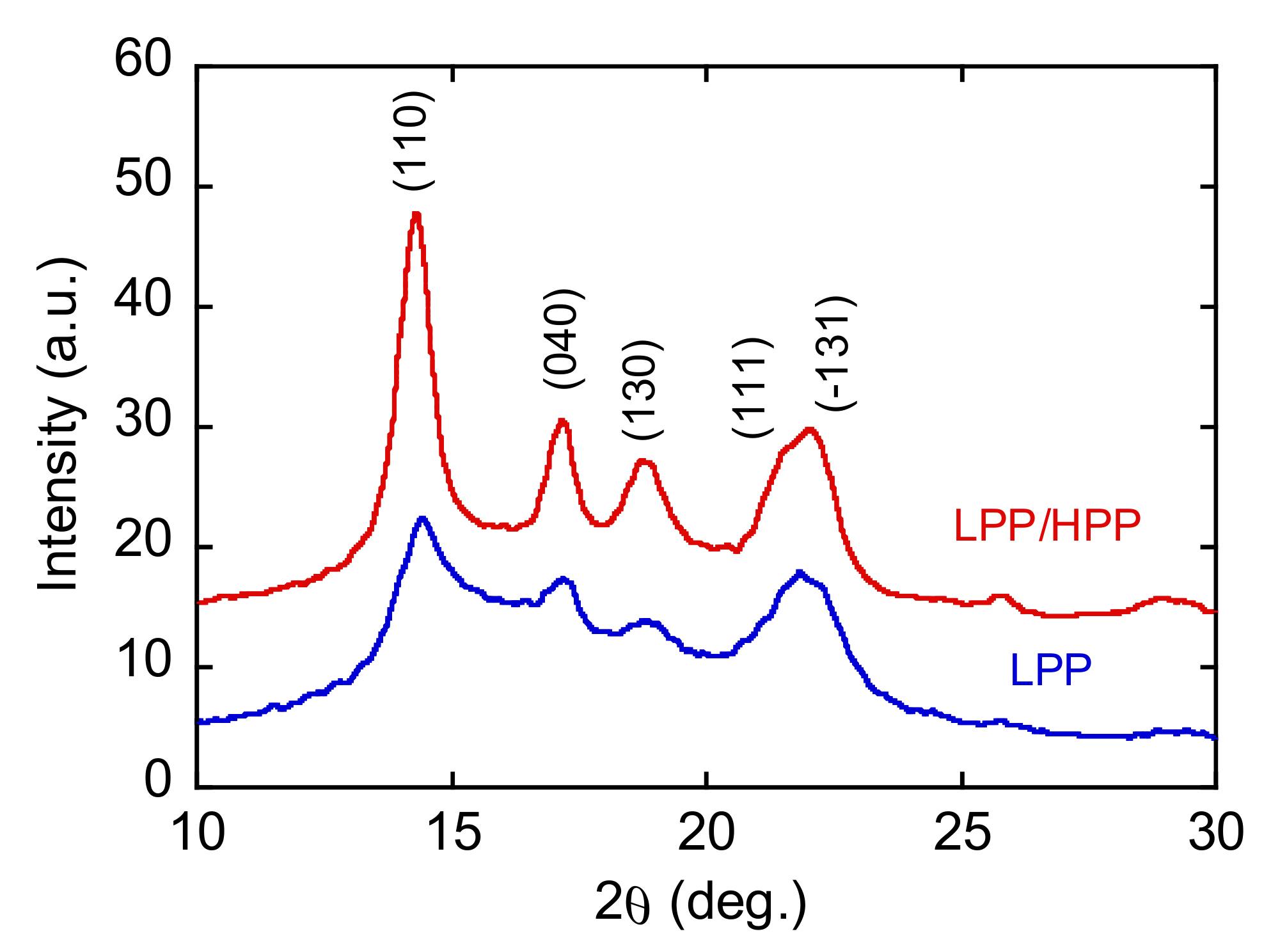
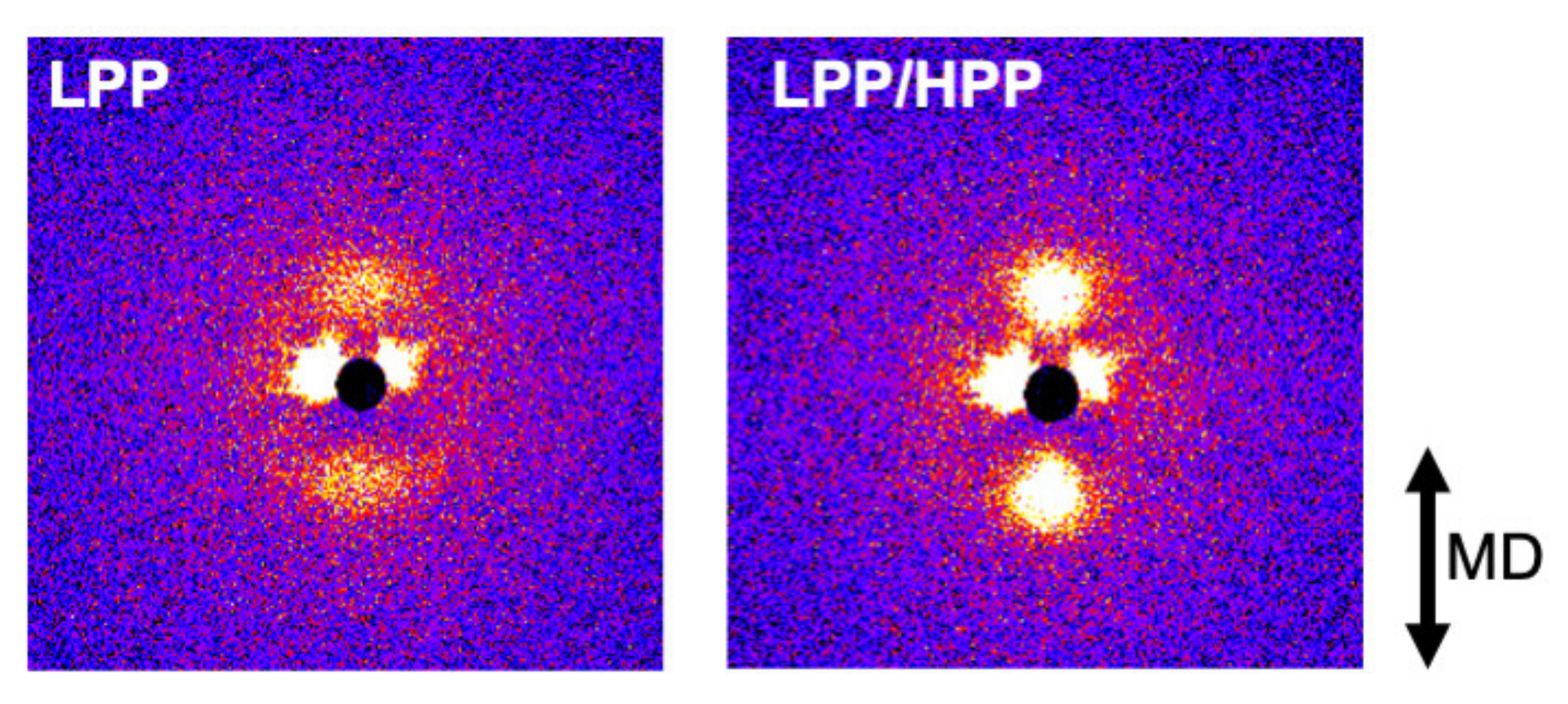
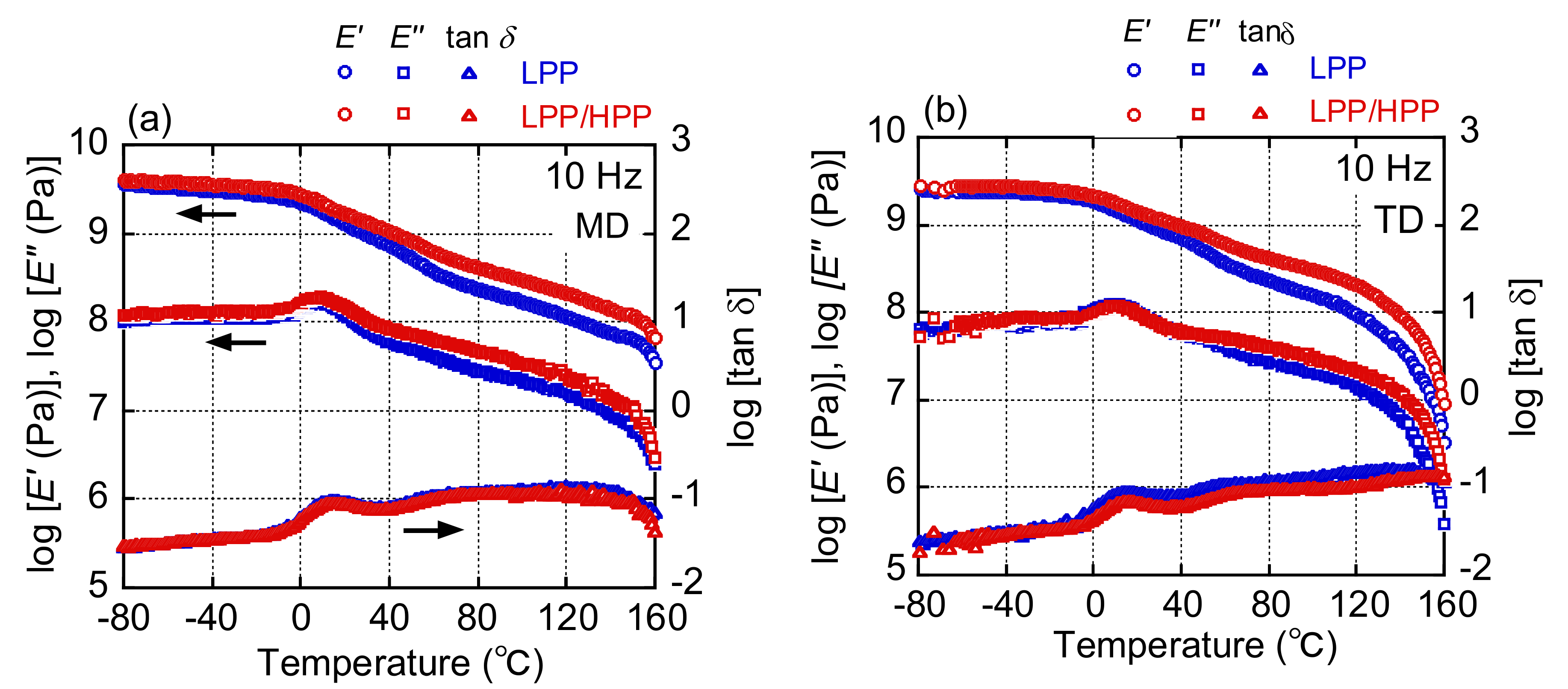
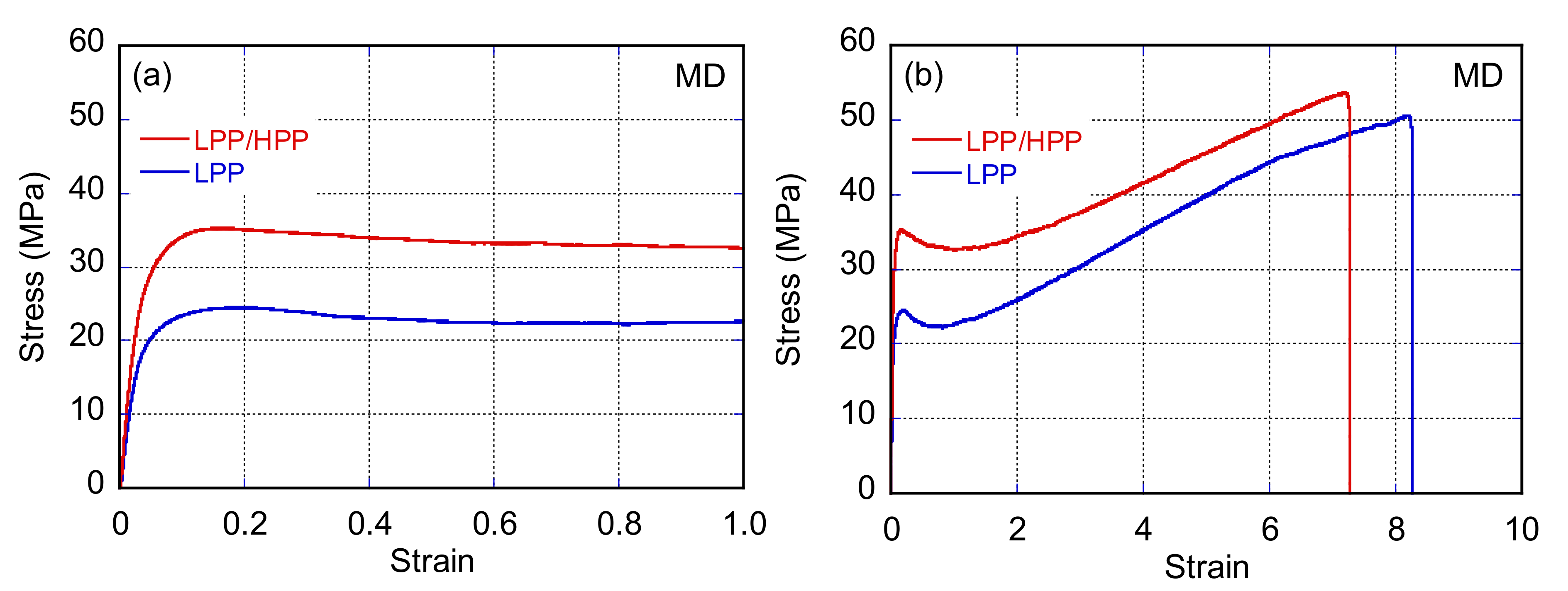
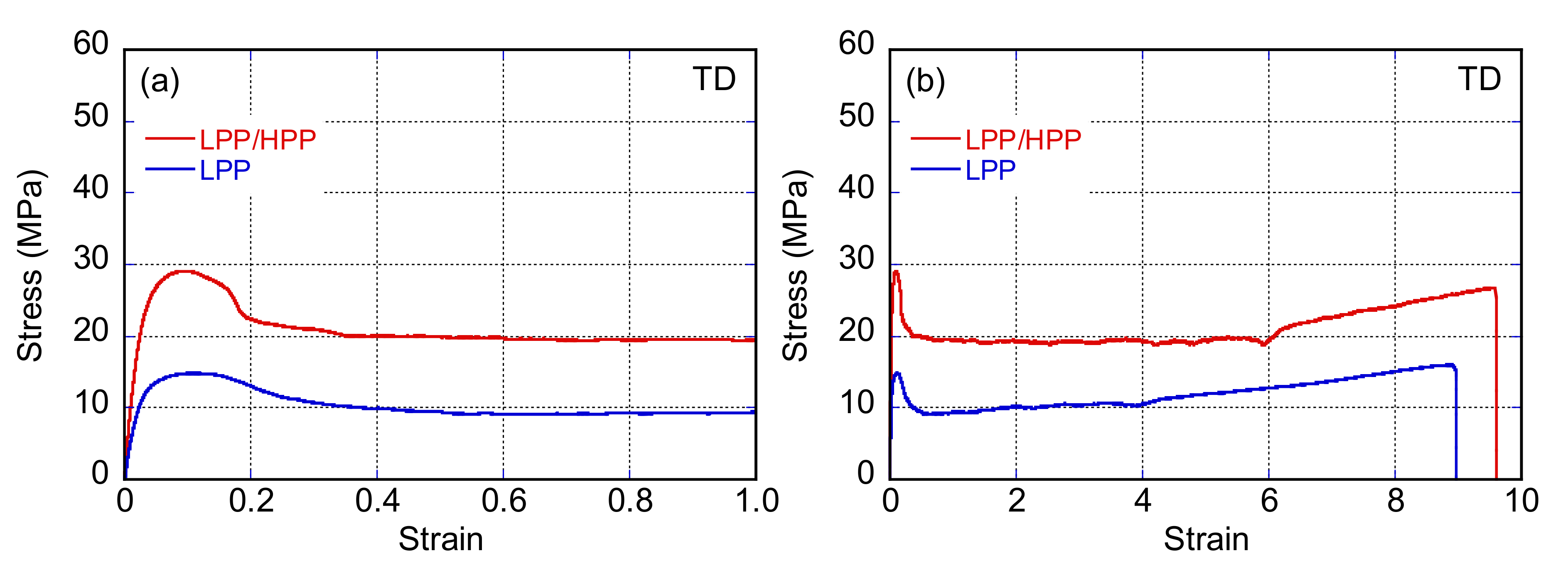
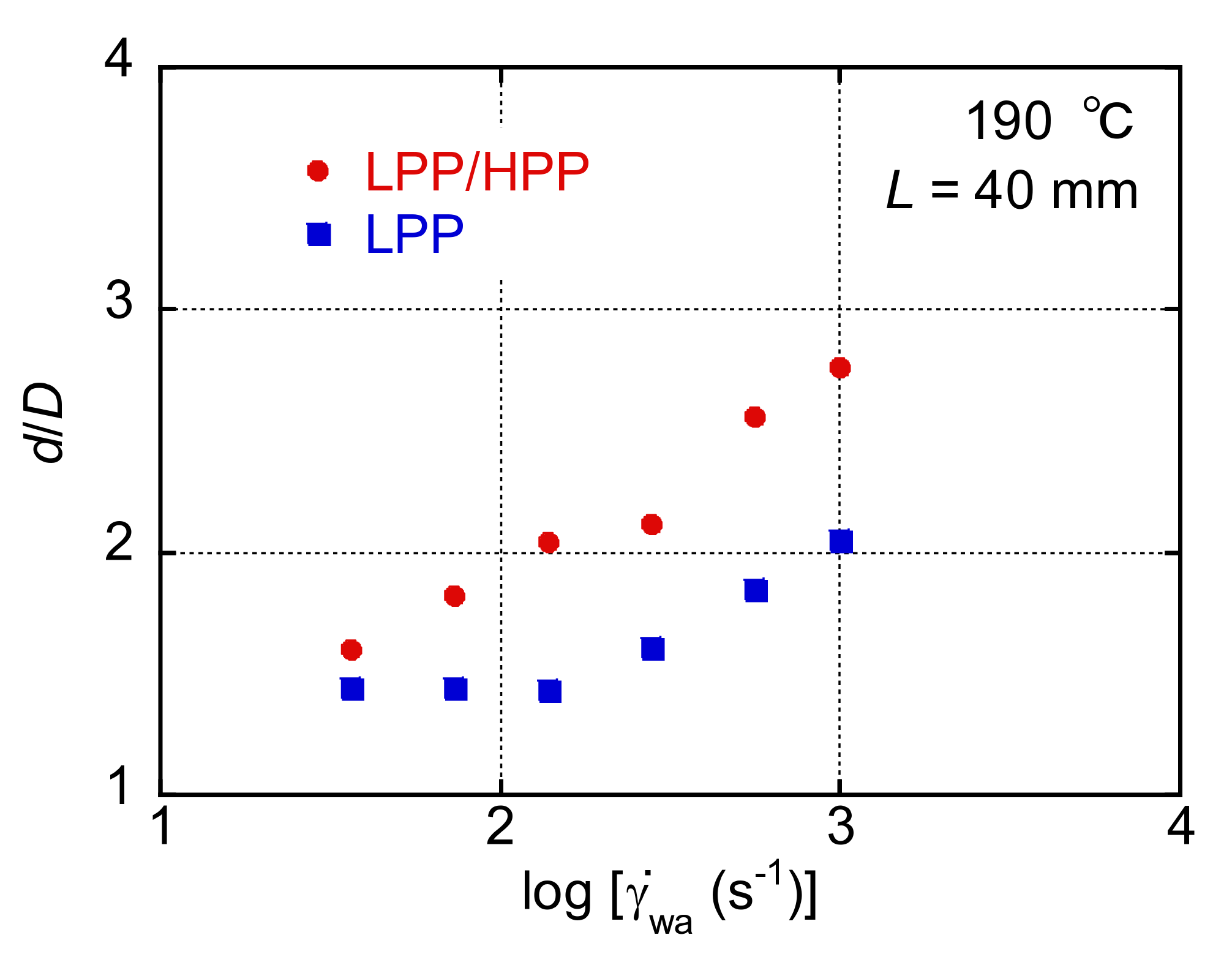
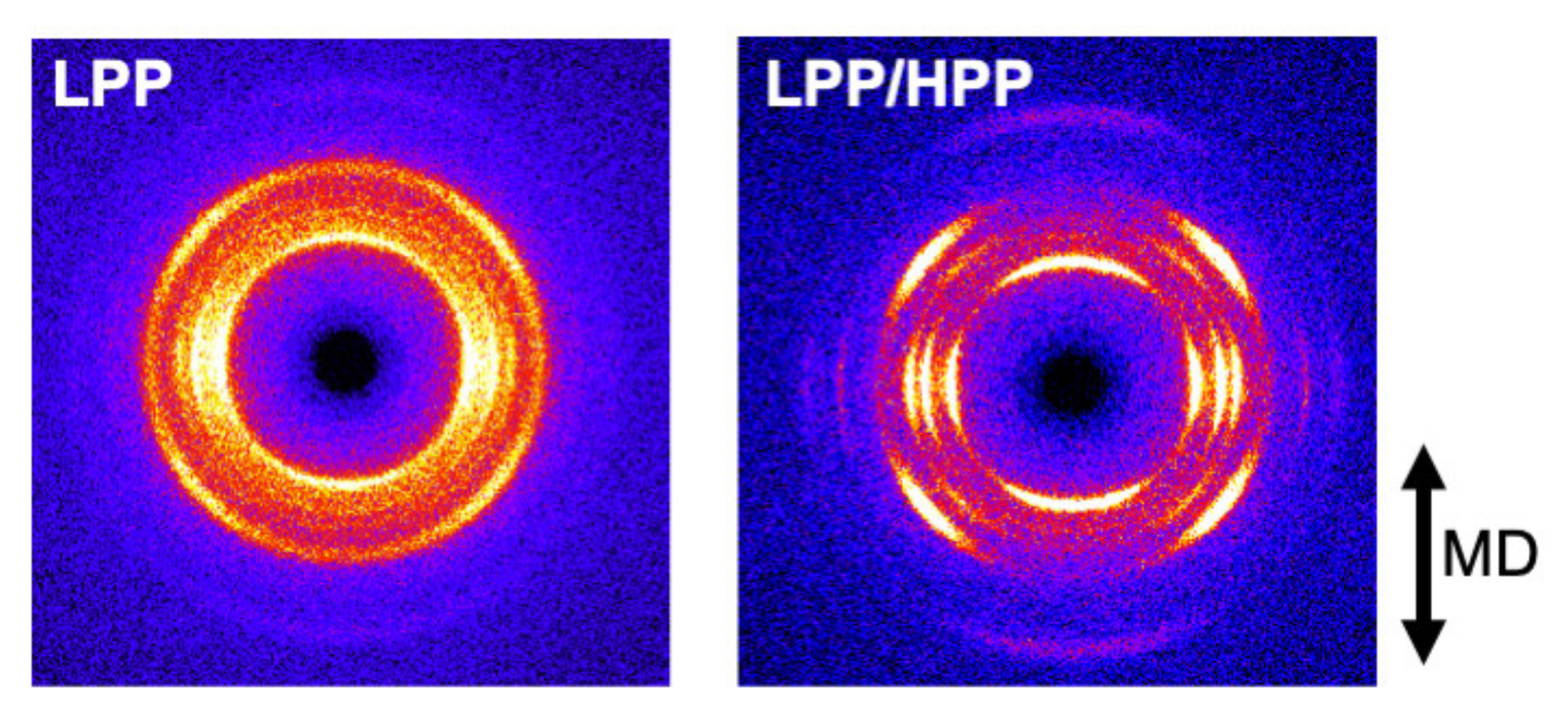
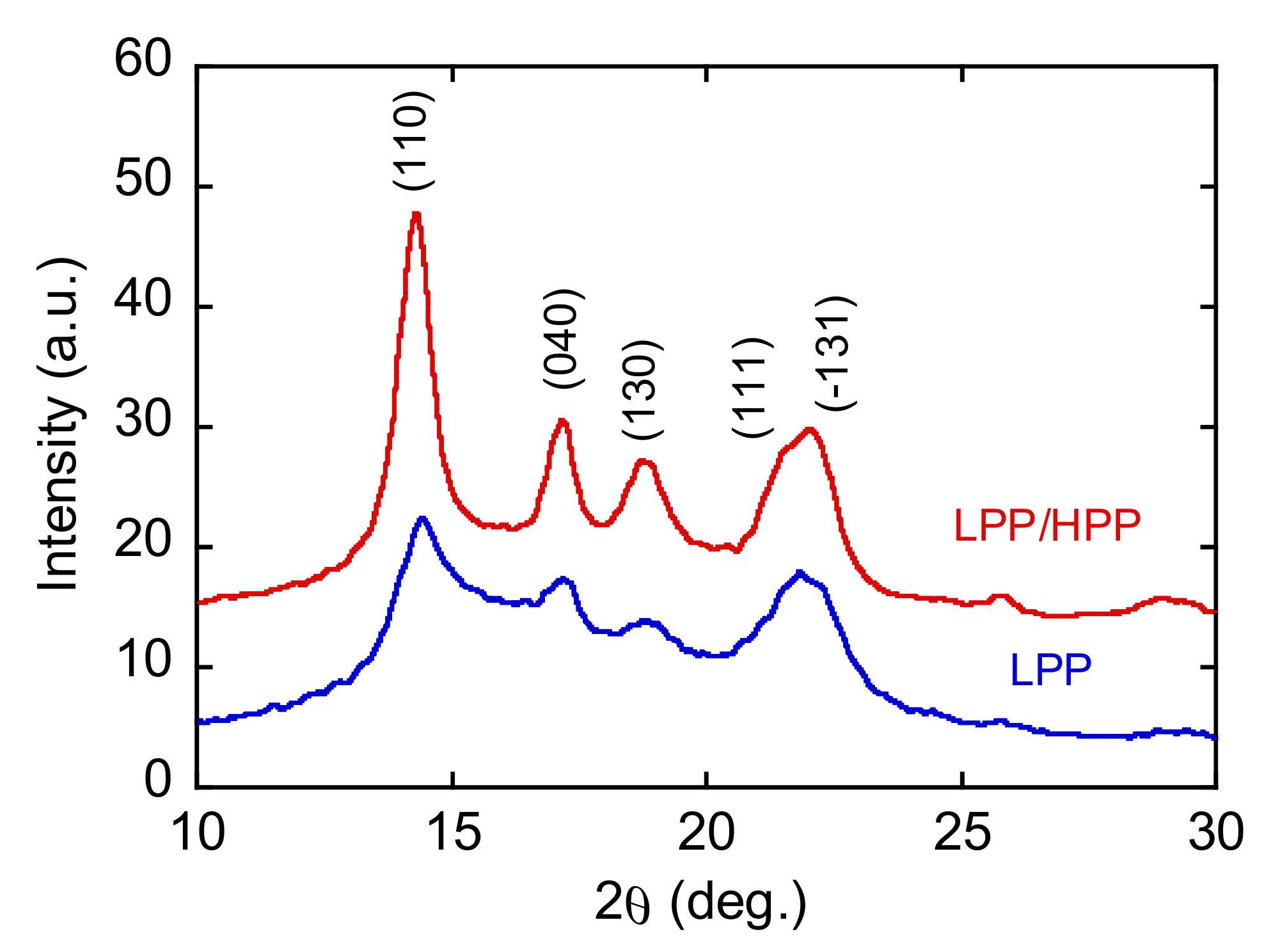
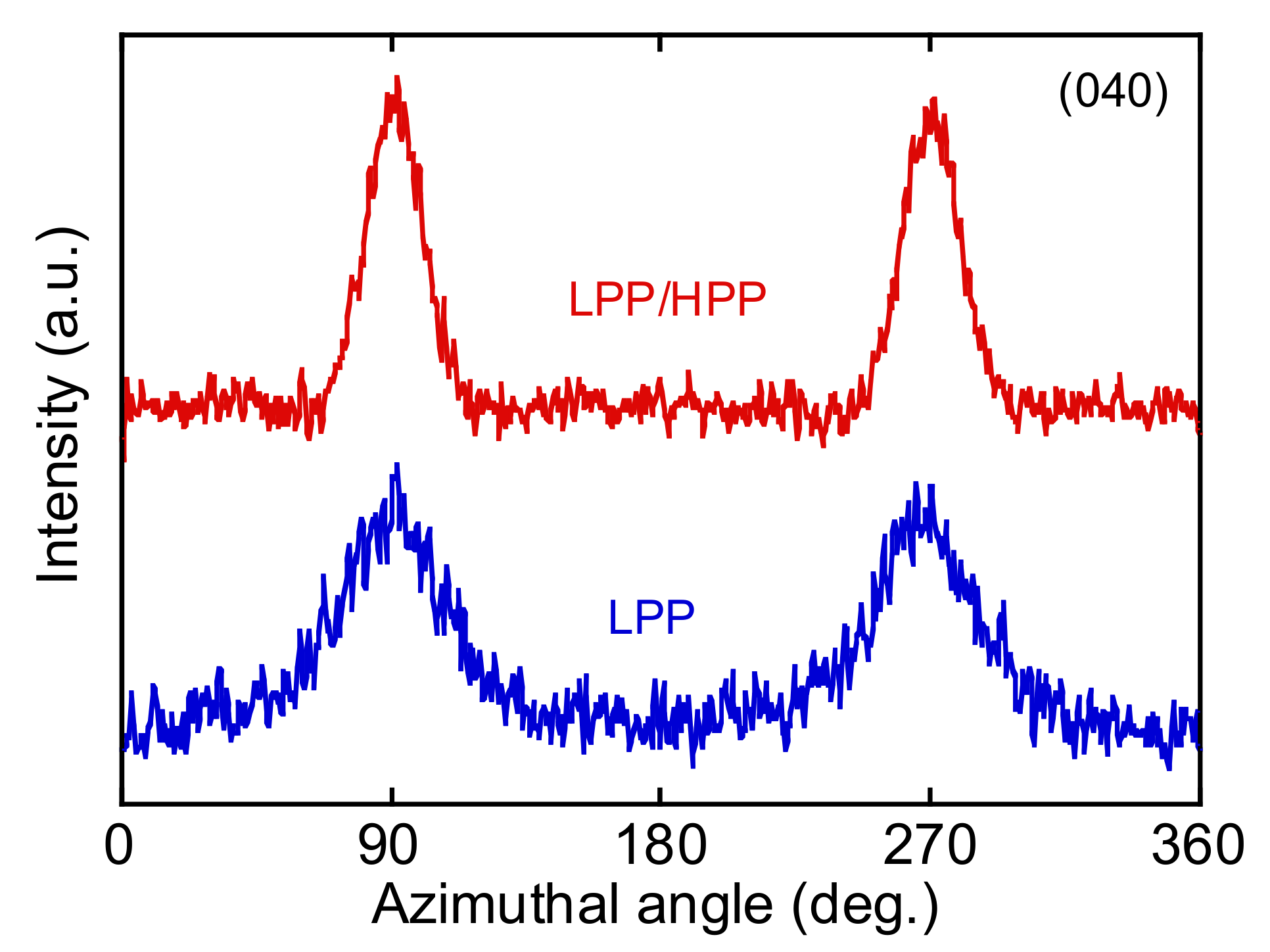
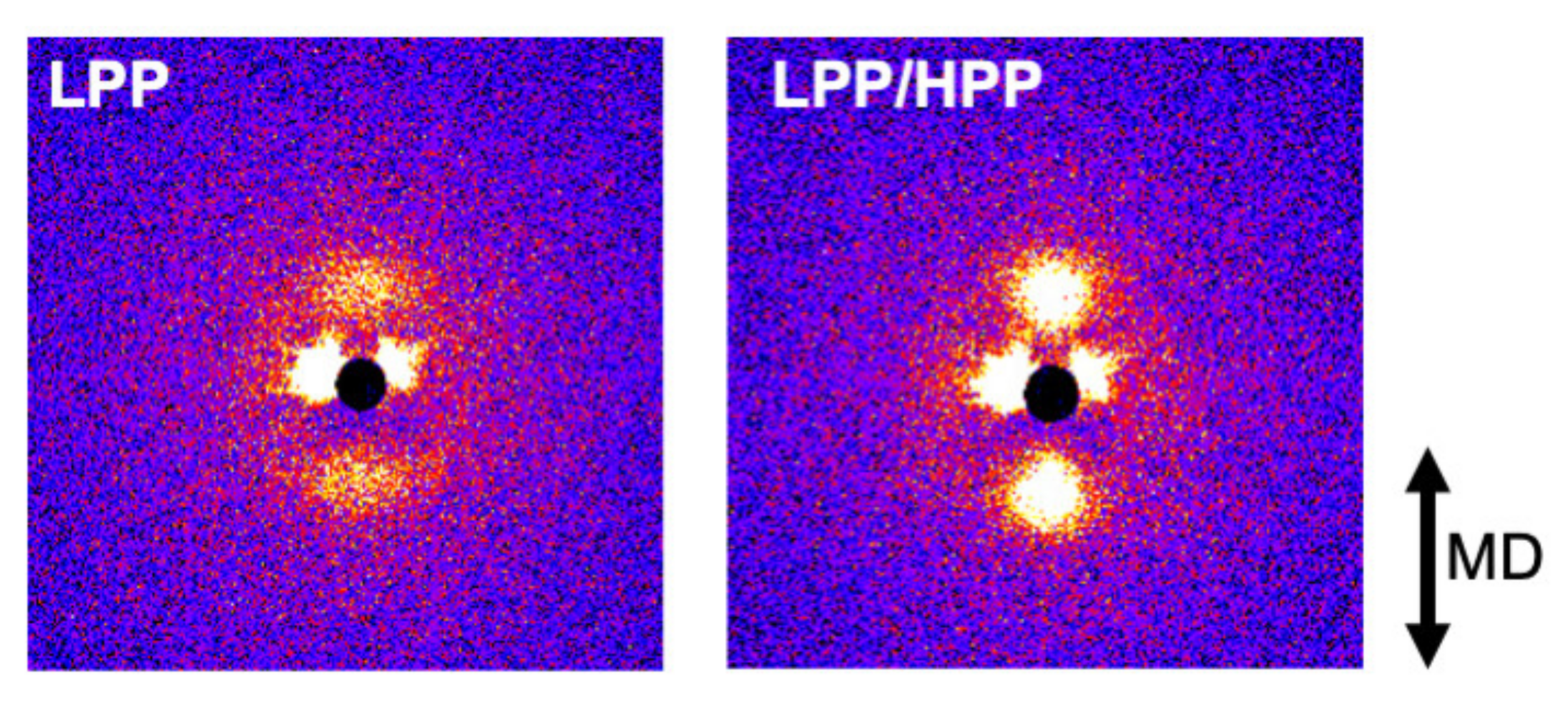
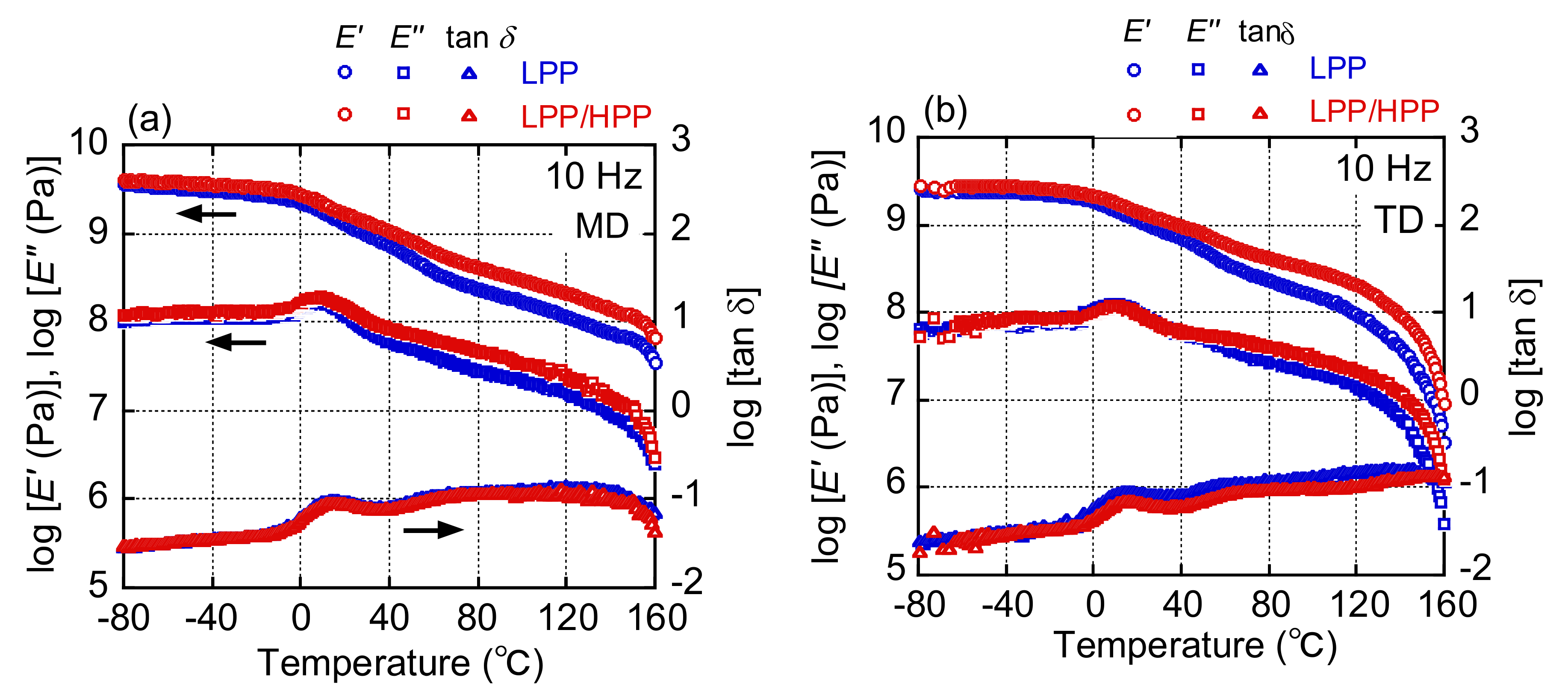
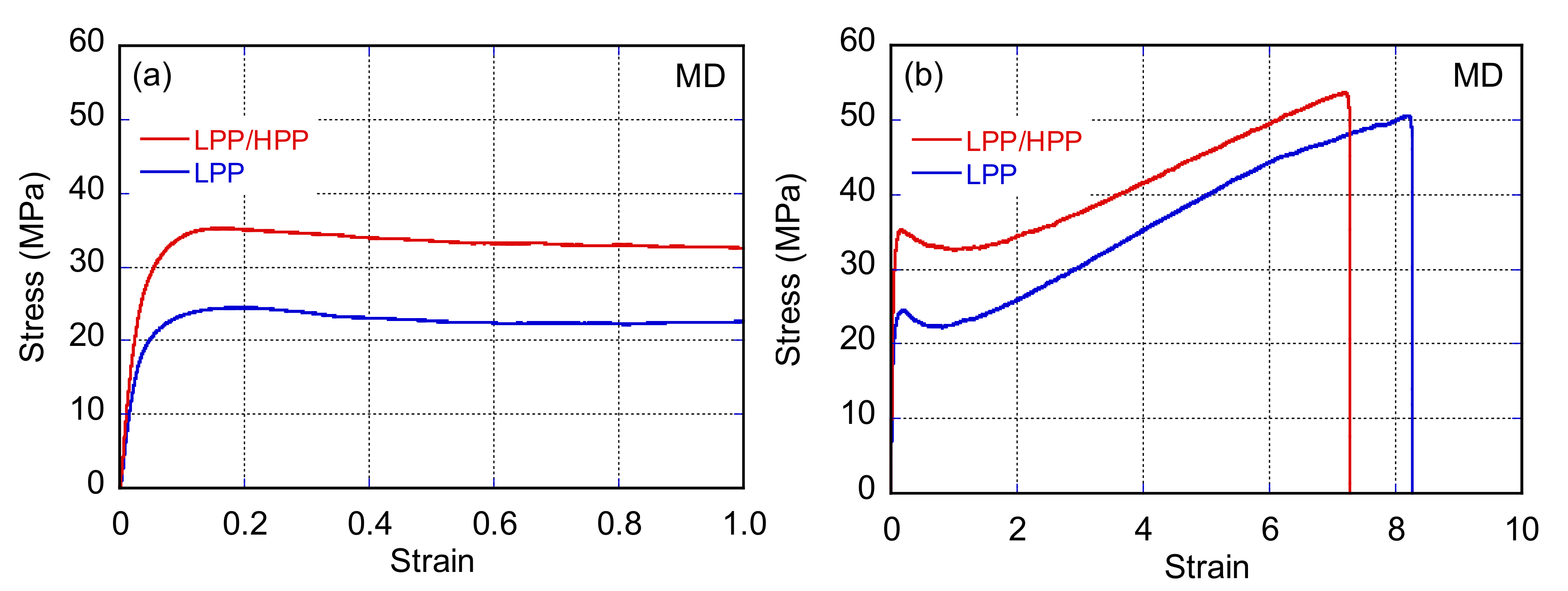
3.3. Morphology and Mechanical Properties of Films
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gentekos, D.T.; Sifri, R.J.; Fors, B.P. Controlling polymer properties through the shape of the molecular-weight distribution. Nat. Rev. Mater. 2019, 4, 761–774. [Google Scholar] [CrossRef]
- Kennedy, M.A.; Peacock, A.J.; Failla, M.D.; Lucasji, J.C.; Mandelkern, L. Tensile Properties of Crystalline Polymers: Random Copolymers of Ethylene. Macromolecules 1995, 28, 1407–1421. [Google Scholar] [CrossRef]
- Hu, Y.; Shao, Y.; Liu, Z.; He, X.; Liu, B. Effect of short-chain branching on the tie chains and dynamics of bimodal polyethylene: Molecular dynamics simulation. Eur. Polym. J. 2018, 103, 312–321. [Google Scholar] [CrossRef]
- Zielińska, D.; Rydzkowski, T.; Thakur, V.K.; Borysiak, S. Enzymatic engineering of nanometric cellulose for sustainable polypropylene nanocomposites. Ind. Crops Prod. 2021, 161, 11388. [Google Scholar] [CrossRef]
- Nishikawa, R.; Aridome, N.; Ojima, N.; Yamaguchi, M. Structure and properties of fiber-reinforced polypropylene prepared by direct incorporation of aqueous solution of poly(vinyl alcohol). Polymer 2020, 199, 122566. [Google Scholar] [CrossRef]
- Iwasaki, S.; Inoue, M.; Takei, Y.; Nishikawa, R.; Yamaguchi, M. Modulus enhancement of polypropylene by sorbitol nucleating agent in flow field. Polym. Cryst. 2021, 4, e10170. [Google Scholar] [CrossRef]
- Phulkerd, P.; Nakabayashi, T.; Iwasaki, S.; Yamaguchi, M. Enhancement of drawdown force in polypropylene containing nucleating agent. J. Appl. Polym. Sci. 2019, 136, 1–7. [Google Scholar] [CrossRef]
- Colby, R.H.; Fetters, L.J.; Graessley, W.W. Melt Viscosity-Molecular Weight Relationship for Linear Polymers. Macromolecules 1987, 20, 2226–2237. [Google Scholar] [CrossRef]
- Pearson, D.S.; Ver Strate, G.; von Meerwall, E.; Schilling, F.C. Viscosity and Self-Diffusion Coefficient of Linear Polyethylene. Macromolecules 1987, 20, 1133–1141. [Google Scholar] [CrossRef]
- Anderews, J.M.; Ward, I.M. The cold-drawing of high density polyethylene. J. Mater. Sci. 1970, 5, 411–417. [Google Scholar] [CrossRef]
- Kennedy, M.A.; Peacock, A.J.; Mandelkern, L. Tensile Properties of Crystalline Polymers: Linear Polyethylene. Macromolecules 1994, 27, 5297–5310. [Google Scholar] [CrossRef]
- Diop, M.F.; Burghardt, W.R.; Torkelson, J.M. Well-mixed blends of HDPE and ultrahigh molecular weight polyethylene with major improvements in impact strength achieved via solid-state shear pulverization. Polymer 2014, 55, 4948–4958. [Google Scholar] [CrossRef]
- Tanaka, H.; Saijo, S.; Kakiage, M.; Yamanobe, T.; Uehara, H. In-situ analysis for melt-drawing behavior of ultra-high molecular weight polyethylene/normal molecular weight polyethylene blend films. Polymer 2021, 213, 123213. [Google Scholar] [CrossRef]
- Capaccio, G.; Ward, I.M. Preparation of ultra-high modulus linear polyethylenes; effect of molecular weight and molecular weight distribution on drawing behaviour and mechanical properties. Polymer 1974, 15, 233–238. [Google Scholar] [CrossRef]
- Sun, X.; Shen, H.; Xie, B.; Yang, W.; Yang, M. Fracture behavior of bimodal polyethylene: Effect of molecular weight distribution characteristics. Polymer 2011, 52, 564–570. [Google Scholar] [CrossRef]
- Kida, T.; Tanaka, R.; Hiejima, Y.; Nitta, K.-H.; Shiono, T. Improving the strength of polyethylene solids by simple controlling of the molecular weight distribution. Polymer 2021, 218, 123526. [Google Scholar] [CrossRef]
- Münstedt, H. Dependence of the Elongational Behavior of Polystyrene Melts on Molecular Weight and Molecular Weight Distribution. J. Rheol. 1980, 24, 847–867. [Google Scholar] [CrossRef]
- Minegishi, A.; Nishioka, A.; Takahashi, T.; Masubuchi, Y.; Takimoto, J.; Koyama, K. Uniaxial elongational viscosity of PS/a small amount of UHMW-PS blends. Rheol. Acta 2001, 40, 329–338. [Google Scholar] [CrossRef]
- Nielsen, J.K.; Rasmussen, H.K.; Hassager, O.; McKinley, G.H. Elongational viscosity of monodisperse and bidisperse polystyrene melts. J. Rheol. 2006, 50, 453–476. [Google Scholar] [CrossRef] [Green Version]
- Wingstrand, S.L.; Shen, B.; Kornfield, J.A.; Mortensen, K.; Parisi, D.; Vlassopoulos, D.; Hassager, O. Rheological Link between Polymer Melts with a High Molecular Weight Tail and Enhanced Formation of Shish-Kebabs. ACS Macro Lett. 2017, 6, 1268–1273. [Google Scholar] [CrossRef]
- Mykhaylyk, O.O.; Chambon, P.; Impradice, C.; Fairclough, J.P.A.; Terrill, N.J.; Ryan, A.J. Control of structural morphology in shear-induced crystallization of polymers. Macromolecules 2010, 43, 2389–2405. [Google Scholar] [CrossRef]
- Hamad, F.G.; Colby, R.H.; Milner, S.T. Onset of Flow-Induced Crystallization Kinetics of Highly Isotactic Polypropylene. Macromolecules 2015, 48, 3725–3738. [Google Scholar] [CrossRef]
- Matsuba, G.; Sakamoto, S.; Ogino, Y.; Nishida, K.; Kanaya, T. Crystallization of polyethylene blends under shear flow. Effects of crystallization temperature and ultrahigh molecular weight component. Macromolecules 2007, 40, 7270–7275. [Google Scholar] [CrossRef]
- Kanaya, T.; Matsuba, G.; Ogino, Y.; Nishida, K.; Shimizu, H.M.; Shinohara, T.; Oku, T.; Suzuki, J.; Otomo, T. Hierarchic structure of shish-kebab by neutron scattering in a wide Q range. Macromolecules 2007, 40, 3650–3654. [Google Scholar] [CrossRef]
- Bashir, Z.; Odell, J.A.; Keller, A. High modulus filaments of polyethylene with lamellar structure by melt processing; the role of the high molecular weight component. J. Mater. Sci. 1984, 19, 3713–3725. [Google Scholar] [CrossRef]
- Bashir, Z.; Odell, J.A.; Keller, A. Stiff and strong polyethylene with shish kebab morphology by continuous melt extrusion. J. Mater. Sci. 1986, 21, 3993–4002. [Google Scholar] [CrossRef]
- Seemork, J.; Siriprumpoonthum, M.; Lee, Y.; Nobukawa, S.; Yamaguchi, M. Effect of die geometry on drawdown force of polypropylene at capillary extrusion. Adv. Polym. Technol. 2015, 34, 1–7. [Google Scholar] [CrossRef]
- Wunderlich, B. Macromolecular Physics, Crystal Structure, Morphology, Defects; Academic Press: New York, NY, USA, 1973. [Google Scholar]
- Zia, Q.; Mileva, D.; Androsch, R. Rigid amorphous fraction in isotactic polypropylene. Macromolecules 2008, 41, 8095–8102. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L.; Righetti, M.C. Crystallization-induced formation of rigid amorphous fraction. Polym. Cryst. 2018, 1, 1–14. [Google Scholar] [CrossRef]
- Wunderlich, B. The Athas database on heat capacities of polymers. Pure Appl. Chem. 1995, 67, 1019–1026. [Google Scholar] [CrossRef]
- Wunderlich, B. Thermal Analysis of Polymeric Materials; Springer: New York, NY, USA, 1995. [Google Scholar]
- Wilchinsky, Z.W. Measurement of Orientation in Polypropylene Film. J. Appl. Phys. 1960, 31, 1969–1972. [Google Scholar] [CrossRef]
- Tanaka, M.; Young, R. Molecular orientation distributions in uniaxially oriented poly (L-lactic acid) films determined by polarized Raman spectroscopy. Macromolecules 2006, 39, 3312–3321. [Google Scholar] [CrossRef]
- Arvidson, S.A.; Khan, S.A.; Gorga, R.E. Mesomorphic-α-monoclinic phase transition in isotactic polypropylene: A study of processing effects on structure and mechanical properties. Macromolecules 2010, 43, 2916–2924. [Google Scholar] [CrossRef]
- Bower, D.I.; Maddams, W.F. The Vibrational Spectroscopy of Polymers; Cambridge University Press: New York, NY, USA, 1992. [Google Scholar]
- Nitta, K.-H.; Sawada, T.; Yoshida, S.; Kawamura, T. Three dimensional molecular orientation of isotactic polypropylene films under biaxial deformation at higher temperatures. Polymer 2015, 74, 30–37. [Google Scholar] [CrossRef]
- Parthasarthy, G.; Sevegney, M.; Kannan, R.M. Rheooptical Fourier transform infrared spectroscopy of the deformation behavior in quenched and slow-cooled isotactic polypropylene films. J. Polym. Sci. Part B Polym. Phys. 2002, 40, 2539–2551. [Google Scholar] [CrossRef]
- Lanyi, F.J.; Wenzke, N.; Kaschta, J.; Schubert, D.W. A method to reveal bulk and surface crystallinity of Polypropylene by FTIR spectroscopy—Suitable for fibers and nonwovens. Polym. Test. 2018, 71, 49–55. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Takahashi, M. Rheological properties of low-density polyethylenes produced by tubular and vessel processes. Polymer 2001, 42, 8663–8670. [Google Scholar] [CrossRef]
- Mieda, N.; Yamaguchi, M. Flow instability for binary blends of linear polyethylene and long-chain branched polyethylene. J. Non-Newton. Fluid Mech. 2011, 166, 231–240. [Google Scholar] [CrossRef]
- Osaki, K.; Murai, A.; Bessho, N.; Kim, B.S. Linear Viscoelastic Relation Concerning Shear Stresses at the Start and Cessation of Steady Shear Flow. Nihon Reoroji Gakkaishi (J. Soc. Rheol. Jpn.) 1976, 4, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, M. Flow instability in capillary extrusion of plasticized poly(vinyl chloride). J. Appl. Polym. Sci. 2001, 82, 1277–1283. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Suzuki, K.I. Enhanced strain hardening in elongational viscosity for HDPE/crosslinked HDPE blend. II. Processability of thermoforming. J. Appl. Polym. Sci. 2002, 86, 79–83. [Google Scholar] [CrossRef]
- Yamaguchi, M. Material Strength in Molten State for Foam. In Foram Extrusion: Principles and Practie; Lee, S.-T., Park, C.B., Eds.; CRC Press: New York, NY, USA, 2014; p. 36. ISBN 9780429184703. [Google Scholar]
- Kugimoto, D.; Kouda, S.; Yamaguchi, M. Modification of Poly(Lactic Acid) Rheological Properties Using Ethylene—Vinyl Acetate Copolymer. J. Polym. Environ. 2021, 29, 121–129. [Google Scholar] [CrossRef]
- Seki, M.; Thurman, D.W.; Oberhauser, J.P.; Kornfield, J.A. Shear-Mediated Crystallization of Isotactic Polypropylene: The Role of Long Chain−Long Chain Overlap. Macromolecules 2002, 35, 2583–2594. [Google Scholar] [CrossRef]
- Koscher, E.; Fulchiron, R. Influence of shear on polypropylene crystallization: Morphology development and kinetics. Polymer 2002, 43, 6931–6942. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: Oxford, UK, 1986. [Google Scholar]
- Fetters, L.J.; Lohse, D.J.; Graessley, W.W. Chain dimensions and entanglement spacings in dense macromolecular systems. J. Polym. Sci. Part B Polym. Phys. 1999, 37, 1023–1033. [Google Scholar] [CrossRef]
- Pasquini, N. Polypropylene Handbook; Carl Hanser Verlag: Munich, Germany, 2005. [Google Scholar]
- Takayanagi, M.; Nitta, K.-H. Application of a tie molecule model to the postyielding deformation of crystalline polymers. Macromol. Theory Simul. 1997, 6, 181–195. [Google Scholar] [CrossRef]
- Nitta, K.-H.; Nomura, H. Stress–strain behavior of cold-drawn isotactic polypropylene subjected to various drawn histories. Polymer 2014, 55, 6614–6622. [Google Scholar] [CrossRef]
- Strobl, G. The Physics of Polymers: Concepts for Understanding Their Structures and Behavior, 3rd ed.; Springer: Berlin, Germany, 2007. [Google Scholar]
- Nitta, K.-H.; Takayanagi, M. Novel proposal of lamellar clustering process for elucidation of tensile yield behavior of linear polyethylenes. J. Macromol. Sci. Phys. 2003, 42, 107–126. [Google Scholar] [CrossRef]
- Kuriyagawa, M.; Nitta, K.-H. Structural explanation on natural draw ratio of metallocene-catalyzed high density polyethylene. Polymer 2011, 52, 3469–3477. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | Drawdown Force (mN) | |
|---|---|---|
| DR = 10 | DR = 30 | |
| LPP | 56 | 70 |
| LPP/HPP | 86 | 96 |
| Sample Code | χDSC (wt%) | χRA (wt%) | χWAXD (wt%) | χIR (wt%) | fWAXD | fIR,c | fIR,a |
|---|---|---|---|---|---|---|---|
| LPP | 34.3 | 59.5 | 49.3 | 51.8 | 0.12 | 0.22 | 0.15 |
| LPP/HPP | 34.5 | 54.1 | 52.1 | 53.6 | 0.18 | 0.28 | 0.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kida, T.; Kimura, T.; Eno, A.; Janchai, K.; Yamaguchi, M.; Otsuki, Y.; Kimura, T.; Mizukawa, T.; Murakami, T.; Hato, K.; et al. Effect of Ultra-High-Molecular-Weight Molecular Chains on the Morphology, Crystallization, and Mechanical Properties of Polypropylene. Polymers 2021, 13, 4222. https://doi.org/10.3390/polym13234222
Kida T, Kimura T, Eno A, Janchai K, Yamaguchi M, Otsuki Y, Kimura T, Mizukawa T, Murakami T, Hato K, et al. Effect of Ultra-High-Molecular-Weight Molecular Chains on the Morphology, Crystallization, and Mechanical Properties of Polypropylene. Polymers. 2021; 13(23):4222. https://doi.org/10.3390/polym13234222
Chicago/Turabian StyleKida, Takumitsu, Takeyoshi Kimura, Ayaka Eno, Khunanya Janchai, Masayuki Yamaguchi, Yasuhiko Otsuki, Tokutaro Kimura, Tomoaki Mizukawa, Tomoya Murakami, Kazuki Hato, and et al. 2021. "Effect of Ultra-High-Molecular-Weight Molecular Chains on the Morphology, Crystallization, and Mechanical Properties of Polypropylene" Polymers 13, no. 23: 4222. https://doi.org/10.3390/polym13234222
APA StyleKida, T., Kimura, T., Eno, A., Janchai, K., Yamaguchi, M., Otsuki, Y., Kimura, T., Mizukawa, T., Murakami, T., Hato, K., & Okawa, T. (2021). Effect of Ultra-High-Molecular-Weight Molecular Chains on the Morphology, Crystallization, and Mechanical Properties of Polypropylene. Polymers, 13(23), 4222. https://doi.org/10.3390/polym13234222