
Connecting Gas-Phase Computational Chemistry to Condensed Phase Kinetic Modeling: The State-of-the-Art



Abstract
1. Introduction
2. Gas Phase QCC Approaches
3. Gas Phase Computational Chemistry Tools
3.1. Wave-Function Based Calculations
3.2. Density Functional Theory
3.3. Semi-Empirical Methods
4. Solution Phase Computational Chemistry Tools
4.1. Transition State Theory
4.2. Solvation Models
4.2.1. Implicit Models
4.2.2. Explicit Models
4.2.3. Hybrid Models
4.2.4. Advantages and Disadvantages
5. Connection of Lower-Scale Modeling with Higher-Scale Modeling
6. Case Studies for Connection of Computational Chemistry and Kinetic Modeling
6.1. Case Study 1: Radical Polymerization
6.1.1. Initiation
6.1.2. Propagation (Non-Aqueous)
6.1.3. Propagation (Aqueous)
6.1.4. Termination
6.1.5. Secondary Reactions
6.1.6. Controlled Radical Polymerization
6.2. Case Study 2: (Metal Complex Catalyzed) Polymerization of Olefins
6.2.1. (Co)monomers
6.2.2. Homogeneous and Heterogeneous Catalysts
6.3. Case Study 3: Step-Growth Polymerization
6.4. Case Study 4: Polymer Environmental or Aging Degradation
6.5. Case Study 5: Material Properties
6.5.1. Polymer Batteries
6.5.2. Capacitors
7. Conclusions
Funding
Conflicts of Interest
References
- Raps, D.; Hossieny, N.; Park, C.B.; Altstädt, V. Past and present developments in polymer bead foams and bead foaming technology. Polymer 2015, 56, 5–19. [Google Scholar] [CrossRef]
- De Keer, L.; Van Steenberge, P.H.; Reyniers, M.F.; Marin, G.B.; Hungenberg, K.D.; Seda, L.; D’hooge, D.R. A complete understanding of the reaction kinetics for the industrial production process of expandable polystyrene. Aiche J. 2017, 63, 2043–2059. [Google Scholar] [CrossRef]
- Godwin, A.; Bolina, K.; Clochard, M.; Dinand, E.; Rankin, S.; Simic, S.; Brocchini, S. New strategies for polymer development in pharmaceutical science—A short review. J. Pharm. Pharmacol. 2001, 53, 1175–1184. [Google Scholar] [CrossRef]
- Girase, M.L.; Patil, P.G.; Ige, P.P. Polymer-drug conjugates as nanomedicine: A review. Int. J. Polym. Mater. Polym. Biomater. 2020, 69, 990–1014. [Google Scholar] [CrossRef]
- Han, J.; Zhao, D.; Li, D.; Wang, X.; Jin, Z.; Zhao, K. Polymer-based nanomaterials and applications for vaccines and drugs. Polymers 2018, 10, 31. [Google Scholar] [CrossRef]
- Jaiswal, M.; Menon, R. Polymer electronic materials: A review of charge transport. Polym. Int. 2006, 55, 1371–1384. [Google Scholar] [CrossRef]
- Sun, W.; Mao, J.; Wang, S.; Zhang, L.; Cheng, Y. Review of recent advances of polymer based dielectrics for high-energy storage in electronic power devices from the perspective of target applications. Front. Chem. Sci. Eng. 2021, 15, 18–34. [Google Scholar] [CrossRef]
- Zhan, C.; Yu, G.; Lu, Y.; Wang, L.; Wujcik, E.; Wei, S. Conductive polymer nanocomposites: A critical review of modern advanced devices. J. Mater. Chem. C 2017, 5, 1569–1585. [Google Scholar] [CrossRef]
- Gao, J. Polymer light-emitting electrochemical cells—Recent advances and future trends. Curr. Opin. Electrochem. 2018, 7, 87–94. [Google Scholar] [CrossRef]
- de Brito, E.B.; Valaski, R.; Marques, M.d.F.V. Development of polymeric active layer for RGB light-emitting devices: A review. J. Mater. Sci. Mater. Electron. 2020, 31, 21856–21895. [Google Scholar] [CrossRef]
- Rose, J.C.; Fölster, M.; Kivilip, L.; Gerardo-Nava, J.L.; Jaekel, E.E.; Gehlen, D.B.; Rohlfs, W.; De Laporte, L. Predicting the orientation of magnetic microgel rods for soft anisotropic biomimetic hydrogels. Polym. Chem. 2020, 11, 496–507. [Google Scholar] [CrossRef]
- Russell, G.T. The kinetics of free radical polymerizing systems at low conversion, 2. On the influence of the monomer and initiator concentrations. Macromol. Theory Simul. 1995, 4, 519–548. [Google Scholar] [CrossRef]
- De Keer, L.; Van Steenberge, P.H.; Reyniers, M.-F.; D’hooge, D.R. Going Beyond the Carothers, Flory and Stockmayer Equation by Including Cyclization Reactions and Mobility Constraints. Polymers 2021, 13, 2410. [Google Scholar] [CrossRef] [PubMed]
- D’hooge, D.R.; Van Steenberge, P.H.M.; Reyniers, M.-F.; Marin, G.B. The strength of multi-scale modeling to unveil the complexity of radical polymerization. Prog. Polym. Sci. 2016, 58, 59–89. [Google Scholar] [CrossRef]
- Toloza Porras, C.; D’hooge, D.R.; Van Steenberge, P.H.; Reyniers, M.-F.O.; Marin, G.B. ICAR ATRP for estimation of intrinsic macro-activation/deactivation arrhenius parameters under polymerization conditions. Ind. Eng. Chem. Res. 2014, 53, 9674–9685. [Google Scholar] [CrossRef]
- Ponnuswamy, S.; Penlidis, A.; Kiparissides, C. Batch solution polymerization of methyl methacrylate: Parameter estimation. Chem. Eng. J. 1988, 39, 175–183. [Google Scholar] [CrossRef]
- Coote, M.L. Computational Quantum Chemistry for Free-Radical Polymerization. Encycl. Polym. Sci. Technol. 2002. [Google Scholar] [CrossRef]
- Charpentier, J.; McKenna, T. Managing complex systems: Some trends for the future of chemical and process engineering. Chem. Eng. Sci. 2004, 59, 1617–1640. [Google Scholar] [CrossRef]
- Zhao, S.; Luo, Y. Multiscale Modeling of Lignocellulosic Biomass Thermochemical Conversion Technology: An Overview on the State-of-the-Art. Energy Fuels 2020, 34, 11867–11886. [Google Scholar] [CrossRef]
- Noble, B.B.; Coote, M.L. First principles modelling of free-radical polymerisation kinetics. Int. Rev. Phys. Chem. 2013, 32, 467–513. [Google Scholar] [CrossRef]
- Elliott, J.A. Novel approaches to multiscale modelling in materials science. Int. Mater. Rev. 2011, 56, 207–225. [Google Scholar] [CrossRef]
- Marien, Y.W.; Van Steenberge, P.H.; D’hooge, D.R.; Marin, G.B. Particle by particle kinetic monte carlo tracking of reaction and mass transfer events in miniemulsion free radical polymerization. Macromolecules 2019, 52, 1408–1423. [Google Scholar] [CrossRef]
- D’hooge, D.R.; Reyniers, M.-F.; Stadler, F.J.; Dervaux, B.; Bailly, C.; Du Prez, F.E.; Marin, G.B. Atom transfer radical polymerization of isobornyl acrylate: A kinetic modeling study. Macromolecules 2010, 43, 8766–8781. [Google Scholar] [CrossRef]
- Krallis, A.; Kotoulas, C.; Papadopoulos, S.; Kiparissides, C.; Bousquet, J.; Bonardi, C. A comprehensive kinetic model for the free-radical polymerization of vinyl chloride in the presence of monofunctional and bifunctional initiators. Ind. Eng. Chem. Res. 2004, 43, 6382–6399. [Google Scholar] [CrossRef]
- Peklak, A.D.; Butté, A.; Storti, G.; Morbidelli, M. Gel effect in the bulk reversible addition–fragmentation chain transfer polymerization of methyl methacrylate: Modeling and experiments. J. Polym. Sci. Part A: Polym. Chem. 2006, 44, 1071–1085. [Google Scholar] [CrossRef]
- Johnston-Hall, G.; Monteiro, M.J. Kinetic simulations of atom transfer radical polymerization (ATRP) in light of chain length dependent termination. Macromol. Theory Simul. 2010, 19, 387–393. [Google Scholar] [CrossRef]
- Iedema, P.D.; Wulkow, M.; Hoefsloot, H.C. Modeling molecular weight and degree of branching distribution of low-density polyethylene. Macromolecules 2000, 33, 7173–7184. [Google Scholar] [CrossRef]
- Al-Harthi, M.; Soares, J.B.; Simon, L.C. Dynamic Monte Carlo simulation of atom-transfer radical polymerization. Macromol. Mater. Eng. 2006, 291, 993–1003. [Google Scholar] [CrossRef]
- Purmova, J.; Pauwels, K.F.; Van Zoelen, W.; Vorenkamp, E.J.; Schouten, A.J.; Coote, M.L. New insight into the formation of structural defects in poly (vinyl chloride). Macromolecules 2005, 38, 6352–6366. [Google Scholar] [CrossRef][Green Version]
- De Rybel, N.; Van Steenberge, P.H.; Reyniers, M.-F.; D’hooge, D.R.; Marin, G.B. How chain length dependencies interfere with the bulk RAFT polymerization rate and microstructural control. Chem. Eng. Sci. 2018, 177, 163–179. [Google Scholar] [CrossRef]
- Wieme, J.; Reyniers, M.-F.O.; Marin, G.B. Microkinetic modeling of structural properties of poly (vinyl chloride). Macromolecules 2009, 42, 7797–7810. [Google Scholar] [CrossRef]
- Ruiperez, F. Application of quantum chemical methods in polymer chemistry. Int. Rev. Phys. Chem. 2019, 38, 343–403. [Google Scholar] [CrossRef]
- Jensen, F. An introduction to the state of the art in quantum chemistry. Annu. Rep. Comput. Chem. 2005, 1, 3–17. [Google Scholar]
- Szabo, A.; Ostlund, N.S. Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory; Courier Corporation: Chelmsford, MA, USA, 2012. [Google Scholar]
- Sen, K.D. Reviews of Modern Quantum Chemistry: A Celebration of the Contributions of Robert G. Parr; World Scientific: Singapore, 2002. [Google Scholar]
- Lowe, J.P.; Peterson, K. Quantum Chemistry; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Hartree, D.R. The wave mechanics of an atom with a non-Coulomb central field. Part I. Theory and methods. In Mathematical Proceedings of the Cambridge Philosophical Society; Cambridge University Press: Cambridge, UK, 1928; Volume 24, pp. 89–110. [Google Scholar] [CrossRef]
- Fock, V. Näherungsmethode zur Lösung des quantenmechanischen Mehrkörperproblems. Z. Für Phys. 1930, 61, 126–148. [Google Scholar] [CrossRef]
- Kutzelnigg, W. Present and future trends in quantum chemical calculations. J. Mol. Struct. 1988, 181, 33–54. [Google Scholar] [CrossRef]
- Bartlett, R.J. Recent Advances in Coupled-Cluster Methods; World Scientificl: Singapore, 1997. [Google Scholar]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Zhao, Y.; Pu, J.; Lynch, B.J.; Truhlar, D.G. Tests of second-generation and third-generation density functionals for thermochemical kinetics. Phys. Chem. Chem. Phys. 2004, 6, 673–676. [Google Scholar] [CrossRef]
- Schenker, S.; Schneider, C.; Tsogoeva, S.B.; Clark, T. Assessment of popular DFT and semiempirical molecular orbital techniques for calculating relative transition state energies and kinetic product distributions in enantioselective organocatalytic reactions. J. Chem. Theory Comput. 2011, 7, 3586–3595. [Google Scholar] [CrossRef]
- Godbout, N.; Salahub, D.R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-type basis sets for local spin density functional calculations. Part I. Boron through neon, optimization technique and validation. Can. J. Chem. 1992, 70, 560–571. [Google Scholar] [CrossRef]
- de la Vega, J.G.; Miguel, B. Basis sets for computational chemistry. In Introduction to Advanced Topics of Computational Chemistry; Montero, L.A., Dıaz, L.A., Bader, R., Eds.; Wiley: Hoboken, NJ, USA, 2003; pp. 41–80. [Google Scholar]
- Koch, W.; Holthausen, M.C. A Chemist’s Guide to Density Functional Theory; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Christopher, J.C. Essentials of Computational Chemistry: Theories and Models; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Hermosilla, L.; Calle, P.; García De La Vega, J.; Sieiro, C. Density functional theory predictions of isotropic hyperfine coupling constants. J. Phys. Chem. A 2005, 109, 1114–1124. [Google Scholar] [CrossRef]
- Hermosilla, L.; Calle, P.; Sieiro, C. Assignments of hyperfine splittings by DFT methods of radicals containing 33S (I = 3/2), 31P (I = 1/2), and 29Si (I = 1/2) Nuclei. Phosphorussulfurand Silicon Relat. Elem. 2005, 180, 1421–1422. [Google Scholar] [CrossRef]
- Hermosilla, L.; Calle, P.; Garcia de la Vega, J.; Sieiro, C. Theoretical isotropic hyperfine coupling constants of third-row nuclei (29Si, 31P, and 33S). J. Phys. Chem. A 2005, 109, 7626–7635. [Google Scholar] [CrossRef]
- Hermosilla, L.; Calle, P.; García de La Vega, J.; Sieiro, C. Density functional theory study of 14N isotropic hyperfine coupling constants of organic radicals. J. Phys. Chem. A 2006, 110, 13600–13608. [Google Scholar] [CrossRef] [PubMed]
- Engels, B.; Eriksson, L.A.; Lunell, S. Recent Developments in Configuration Interaction and Density Functional Theory Calculations of Radical Hyperfine Structure. In Advances in Quantum Chemistry; Elsevier: Amsterdam, The Netherlands, 1996; Volume 27, pp. 297–369. [Google Scholar]
- Barone, V.; Cimino, P. Accurate and feasible computations of structural and magnetic properties of large free radicals: The PBE0/N07D model. Chem. Phys. Lett. 2008, 454, 139–143. [Google Scholar] [CrossRef]
- Barone, V.; Biczysko, M.; Bloino, J.; Egidi, F.; Puzzarini, C. Accurate structure, thermodynamics, and spectroscopy of medium-sized radicals by hybrid coupled cluster/density functional theory approaches: The case of phenyl radical. J. Chem. Phys. 2013, 138, 234303. [Google Scholar] [CrossRef]
- Puzzarini, C.; Biczysko, M.; Barone, V. Accurate harmonic/anharmonic vibrational frequencies for open-shell systems: Performances of the B3LYP/N07D model for semirigid free radicals benchmarked by CCSD (T) computations. J. Chem. Theory Comput. 2010, 6, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Bryce, R.A.; Hillier, H.I. Quantum chemical approaches: Semiempirical molecular orbital and hybrid quantum mechanical/molecular mechanical techniques. Curr. Pharm. Des. 2014, 20, 3293–3302. [Google Scholar] [CrossRef][Green Version]
- Dewar, M.; Thiel, W. The MNDO method, approximation and parameters. J. Am. Chem. Soc. 1977, 90, 4899. [Google Scholar] [CrossRef]
- Dewar, M.; Zoebisch, E.; Healy, E.; Stewart, J. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1993, 115, 5348. [Google Scholar] [CrossRef]
- Stewart, J.J. Optimization of parameters for semiempirical methods I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Stewart, J.J. Optimization of parameters for semiempirical methods II. Applications. J. Comput. Chem. 1989, 10, 221–264. [Google Scholar] [CrossRef]
- Chung, L.W.; Sameera, W.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F. The ONIOM method and its applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef]
- Zhang, G.; Konstantinov, I.A.; Arturo, S.G.; Yu, D.; Broadbelt, L.J. Assessment of a cost-effective approach to the calculation of kinetic and thermodynamic properties of methyl methacrylate homopolymerization: A comprehensive theoretical study. J. Chem. Theory Comput. 2014, 10, 5668–5676. [Google Scholar] [CrossRef] [PubMed]
- Morokuma, K. ONIOM and its applications to material chemistry and catalyses. Bull. Korean Chem. Soc. 2003, 24, 797–801. [Google Scholar]
- Fernández-Ramos, A.; Miller, J.A.; Klippenstein, S.J.; Truhlar, D.G. Modeling the kinetics of bimolecular reactions. Chem. Rev. 2006, 106, 4518–4584. [Google Scholar] [CrossRef]
- Desmet, G.B.; Marien, Y.W.; Van Steenberge, P.H.; D’hooge, D.R.; Reyniers, M.-F.; Marin, G.B. Ab initio based kinetic Monte Carlo analysis to unravel the propagation kinetics in vinyl acetate pulsed laser polymerization. Polym. Chem. 2017, 8, 7143–7150. [Google Scholar] [CrossRef]
- Vandeputte, A.G.; Sabbe, M.K.; Reyniers, M.-F.; Van Speybroeck, V.; Waroquier, M.; Marin, G.B. Theoretical study of the thermodynamics and kinetics of hydrogen abstractions from hydrocarbons. J. Phys. Chem. A 2007, 111, 11771–11786. [Google Scholar] [CrossRef]
- Sabbe, M.; Reyniers, M.-F.; Waroquier, M.; Marin, G. Hydrogen radical additions to unsaturated hydrocarbons and the reverse β-scission reactions: Modeling of activation energies and pre-exponential factors. ChemPhysChem 2010, 11, 195–210. [Google Scholar] [CrossRef]
- Heuts, J.P.; Gilbert, R.G.; Radom, L. A priori prediction of propagation rate coefficients in free-radical polymerizations: Propagation of ethylene. Macromolecules 1995, 28, 8771–8781. [Google Scholar] [CrossRef]
- Heuts, J.P.; Gilbert, R.G.; Radom, L. Determination of arrhenius parameters for propagation in free-radical polymerizations: An assessment of ab initio procedures. J. Phys. Chem. 1996, 100, 18997–19006. [Google Scholar] [CrossRef]
- Meier, R.J. Are DFT level calculations the answer to real-world molecular systems? Comput. Mater. Sci. 2003, 27, 219–223. [Google Scholar] [CrossRef]
- Green, W.H. Predictive chemical kinetics: Density functional and hartree–fock calculations on free-radial reaction transition states. Int. J. Quantum Chem. 1994, 52, 837–847. [Google Scholar] [CrossRef]
- Susnow, R.G.; Dean, A.M.; Green, W.H., Jr. Hydrogen abstraction rates via density functional theory. Chem. Phys. Lett. 1999, 312, 262–268. [Google Scholar] [CrossRef]
- Sabbe, M.K.; Vandeputte, A.G.; Reyniers, M.-F.; Van Speybroeck, V.; Waroquier, M.; Marin, G.B. Ab Initio Thermochemistry and Kinetics for Carbon-Centered Radical Addition and β-Scission Reactions. J. Phys. Chem. A 2007, 111, 8416–8428. [Google Scholar] [CrossRef] [PubMed]
- Van Speybroeck, V.; Van Neck, D.; Waroquier, M.; Wauters, S.; Saeys, M.; Marin, G. Ab initio study of radical addition reactions: Addition of a primary ethylbenzene radical to ethene (I). J. Phys. Chem. A 2000, 104, 10939–10950. [Google Scholar] [CrossRef]
- Vansteenkiste, P.; Van Speybroeck, V.; Marin, G.; Waroquier, M. Ab initio calculation of entropy and heat capacity of gas-phase n-alkanes using internal rotations. J. Phys. Chem. A 2003, 107, 3139–3145. [Google Scholar] [CrossRef]
- Berns, V.M.; Engelkemier, J.; Guo, Y.; Kilduff, B.J.; Fredrickson, D.C. Progress in visualizing atomic size effects with DFT-Chemical pressure analysis: From isolated atoms to trends in AB5 intermetallics. J. Chem. Theory Comput. 2014, 10, 3380–3392. [Google Scholar] [CrossRef]
- Wu, Q.; Zhu, W.; Xiao, H. Comparative DFT-and DFT-D-based molecular dynamics studies of pressure effects in crystalline 1, 3,5-triamino-2,4,6-trinitrobenzene at room temperature. RSC Adv. 2014, 4, 53149–53156. [Google Scholar] [CrossRef]
- Dutta, P.; Pandey, S.K. Understanding the temperature-and pressure-dependent electronic properties of FeSi: DFT + DMFT study. EPL (Europhys. Lett.) 2020, 132, 37003. [Google Scholar] [CrossRef]
- de Lázaro, S.R. Introductory Chapter: A Brief Mention for High-Pressure in Oxides from DFT Simulations. In Density Functional Theory Calculations; de Lázaro, S.R., Ed.; IntechOpen: London, UK, 2021; pp. 3–7. [Google Scholar]
- Nazir, G.; Ahmad, A.; Khan, M.F.; Tariq, S. Putting DFT to the trial: First principles pressure dependent analysis on optical properties of cubic perovskite SrZrO3. Comput. Condens. Matter 2015, 4, 32–39. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G. Implicit solvation models: Equilibria, structure, spectra, and dynamics. Chem. Rev. 1999, 99, 2161–2200. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Sato, H. A modern solvation theory: Quantum chemistry and statistical chemistry. Phys. Chem. Chem. Phys. 2013, 15, 7450–7465. [Google Scholar] [CrossRef] [PubMed]
- Mathew, K.; Sundararaman, R.; Letchworth-Weaver, K.; Arias, T.; Hennig, R.G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106. [Google Scholar] [CrossRef] [PubMed]
- Roux, B.; Simonson, T. Implicit solvent models. Biophys. Chem. 1999, 78, 1–20. [Google Scholar] [CrossRef]
- Kleinjung, J.; Fraternali, F. Design and application of implicit solvent models in biomolecular simulations. Curr. Opin. Struct. Biol. 2014, 25, 126–134. [Google Scholar] [CrossRef]
- Dong, F.; Wagoner, J.A.; Baker, N.A. Assessing the performance of implicit solvation models at a nucleic acid surface. Phys. Chem. Chem. Phys. 2008, 10, 4889–4902. [Google Scholar] [CrossRef][Green Version]
- Skyner, R.; McDonagh, J.; Groom, C.; Van Mourik, T.; Mitchell, J. A review of methods for the calculation of solution free energies and the modelling of systems in solution. Phys. Chem. Chem. Phys. 2015, 17, 6174–6191. [Google Scholar] [CrossRef]
- Alibakhshi, A.; Hartke, B. Improved prediction of solvation free energies by machine-learning polarizable continuum solvation model. Nat. Commun. 2021, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Basdogan, Y.; Maldonado, A.M.; Keith, J.A. Advances and challenges in modeling solvated reaction mechanisms for renewable fuels and chemicals. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, e1446. [Google Scholar] [CrossRef]
- Baker, N.A.; Bashford, D.; Case, D.A. Implicit solvent electrostatics in biomolecular simulation. In New Algorithms for Macromolecular Simulation; Springer: Berlin/Heidelberg, Germany, 2006; pp. 263–295. [Google Scholar]
- Mennucci, B. Polarizable continuum model. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 386–404. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V. Solvent effect on vertical electronic transitions by the polarizable continuum model. J. Chem. Phys. 2000, 112, 2427–2435. [Google Scholar] [CrossRef]
- Amovilli, C.; Barone, V.; Cammi, R.; Cancès, E.; Cossi, M.; Mennucci, B.; Pomelli, C.S.; Tomasi, J. Recent advances in the description of solvent effects with the polarizable continuum model. Adv. Quantum Chem. 1998, 32, 227–261. [Google Scholar]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- York, D.M.; Karplus, M. A smooth solvation potential based on the conductor-like screening model. J. Phys. Chem. A 1999, 103, 11060–11079. [Google Scholar] [CrossRef]
- Truong, T.N.; Nguyen, U.N.; Stefanovich, E.V. Generalized conductor-like screening model (GCOSMO) for solvation: An assessment of its accuracy and applicability. Int. J. Quantum Chem. 1996, 60, 1615–1622. [Google Scholar] [CrossRef]
- Lunkenheimer, B.; Köhn, A. Solvent effects on electronically excited states using the conductor-like screening model and the second-order correlated method ADC (2). J. Chem. Theory Comput. 2013, 9, 977–994. [Google Scholar] [CrossRef]
- Grochowski, P.; Trylska, J. Continuum molecular electrostatics, salt effects, and counterion binding—A review of the Poisson–Boltzmann theory and its modifications. Biopolym. Orig. Res. Biomol. 2008, 89, 93–113. [Google Scholar] [CrossRef]
- Vlachy, V. Ionic effects beyond Poisson-Boltzmann theory. Annu. Rev. Phys. Chem. 1999, 50, 145–165. [Google Scholar] [CrossRef]
- Lu, B.; Zhou, Y.; Holst, M.; McCammon, J. Recent progress in numerical methods for the Poisson-Boltzmann equation in biophysical applications. Commun. Comput. Phys. 2008, 3, 973–1009. [Google Scholar]
- Lamm, G. The poisson-boltzmann equation. Rev. Comput. Chem. 2003, 19, 147–333. [Google Scholar]
- Su, M.; Wang, Y. A brief review of continuous models for ionic solutions: The Poisson–Boltzmann and related theories. Commun. Theor. Phys. 2020, 72, 067601. [Google Scholar] [CrossRef]
- Wojciechowski, M.; Lesyng, B. Generalized Born model: Analysis, refinement, and applications to proteins. J. Phys. Chem. B 2004, 108, 18368–18376. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Mirzaei, S.; Ivanov, M.V.; Timerghazin, Q.K. Improving Performance of the SMD Solvation Model: Bondi Radii Improve Predicted Aqueous Solvation Free Energies of Ions and p K a Values of Thiols. J. Phys. Chem. A 2019, 123, 9498–9504. [Google Scholar] [CrossRef]
- Klamt, A.; Schuurmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin. Trans. 2 1993, 5, 799. [Google Scholar] [CrossRef]
- Klamt, A. Conductor-like screening model for real solvents: A new approach to the quantitative calculation of solvation phenomena. J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- Klamt, A.; Jonas, V. Treatment of the outlying charge in continuum solvation models. J. Chem. Phys. 1996, 105, 9972–9981. [Google Scholar] [CrossRef]
- Baldridge, K.; Klamt, A. First principles implementation of solvent effects without outlying charge error. J. Chem. Phys. 1997, 106, 6622. [Google Scholar] [CrossRef]
- Leung, B.O.; Reid, D.L.; Armstrong, D.A.; Rauk, A. Entropies in solution from entropies in the gas phase. J. Phys. Chem. A 2004, 108, 2720–2725. [Google Scholar] [CrossRef]
- Gilson, M.K.; Honig, B.H. Energetics of charge–charge interactions in proteins. Proteins Struct. Funct. Bioinform. 1988, 3, 32–52. [Google Scholar] [CrossRef]
- Zhou, H.X. Macromolecular electrostatic energy within the nonlinear Poisson–Boltzmann equation. J. Chem. Phys. 1994, 100, 3152–3162. [Google Scholar] [CrossRef]
- Labat, F.; Civalleri, B.; Dovesi, R. Implicit Solvation Using a Generalized Finite-Difference Approach in CRYSTAL: Implementation and Results for Molecules, Polymers, and Surfaces. J. Chem. Theory Comput. 2018, 14, 5969–5983. [Google Scholar] [CrossRef]
- Zhulina, E.; Borisov, O. Poisson–Boltzmann theory of pH-sensitive (annealing) polyelectrolyte brush. Langmuir 2011, 27, 10615–10633. [Google Scholar] [CrossRef]
- Wang, L.; Lin, J.; Zhang, Q. Self-consistent field theory study of the solvation effect in polyelectrolyte solutions: Beyond the Poisson–Boltzmann model. Soft Matter 2013, 9, 4015–4025. [Google Scholar] [CrossRef]
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Onufriev, A.V.; Case, D.A. Generalized Born implicit solvent models for biomolecules. Annu. Rev. Biophys. 2019, 48, 275–296. [Google Scholar] [CrossRef]
- Nguyen, H.; Pérez, A.; Bermeo, S.; Simmerling, C. Refinement of generalized born implicit solvation parameters for nucleic acids and their complexes with proteins. J. Chem. Theory Comput. 2015, 11, 3714–3728. [Google Scholar] [CrossRef]
- Boereboom, J.M.; Fleurat-Lessard, P.; Bulo, R.E. Explicit solvation matters: Performance of QM/MM solvation models in nucleophilic addition. J. Chem. Theory Comput. 2018, 14, 1841–1852. [Google Scholar] [CrossRef]
- Chen, J.; Chan, B.; Shao, Y.; Ho, J. How accurate are approximate quantum chemical methods at modelling solute–solvent interactions in solvated clusters? Phys. Chem. Chem. Phys. 2020, 22, 3855–3866. [Google Scholar] [CrossRef]
- Moors, S.L.; Brigou, B.; Hertsen, D.; Pinter, B.; Geerlings, P.; Van Speybroeck, V.; Catak, S.; De Proft, F. Influence of solvation and dynamics on the mechanism and kinetics of nucleophilic aromatic substitution reactions in liquid ammonia. J. Org. Chem. 2016, 81, 1635–1644. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Koper, M.T. First-principles computational electrochemistry: Achievements and challenges. Electrochim. Acta 2012, 84, 3–11. [Google Scholar] [CrossRef]
- Wang, L.-P.; Van Voorhis, T. A polarizable QM/MM explicit solvent model for computational electrochemistry in water. J. Chem. Theory Comput. 2012, 8, 610–617. [Google Scholar] [CrossRef]
- Marenich, A.V.; Ho, J.; Coote, M.L.; Cramer, C.J.; Truhlar, D.G. Computational electrochemistry: Prediction of liquid-phase reduction potentials. Phys. Chem. Chem. Phys. 2014, 16, 15068–15106. [Google Scholar] [CrossRef] [PubMed]
- Blumberger, J.; Bernasconi, L.; Tavernelli, I.; Vuilleumier, R.; Sprik, M. Electronic structure and solvation of copper and silver ions: A theoretical picture of a model aqueous redox reaction. J. Am. Chem. Soc. 2004, 126, 3928–3938. [Google Scholar] [CrossRef] [PubMed]
- Sterling, C.M.; Bjornsson, R. Multistep explicit solvation protocol for calculation of redox potentials. J. Chem. Theory Comput. 2018, 15, 52–67. [Google Scholar] [CrossRef]
- Schilling, M.; Luber, S. Determination of pKa values via ab initio molecular dynamics and its application to transition metal-based water oxidation catalysts. Inorganics 2019, 7, 73. [Google Scholar] [CrossRef]
- Uddin, N.; Choi, T.H.; Choi, C.H. Direct Absolute p K a Predictions and Proton Transfer Mechanisms of Small Molecules in Aqueous Solution by QM/MM-MD. J. Phys. Chem. B 2013, 117, 6269–6275. [Google Scholar] [CrossRef]
- Tummanapelli, A.K.; Vasudevan, S. Dissociation constants of weak acids from ab initio molecular dynamics using metadynamics: Influence of the inductive effect and hydrogen bonding on p K a Values. J. Phys. Chem. B 2014, 118, 13651–13657. [Google Scholar] [CrossRef]
- Cheng, J.; Sulpizi, M.; Sprik, M. Redox potentials and p K a for benzoquinone from density functional theory based molecular dynamics. J. Chem. Phys. 2009, 131, 154504. [Google Scholar] [CrossRef]
- De Meyer, T.; Ensing, B.; Rogge, S.M.; De Clerck, K.; Meijer, E.J.; Van Speybroeck, V. Acidity Constant (pK a) Calculation of Large Solvated Dye Molecules: Evaluation of Two Advanced Molecular Dynamics Methods. ChemPhysChem 2016, 17, 3447. [Google Scholar] [CrossRef]
- De Keer, L.; Kilic, K.I.; Van Steenberge, P.H.; Daelemans, L.; Kodura, D.; Frisch, H.; De Clerck, K.; Reyniers, M.-F.; Barner-Kowollik, C.; Dauskardt, R.H. Computational prediction of the molecular configuration of three-dimensional network polymers. Nat. Mater. 2021, 1–9. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Field, M.J.; Bash, P.A.; Karplus, M. A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comput. Chem. 1990, 11, 700–733. [Google Scholar] [CrossRef]
- Kerdcharoen, T.; Liedl, K.R.; Rode, B.M. A QM/MM simulation method applied to the solution of Li+ in liquid ammonia. Chem. Phys. 1996, 211, 313–323. [Google Scholar] [CrossRef]
- Kerdcharoen, T.; Morokuma, K. ONIOM-XS: An extension of the ONIOM method for molecular simulation in condensed phase. Chem. Phys. Lett. 2002, 355, 257–262. [Google Scholar] [CrossRef]
- Heyden, A.; Lin, H.; Truhlar, D.G. Adaptive partitioning in combined quantum mechanical and molecular mechanical calculations of potential energy functions for multiscale simulations. J. Phys. Chem. B 2007, 111, 2231–2241. [Google Scholar] [CrossRef]
- Bulo, R.E.; Ensing, B.; Sikkema, J.; Visscher, L. Toward a practical method for adaptive QM/MM simulations. J. Chem. Theory Comput. 2009, 5, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, N.; Várnai, C.; Solt, I.; Winfield, S.A.; Payne, M.C.; Simon, I.; Fuxreiter, M.; Csányi, G. QM/MM simulation of liquid water with an adaptive quantum region. Phys. Chem. Chem. Phys. 2012, 14, 646–656. [Google Scholar] [CrossRef]
- Rowley, C.N.; Roux, B. The solvation structure of Na+ and K+ in liquid water determined from high level ab initio molecular dynamics simulations. J. Chem. Theory Comput. 2012, 8, 3526–3535. [Google Scholar] [CrossRef] [PubMed]
- Pezeshki, S.; Lin, H. Molecular dynamics simulations of ion solvation by flexible-boundary QM/MM: On-the-fly partial charge transfer between QM and MM subsystems. J. Comput. Chem. 2014, 35, 1778–1788. [Google Scholar] [CrossRef]
- Bulo, R.E.; Michel, C.; Fleurat-Lessard, P.; Sautet, P. Multiscale modeling of chemistry in water: Are we there yet? J. Chem. Theory Comput. 2013, 9, 5567–5577. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A.; Eckert, F. COSMO-RS: A novel and efficient method for the a priori prediction of thermophysical data of liquids. Fluid Phase Equilibria 2000, 172, 43–72. [Google Scholar] [CrossRef]
- D’hooge, D.R.; Reyniers, M.F.; Marin, G.B. The crucial role of diffusional limitations in controlled radical polymerization. Macromol. React. Eng. 2013, 7, 362–379. [Google Scholar] [CrossRef]
- Achilias, D.; Kiparissides, C. Development of a general mathematical framework for modeling diffusion-controlled free-radical polymerization reactions. Macromolecules 1992, 25, 3739–3750. [Google Scholar] [CrossRef]
- Achilias, D.S. A review of modeling of diffusion controlled polymerization reactions. Macromol. Theory Simul. 2007, 16, 319–347. [Google Scholar] [CrossRef]
- Derboven, P.; D’hooge, D.R.; Stamenovic, M.M.; Espeel, P.; Marin, G.B.; Du Prez, F.E.; Reyniers, M.-F.O. Kinetic modeling of radical thiol–ene chemistry for macromolecular design: Importance of side reactions and diffusional limitations. Macromolecules 2013, 46, 1732–1742. [Google Scholar] [CrossRef]
- Wieme, J.; D’hooge, D.R.; Reyniers, M.F.; Marin, G.B. Importance of radical transfer in precipitation polymerization: The case of vinyl chloride suspension polymerization. Macromol. React. Eng. 2009, 3, 16–35. [Google Scholar] [CrossRef]
- Edeleva, M.; Marien, Y.W.; Van Steenberge, P.H.; D’hooge, D.R. Jacket temperature regulation allowing well-defined non-adiabatic lab-scale solution free radical polymerization of acrylates. React. Chem. Eng. 2021, 6, 1053–1069. [Google Scholar] [CrossRef]
- D’hooge, D.R.; Fantin, M.; Magenau, A.J.; Konkolewicz, D.; Matyjaszewski, K. Two-compartment kinetic Monte Carlo modelling of electrochemically mediated ATRP. React. Chem. Eng. 2018, 3, 866–874. [Google Scholar] [CrossRef]
- Dompazis, G.; Kanellopoulos, V.; Kiparissides, C. Development of a multi-compartment dynamic model for the prediction of particle size distribution and particle segregation in a catalytic olefin polymerization FBR. In Computer Aided Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2006; Volume 21, pp. 345–350. [Google Scholar]
- Drikakis, D.; Frank, M.; Tabor, G. Multiscale computational fluid dynamics. Energies 2019, 12, 3272. [Google Scholar] [CrossRef]
- Pan, H.; Chen, X.-Z.; Liang, X.-F.; Zhu, L.-T.; Luo, Z.-H. CFD simulations of gas–liquid–solid flow in fluidized bed reactors—A review. Powder Technol. 2016, 299, 235–258. [Google Scholar] [CrossRef]
- Xie, L.; Luo, Z.-H. Multiscale computational fluid dynamics–population balance model coupled system of atom transfer radical suspension polymerization in stirred tank reactors. Ind. Eng. Chem. Res. 2017, 56, 4690–4702. [Google Scholar] [CrossRef]
- Xie, L.; Zhu, L.T.; Luo, Z.H. Computational fluid dynamics simulation of multiscale mixing in anionic polymerization tubular reactors. Chem. Eng. Technol. 2016, 39, 857–864. [Google Scholar] [CrossRef]
- Lemos, T.; Melo, P.A.; Pinto, J.C. Stochastic modeling of polymer microstructure from residence time distribution. Macromol. React. Eng. 2015, 9, 259–270. [Google Scholar] [CrossRef]
- Van Steenberge, P.H.; Hutchinson, R.A. Design of 2-hydroxyethyl methacrylate-functional macromonomer dispersants by semi-batch cobalt chain transfer polymerization. AIChE J. 2019, 65, e16723. [Google Scholar] [CrossRef]
- Nasresfahani, A.; Schiavi, D.; Grady, M.C.; Hutchinson, R.A. An automated recipe generator for semi-batch solution radical copolymerization via comprehensive stochastic modeling and derivative-free algorithms. Chem. Eng. J. 2021, 417, 127920. [Google Scholar] [CrossRef]
- Heidarzadeh, N.; Hutchinson, R.A. Maximizing macromonomer content produced by starved-feed high temperature acrylate/methacrylate semi-batch polymerization. Polym. Chem. 2020, 11, 2137–2146. [Google Scholar] [CrossRef]
- Nasresfahani, A.; Heidarzadeh, N.; Bygott, E.G.; Hutchinson, R.A. Stochastic Modeling of Poly(acrylate) Distributions Obtained by Radical Polymerization under High-Temperature Semi-Batch Starved-Feed Conditions: Investigation of Model Predictions versus Experimental Data. Macromol. Theory Simul. 2021, 30, 2000093. [Google Scholar] [CrossRef]
- Schier, J.E.; Cohen-Sacal, D.; Larsen, O.R.; Hutchinson, R.A. The effect of hydrogen bonding on radical semi-batch copolymerization of butyl acrylate and 2-hydroxyethyl acrylate. Polymers 2017, 9, 368. [Google Scholar] [CrossRef]
- Nasresfahani, A.; Hutchinson, R.A. Distribution of functional groups in starved-feed semi-batch free radical copolymerization: An accelerated stochastic modeling approach. In Proceedings of the Polymer Reaction Engineering Conference, Punta Cana, Dominican Republic, 20–25 May 2018. [Google Scholar]
- D’hooge, D.R.; Van Steenberge, P.H.; Derboven, P.; Reyniers, M.-F.; Marin, G.B. Model-based design of the polymer microstructure: Bridging the gap between polymer chemistry and engineering. Polym. Chem. 2015, 6, 7081–7096. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, R.; Yang, L.; Yu, B.; Li, B.; Zhu, S. Effect of reversible addition−fragmentation transfer (RAFT) reactions on (mini) emulsion polymerization kinetics and estimate of RAFT equilibrium constant. Macromolecules 2006, 39, 1328–1337. [Google Scholar] [CrossRef]
- Senftle, T.P.; Hong, S.; Islam, M.M.; Kylasa, S.B.; Zheng, Y.; Shin, Y.K.; Junkermeier, C.; Engel-Herbert, R.; Janik, M.J.; Aktulga, H.M. The ReaxFF reactive force-field: Development, applications and future directions. NPJ Comput. Mater. 2016, 2, 1–14. [Google Scholar] [CrossRef]
- Mackerell, A.D., Jr.; Feig, M.; Brooks, C.L., III. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Shin, Y.K.; Ashraf, C.M.; van Duin, A.C. Development and Applications of the ReaxFF Reactive Force Field for Biological Systems. In Computational Materials, Chemistry, and Biochemistry: From Bold Initiatives to the Last Mile; Springer: Berlin/Heidelberg, Germany, 2021; pp. 157–182. [Google Scholar]
- Hatzell, M.C.; Raju, M.; Watson, V.J.; Stack, A.G.; Van Duin, A.C.; Logan, B.E. Effect of strong acid functional groups on electrode rise potential in capacitive mixing by double layer expansion. Environ. Sci. Technol. 2014, 48, 14041–14048. [Google Scholar] [CrossRef]
- Senftle, T.P.; Van Duin, A.C.; Janik, M.J. Determining in situ phases of a nanoparticle catalyst via grand canonical Monte Carlo simulations with the ReaxFF potential. Catal. Commun. 2014, 52, 72–77. [Google Scholar] [CrossRef]
- Pai, S.J.; Yeo, B.C.; Han, S.S. Development of the ReaxFF CBN reactive force field for the improved design of liquid CBN hydrogen storage materials. Phys. Chem. Chem. Phys. 2016, 18, 1818–1827. [Google Scholar] [CrossRef]
- Raju, M.; Ganesh, P.; Kent, P.R.; van Duin, A.C. Reactive force field study of Li/C systems for electrical energy storage. J. Chem. Theory Comput. 2015, 11, 2156–2166. [Google Scholar] [CrossRef]
- Van Steenberge, P.H.; D’hooge, D.R.; Reyniers, M.-F.O.; Marin, G.B.; Cunningham, M.F. 4-Dimensional modeling strategy for an improved understanding of miniemulsion NMP of acrylates initiated by SG1-macroinitiator. Macromolecules 2014, 47, 7732–7741. [Google Scholar] [CrossRef]
- Edeleva, M.V.; Kirilyuk, I.A.; Zhurko, I.F.; Parkhomenko, D.A.; Tsentalovich, Y.P.; Bagryanskaya, E.G. pH-Sensitive C–ON bond homolysis of alkoxyamines of imidazoline series with multiple ionizable groups as an approach for control of Nitroxide Mediated Polymerization. J. Org. Chem. 2011, 76, 5558–5573. [Google Scholar] [CrossRef] [PubMed]
- Edeleva, M.V.; Marque, S.R.; Bagryanskaya, E.G. Imidazoline and imidazolidine nitroxides as controlling agents in nitroxide-mediated pseudoliving radical polymerization. Russ. Chem. Rev. 2018, 87, 328. [Google Scholar] [CrossRef]
- Edeleva, M.; Audran, G.; Marque, S.; Bagryanskaya, E. Smart control of nitroxide-mediated polymerization initiators’ reactivity by pH, complexation with metals, and chemical transformations. Materials 2019, 12, 688. [Google Scholar] [CrossRef]
- Bagryanskaya, E.; Brémond, P.; Edeleva, M.; Marque, S.R.; Parkhomenko, D.; Roubaud, V.; Siri, D. Chemically triggered C–ON Bond homolysis in alkoxyamines. Part 2: DFT Investigation and application of the pH Effect on NMP. Macromol. Rapid Commun. 2012, 33, 152–157. [Google Scholar] [CrossRef]
- Edeleva, M.; Marque, S.R.; Kabytaev, K.; Guillaneuf, Y.; Gigmes, D.; Bagryanskaya, E. H-transfer reaction during decomposition of N-(2-methylpropyl)-N-(1-diethylphosphono-2,2-dimethylpropyl)-N-oxyl (SG1)-based alkoxyamines. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 1323–1336. [Google Scholar] [CrossRef]
- Konkolewicz, D.; Sosnowski, S.; D’hooge, D.R.; Szymanski, R.; Reyniers, M.-F.; Marin, G.B.; Matyjaszewski, K. Origin of the difference between branching in acrylates polymerization under controlled and free radical conditions: A computational study of competitive processes. Macromolecules 2011, 44, 8361–8373. [Google Scholar] [CrossRef]
- D’hooge, D.R.; Reyniers, M.F.; Marin, G.B. Methodology for kinetic modeling of atom transfer radical polymerization. Macromol. React. Eng. 2009, 3, 185–209. [Google Scholar] [CrossRef]
- Payne, K.A.; Van Steenberge, P.H.; D’hooge, D.R.; Reyniers, M.F.; Marin, G.B.; Hutchinson, R.A.; Cunningham, M.F. Controlled synthesis of poly [(butyl methacrylate)-co-(butyl acrylate)] via activator regenerated by electron transfer atom transfer radical polymerization: Insights and improvement. Polym. Int. 2014, 63, 848–857. [Google Scholar] [CrossRef]
- Derboven, P.; Van Steenberge, P.H.; Vandenbergh, J.; Reyniers, M.F.; Junkers, T.; D’hooge, D.R.; Marin, G.B. Improved livingness and control over branching in RAFT polymerization of acrylates: Could microflow synthesis make the difference? Macromol. Rapid Commun. 2015, 36, 2149–2155. [Google Scholar] [CrossRef]
- Dossi, M.; Storti, G.; Moscatelli, D. Initiation kinetics in free-radical polymerization: Prediction of thermodynamic and kinetic parameters based on ab initio calculations. Macromol. Theory Simul. 2010, 19, 170–178. [Google Scholar] [CrossRef]
- Srinivasan, S.; Lee, M.W.; Grady, M.C.; Soroush, M.; Rappe, A.M. Computational evidence for self-initiation in spontaneous high-temperature polymerization of methyl methacrylate. J. Phys. Chem. A 2011, 115, 1125–1132. [Google Scholar] [CrossRef]
- Srinivasan, S.; Lee, M.W.; Grady, M.C.; Soroush, M.; Rappe, A.M. Self-initiation mechanism in spontaneous thermal polymerization of ethyl and n-butyl acrylate: A theoretical study. J. Phys. Chem. A 2010, 114, 7975–7983. [Google Scholar] [CrossRef]
- Srinivasan, S.; Lee, M.W.; Grady, M.C.; Soroush, M.; Rappe, A.M. Computational study of the self-initiation mechanism in thermal polymerization of methyl acrylate. J. Phys. Chem. A 2009, 113, 10787–10794. [Google Scholar] [CrossRef]
- Liu, S.; Srinivasan, S.; Tao, J.; Grady, M.C.; Soroush, M.; Rappe, A.M. Modeling spin-forbidden monomer self-initiation reactions in spontaneous free-radical polymerization of acrylates and methacrylates. J. Phys. Chem. A 2014, 118, 9310–9318. [Google Scholar] [CrossRef]
- Laki, S.; Shamsabadi, A.A.; Riazi, H.; Grady, M.C.; Rappe, A.M.; Soroush, M. Experimental and mechanistic modeling study of self-initiated high-temperature polymerization of ethyl acrylate. Ind. Eng. Chem. Res. 2019, 59, 2621–2630. [Google Scholar] [CrossRef]
- Drawe, P.; Buback, M. The PLP-SEC Method: Perspectives and Limitations. Macromol. Theory Simul. 2016, 25, 74–84. [Google Scholar] [CrossRef]
- Kornherr, A.; Olaj, O.F.; Schnöll-Bitai, I.; Zifferer, G. A Method of Improving the Theoretical Basis of kp Determination from PLP-SEC Measurements. Macromol. Theory Simul. 2003, 12, 332–338. [Google Scholar] [CrossRef]
- Marien, Y.W.; Van Steenberge, P.H.; Barner-Kowollik, C.; Reyniers, M.-F.o.; Marin, G.B.; D’hooge, D.R. Kinetic Monte Carlo modeling extracts information on chain initiation and termination from complete PLP-SEC traces. Macromolecules 2017, 50, 1371–1385. [Google Scholar] [CrossRef]
- Marien, Y.W.; Van Steenberge, P.H.; Kockler, K.B.; Barner-Kowollik, C.; Reyniers, M.-F.; Marin, G.B.; D’hooge, D.R. Estimating the photodissociation quantum yield from PLP-SEC peak heights. Polym. Chem. 2017, 8, 3124–3128. [Google Scholar] [CrossRef]
- Marien, Y.W.; Van Steenberge, P.H.; Kockler, K.B.; Barner-Kowollik, C.; Reyniers, M.-F.; D’hooge, D.R.; Marin, G.B. An alternative method to estimate the bulk backbiting rate coefficient in acrylate radical polymerization. Polym. Chem. 2016, 7, 6521–6528. [Google Scholar] [CrossRef]
- Vir, A.B.; Marien, Y.; Van Steenberge, P.H.; Barner-Kowollik, C.; Reyniers, M.-F.; Marin, G.B.; D’hooge, D.R. Access to the β-scission rate coefficient in acrylate radical polymerization by careful scanning of pulse laser frequencies at elevated temperature. React. Chem. Eng. 2018, 3, 807–815. [Google Scholar] [CrossRef]
- Vir, A.B.; Marien, Y.W.; Van Steenberge, P.H.; Barner-Kowollik, C.; Reyniers, M.-F.; Marin, G.B.; D’hooge, D.R. From n-butyl acrylate Arrhenius parameters for backbiting and tertiary propagation to β-scission via stepwise pulsed laser polymerization. Polym. Chem. 2019, 10, 4116–4125. [Google Scholar] [CrossRef]
- Nikitin, A.N.; Lacık, I.; Hutchinson, R.A.; Buback, M.; Russell, G.T. Detection of PLP structure for accurate determination of propagation rate coefficients over an enhanced range of PLP-SEC conditions. Macromolecules 2018, 52, 55–71. [Google Scholar] [CrossRef]
- Van Speybroeck, V.; Van Neck, D.; Waroquier, M. Ab initio study of radical reactions: Role of coupled internal rotations on the reaction kinetics (III). J. Phys. Chem. A 2002, 106, 8945–8950. [Google Scholar] [CrossRef]
- Van Speybroeck, V.; Vansteenkiste, P.; Van Neck, D.; Waroquier, M. Why does the uncoupled hindered rotor model work well for the thermodynamics of n-alkanes? Chem. Phys. Lett. 2005, 402, 479–484. [Google Scholar] [CrossRef]
- Deglmann, P.; Müller, I.; Becker, F.; Schäfer, A.; Hungenberg, K.D.; Weiß, H. Prediction of propagation rate coefficients in free radical solution polymerization based on accurate quantum chemical methods: Vinylic and related monomers, including acrylates and acrylic acid. Macromol. React. Eng. 2009, 3, 496–515. [Google Scholar] [CrossRef]
- Izgorodina, E.I.; Coote, M.L. Accurate ab initio prediction of propagation rate coefficients in free-radical polymerization: Acrylonitrile and vinyl chloride. Chem. Phys. 2006, 324, 96–110. [Google Scholar] [CrossRef]
- Lin, C.Y.; Izgorodina, E.I.; Coote, M.L. First principles prediction of the propagation rate coefficients of acrylic and vinyl esters: Are we there yet? Macromolecules 2010, 43, 553–560. [Google Scholar] [CrossRef]
- Van Cauter, K.; Van Speybroeck, V.; Waroquier, M. Ab Initio Study of Poly (vinyl chloride) Propagation Kinetics: Head-to-Head versus Head-to-Tail Additions. ChemPhysChem 2007, 8, 541–552. [Google Scholar] [CrossRef]
- Dossi, M.; Storti, G.; Moscatelli, D. Quantum chemistry: A powerful tool in polymer reaction engineering. In Macromolecular Symposia; Wiley-VCH: Weinheim, Germany, 2011; pp. 16–25. [Google Scholar]
- Liang, K.; Dossi, M.; Moscatelli, D.; Hutchinson, R.A. An investigation of free-radical copolymerization propagation kinetics of styrene and 2-hydroxyethyl methacrylate. Macromolecules 2009, 42, 7736–7744. [Google Scholar] [CrossRef]
- Lin, C.Y.; Coote, M.L.; Petit, A.; Richard, P.; Poli, R.; Matyjaszewski, K. Ab initio study of the penultimate effect for the ATRP activation step using propylene, methyl acrylate, and methyl methacrylate monomers. Macromolecules 2007, 40, 5985–5994. [Google Scholar] [CrossRef]
- Degirmenci, I.; Aviyente, V.; Van Speybroeck, V.; Waroquier, M. DFT study on the propagation kinetics of free-radical polymerization of α-substituted acrylates. Macromolecules 2009, 42, 3033–3041. [Google Scholar] [CrossRef]
- Yu, X.; Pfaendtner, J.; Broadbelt, L.J. Ab initio study of acrylate polymerization reactions: Methyl methacrylate and methyl acrylate propagation. J. Phys. Chem. A 2008, 112, 6772–6782. [Google Scholar] [CrossRef]
- Idowu, L.A.; Hutchinson, R.A. Solvent effects on radical copolymerization kinetics of 2-hydroxyethyl methacrylate and butyl methacrylate. Polymers 2019, 11, 487. [Google Scholar] [CrossRef]
- Mavroudakis, E.; Cuccato, D.; Moscatelli, D. On the use of quantum chemistry for the determination of propagation, copolymerization, and secondary reaction kinetics in free radical polymerization. Polymers 2015, 7, 1789–1819. [Google Scholar] [CrossRef]
- Rooney, T.R.; Mavroudakis, E.; Lacík, I.; Hutchinson, R.A.; Moscatelli, D. Pulsed-laser and quantum mechanics study of n-butyl cyanoacrylate and methyl methacrylate free-radical copolymerization. Polym. Chem. 2015, 6, 1594–1603. [Google Scholar] [CrossRef]
- Dossi, M.; Liang, K.; Hutchinson, R.A.; Moscatelli, D. Investigation of free-radical copolymerization propagation kinetics of vinyl acetate and methyl methacrylate. J. Phys. Chem. B 2010, 114, 4213–4222. [Google Scholar] [CrossRef] [PubMed]
- Mavroudakis, E.; Cuccato, D.; Dossi, M.; Comino, G.; Moscatelli, D. Quantum chemistry investigation of fluorinated polymer systems of industrial interest. J. Phys. Chem. A 2014, 118, 238–247. [Google Scholar] [CrossRef]
- Thickett, S.C.; Gilbert, R.G. Propagation rate coefficient of acrylic acid: Theoretical investigation of the solvent effect. Polymer 2004, 45, 6993–6999. [Google Scholar] [CrossRef]
- Dogan, B.; Catak, S.; Van Speybroeck, V.; Waroquier, M.; Aviyente, V. Free radical polymerization of ethyl methacrylate and ethyl α-hydroxy methacrylate: A computational approach to the propagation kinetics. Polymer 2012, 53, 3211–3219. [Google Scholar] [CrossRef]
- Kayık, G.; Tüzün, N.Ş. Stereoselective propagation in free radical polymerization of acrylamides: A DFT study. J. Mol. Graph. Model. 2014, 49, 55–67. [Google Scholar] [CrossRef]
- Özaltın, T.F.; Değirmenci, İ.; Aviyente, V.; Atılgan, C.; De Sterck, B.; Van Speybroeck, V.; Waroquier, M. Controlling the tacticity in the polymerization of N-isopropylacrylamide: A computational study. Polymer 2011, 52, 5503–5512. [Google Scholar] [CrossRef]
- Özaltın, T.F.; Kura, B.; Catak, S.; Goossens, H.; Van Speybroeck, V.; Waroquier, M.; Aviyente, V. Effect of Lewis acids on the stereoregularity of N, N-dimethyl acrylamide: A computational approach. Eur. Polym. J. 2016, 83, 67–76. [Google Scholar] [CrossRef]
- Konstantinov, I.; Ewart, S.; Brown, H.; Eddy, C.; Mendenhall, J.; Munjal, S. Accurate density functional theory (DFT) protocol for screening and designing chain transfer and branching agents for LDPE systems. Mol. Syst. Des. Eng. 2018, 3, 228–242. [Google Scholar] [CrossRef]
- Beuermann, S.; Buback, M. Rate coefficients of free-radical polymerization deduced from pulsed laser experiments. Prog. Polym. Sci. 2002, 27, 191–254. [Google Scholar] [CrossRef]
- Goto, S. Computer model for commercial high-pressure polyethylene reactor based on elementary reaction rates obtained experimentally. Appl. Polym. Symp. 1981, 36, 21–40. [Google Scholar]
- Van Cauter, K.; Van Speybroeck, V.; Vansteenkiste, P.; Reyniers, M.-F.; Waroquier, M. Ab Initio Study of Free-Radical Polymerization: Polyethylene Propagation Kinetics. ChemPhysChem 2006, 7, 131–140. [Google Scholar] [CrossRef]
- Van Speybroeck, V.; Van Cauter, K.; Coussens, B.; Waroquier, M. Ab Initio Study of Free-Radical Polymerizations: Cost-Effective Methods to Determine the Reaction Rates. ChemPhysChem 2005, 6, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Filley, J.; McKinnon, J.T.; Wu, D.T.; Ko, G.H. Theoretical Study of Ethylene-Vinyl Acetate Free-Radical Copolymerization: Reactivity Ratios, Penultimate Effects, and Relative Rates of Chain Transfer to Polymer. Macromolecules 2002, 35, 3731–3738. [Google Scholar] [CrossRef]
- Weiss, H.; Deglmann, P. Needs and Opportunities-Molecular Modeling Meets Polymer Process Modeling. In Macromolecular Symposia; Wiley-VCH: Weinheim, Germany, 2011; pp. 6–15. [Google Scholar]
- Lazzari, S.; Lischewski, A.; Orlov, Y.; Deglmann, P.; Daiss, A.; Schreiner, E.; Vale, H. Toward a digital polymer reaction engineering. Adv. Chem. Eng 2020, 56, 187–230. [Google Scholar]
- Kröger, L.C.; Kopp, W.A.; Leonhard, K. Prediction of chain propagation rate constants of polymerization reactions in aqueous NIPAM/BIS and VCL/BIS systems. J. Phys. Chem. B 2017, 121, 2887–2895. [Google Scholar] [CrossRef]
- Camaioni, D.M.; Autrey, S.T.; Salinas, T.B.; Franz, J.A. Calculation of the effects of branching and conjugation on intrinsic barriers for H atom transfer reactions involving hydrocarbons. J. Am. Chem. Soc. 1996, 118, 2013–2022. [Google Scholar] [CrossRef]
- Ammon, H.L. New atom/functional group volume additivity data bases for the calculation of the crystal densities of C-, H-, N-, O-, F-, S-, P-, Cl-, and Br-containing compounds. Struct. Chem. 2001, 12, 205–212. [Google Scholar] [CrossRef]
- Bamford, C.; Dyson, R.; Eastmond, G. Network formation IV. The nature of the termination reaction in free-radical polymerization. Polymer 1969, 10, 885–899. [Google Scholar] [CrossRef]
- Nakamura, Y.; Lee, R.; Coote, M.L.; Yamago, S. Termination mechanism of the radical polymerization of acrylates. Macromol. Rapid Commun. 2016, 37, 506–513. [Google Scholar] [CrossRef]
- Nakamura, Y.; Ogihara, T.; Hatano, S.; Abe, M.; Yamago, S. Control of the termination mechanism in radical polymerization by viscosity: Selective disproportionation in viscous media. Chem. A Eur. J. 2017, 23, 1299–1305. [Google Scholar] [CrossRef]
- Cuccato, D.; Mavroudakis, E.; Moscatelli, D. Quantum chemistry investigation of secondary reaction kinetics in acrylate-based copolymers. J. Phys. Chem. A 2013, 117, 4358–4366. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Broadbelt, L.J. Kinetic study of 1, 5-hydrogen transfer reactions of methyl acrylate and butyl acrylate using quantum chemistry. Macromol. Theory Simul. 2012, 21, 461–469. [Google Scholar] [CrossRef]
- Dossi, M.; Storti, G.; Moscatelli, D. Relevance of backbiting and beta-scission reactions in the free radical polymerization of Acrylonitrile. In Macromolecular Symposia; Wiley-VCH: Weinheim, Germany, 2010; pp. 119–123. [Google Scholar]
- Moghadam, N.; Liu, S.; Srinivasan, S.; Grady, M.C.; Rappe, A.M.; Soroush, M. Theoretical study of intermolecular chain transfer to polymer reactions of alkyl acrylates. Ind. Eng. Chem. Res. 2015, 54, 4148–4165. [Google Scholar] [CrossRef]
- Mavroudakis, E.; Cuccato, D.; Moscatelli, D. Determination of Reaction Rate Coefficients in Free-Radical Polymerization Using Density Functional Theory. In Computational Quantum Chemistry; Elsevier: Amsterdam, The Netherlands, 2019; pp. 47–98. [Google Scholar]
- Luo, Q.; Shi, Z.; Li, D.; Zhu, C.; Wang, M. DFT study on the ionic cyclization mechanism of copolymers of acrylonitrile-itaconic acid: Direct or autocatalytic? Chem. Phys. Lett. 2017, 687, 158–162. [Google Scholar] [CrossRef]
- De Vleeschouwer, F.; Toro-Labbé, A.; Gutiérrez-Oliva, S.; Van Speybroeck, V.; Waroquier, M.; Geerlings, P.; De Proft, F. Reversibility from DFT-based reactivity indices: Intramolecular side reactions in the polymerization of poly (vinyl chloride). J. Phys. Chem. A 2009, 113, 7899–7908. [Google Scholar] [CrossRef]
- Cuccato, D.; Dossi, M.; Moscatelli, D.; Storti, G. Quantum Chemical Investigation of Secondary Reactions in Poly (vinyl chloride) Free-Radical Polymerization. Macromol. React. Eng. 2012, 6, 330–345. [Google Scholar] [CrossRef]
- Poli, R. Relationship Between One-Electron Transition-Metal Reactivity and Radical Polymerization Processes. Angew. Chem. Int. Ed. 2006, 45, 5058–5070. [Google Scholar] [CrossRef]
- Coote, M.L. Quantum-Chemical Modeling of Free-Radical Polymerization. Macromol. Theory Simul. 2009, 18, 388–400. [Google Scholar] [CrossRef]
- Rosen, B.M.; Percec, V. Single-electron transfer and single-electron transfer degenerative chain transfer living radical polymerization. Chem. Rev. 2009, 109, 5069–5119. [Google Scholar] [CrossRef]
- Lligadas, G.; Grama, S.; Percec, V. Single-electron transfer living radical polymerization platform to practice, develop, and invent. Biomacromolecules 2017, 18, 2981–3008. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process. Aust. J. Chem. 2005, 58, 379–410. [Google Scholar] [CrossRef]
- Coote, M.L.; Krenske, E.H.; Izgorodina, E.I. Computational studies of RAFT polymerization–mechanistic insights and practical applications. Macromol. Rapid Commun. 2006, 27, 473–497. [Google Scholar] [CrossRef]
- Coote, M.L. Ab initio study of the addition−fragmentation equilibrium in raft polymerization: When is polymerization retarded? Macromolecules 2004, 37, 5023–5031. [Google Scholar] [CrossRef]
- Izgorodina, E.I.; Coote, M.L. Reliable Low-Cost Theoretical Procedures for Studying Addition−Fragmentation in RAFT Polymerization. J. Phys. Chem. A 2006, 110, 2486–2492. [Google Scholar] [CrossRef]
- Coote, M.L.; Izgorodina, E.I.; Krenske, E.H.; Busch, M.; Barner-Kowollik, C. Quantum chemical mapping of initialization processes in RAFT polymerization. Macromol. Rapid Commun. 2006, 27, 1015–1022. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Buback, M.; Charleux, B.; Coote, M.L.; Drache, M.; Fukuda, T.; Goto, A.; Klumperman, B.; Lowe, A.B.; Mcleary, J.B. Mechanism and kinetics of dithiobenzoate-mediated RAFT polymerization. I. The current situation. J. Polym. Sci. Part A: Polym. Chem. 2006, 44, 5809–5831. [Google Scholar] [CrossRef]
- Coote, M.; Krenske, E.; Pas, E. Quantum-chemical studies of RAFT polymerization: Methodology, structure-reactivity correlations and kinetic implications. In Handbook of RAFT Polymerization; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 5–49. [Google Scholar]
- Coote, M.L.; Radom, L. Substituent effects in xanthate-mediated polymerization of vinyl acetate: Ab initio evidence for an alternative fragmentation pathway. Macromolecules 2004, 37, 590–596. [Google Scholar] [CrossRef]
- Coote, M.L.; Henry, D.J. Computer-aided design of a destabilized RAFT adduct radical: Toward improved RAFT agents for styrene-block-vinyl acetate copolymers. Macromolecules 2005, 38, 5774–5779. [Google Scholar] [CrossRef]
- Theis, A.; Stenzel, M.H.; Davis, T.P.; Coote, M.L.; Barner-Kowollik, C. A synthetic approach to a novel class of fluorine-bearing reversible addition–fragmentation chain transfer (RAFT) agents: F-RAFT. Aust. J. Chem. 2005, 58, 437–441. [Google Scholar] [CrossRef]
- Derboven, P.; Van Steenberge, P.H.; Reyniers, M.F.; Barner-Kowollik, C.; D’hooge, D.R.; Marin, G.B. Chain transfer in degenerative RAFT polymerization revisited: A comparative study of literature methods. Macromol. Theory Simul. 2016, 25, 104–115. [Google Scholar] [CrossRef]
- De Rybel, N.; Van Steenberge, P.H.; Reyniers, M.F.; Barner-Kowollik, C.; D’hooge, D.R.; Marin, G.B. An update on the pivotal role of kinetic modeling for the mechanistic understanding and design of bulk and solution raft polymerization. Macromol. Theory Simul. 2017, 26, 1600048. [Google Scholar] [CrossRef]
- Chiefari, J.; Mayadunne, R.T.; Moad, C.L.; Moad, G.; Rizzardo, E.; Postma, A.; Skidmore, M.A.; Thang, S.H. Thiocarbonylthio compounds (SC=(Z)S–R) in free radical polymerization with reversible addition-fragmentation chain transfer (RAFT polymerization). Effect of the activating group Z. Macromolecules 2003, 36, 2273–2283. [Google Scholar] [CrossRef]
- YK, C.J.C.; Ercole, F. Living free-radical polymerization by reversible addition−fragmentation chain transfer: The RAFT process. Macromolecules 1998, 31, 5559–5562. [Google Scholar]
- Rodríguez-Sanchez, I.; Glossman-Mitnik, D.; Zaragoza-Contreras, E.A. Theoretical evaluation of the order of reactivity of transfer agents utilized in RAFT polymerization: Group Z. J. Mol. Model. 2009, 15, 1133–1143. [Google Scholar] [CrossRef]
- Rodríguez-Sanchez, I.; Zaragoza-Contreras, E.A.; Glossman-Mitnik, D. Theoretical evaluation of the order of reactivity of transfer agents utilized in RAFT polymerization. J. Mol. Model. 2010, 16, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Devlaminck, D.J.G.; Van Steenberge, P.H.M.; De Keer, L.; Reyniers, M.-F.; D’Hooge, D.R. A detailed mechanistic study of bulk MADIX of styrene and its chain extension. Polym. Chem. 2017, 8, 6948–6963. [Google Scholar] [CrossRef]
- Busch, M.; Roth, M.; Stenzel, M.H.; Davis, T.P.; Barner-Kowollik, C. The use of novel F-RAFT agents in high temperature and high pressure ethene polymerization: Can control be achieved? Aust. J. Chem. 2007, 60, 788–793. [Google Scholar] [CrossRef]
- Benaglia, M.; Chen, M.; Chong, Y.K.; Moad, G.; Rizzardo, E.; Thang, S.H. Polystyrene-block-poly (vinyl acetate) through the Use of a Switchable RAFT Agent. Macromolecules 2009, 42, 9384–9386. [Google Scholar] [CrossRef]
- Keddie, D.J.; Guerrero-Sanchez, C.; Moad, G.; Rizzardo, E.; Thang, S.H. Switchable reversible addition–fragmentation chain transfer (RAFT) polymerization in aqueous solution, N, N-dimethylacrylamide. Macromolecules 2011, 44, 6738–6745. [Google Scholar] [CrossRef]
- Gigmes, D.; Gaudel-Siri, A.; Marque, S.R.; Bertin, D.; Tordo, P.; Astolfi, P.; Greci, L.; Rizzoli, C. Alkoxyamines of Stable Aromatic Nitroxides: N–O vs. C–O Bond Homolysis. Helv. Chim. Acta 2006, 89, 2312–2326. [Google Scholar] [CrossRef]
- Edeleva, M.; Marque, S.R.; Bertin, D.; Gigmes, D.; Guillaneuf, Y.; Morozov, S.V.; Bagryanskaya, E.G. Hydrogen-transfer reaction in nitroxide mediated polymerization of methyl methacrylate: 2,2-Diphenyl-3-phenylimino-2,3-dihydroindol-1-yloxyl nitroxide (DPAIO) vs. TEMPO. J. Polym. Sci. Part A: Polym. Chem. 2008, 46, 6828–6842. [Google Scholar] [CrossRef]
- Gaudel-Siri, A.; Siri, D.; Tordo, P. Homolysis of N-alkoxyamines: A computational study. Chemphyschem A Eur. J. Chem. Phys. Phys. Chem. 2006, 7, 430–438. [Google Scholar] [CrossRef]
- Hodgson, J.L.; Roskop, L.B.; Gordon, M.S.; Lin, C.Y.; Coote, M.L. Side reactions of nitroxide-mediated polymerization: N−O versus O−C cleavage of alkoxyamines. J. Phys. Chem. A 2010, 114, 10458–10466. [Google Scholar] [CrossRef]
- Parkhomenko, D.; Bagryanskaya, E.G.; Marque, S.R.; Siri, D. Intramolecular proton transfer (IPT) in alkoxyamine: A theoretical investigation. Phys. Chem. Chem. Phys. 2013, 15, 13862–13871. [Google Scholar] [CrossRef]
- Gryn’ova, G.; Lin, C.Y.; Coote, M.L. Which side-reactions compromise nitroxide mediated polymerization? Polym. Chem. 2013, 4, 3744–3754. [Google Scholar] [CrossRef]
- Parkhomenko, D.A.; Edeleva, M.V.; Kiselev, V.G.; Bagryanskaya, E.G. pH-sensitive C–ON bond homolysis of alkoxyamines of imidazoline series: A theoretical study. J. Phys. Chem. B 2014, 118, 5542–5550. [Google Scholar] [CrossRef]
- Audran, G.; Bikanga, R.; Brémond, P.; Edeleva, M.; Joly, J.-P.; Marque, S.R.; Nkolo, P.; Roubaud, V. How intramolecular hydrogen bonding (IHB) controls the C–ON bond homolysis in alkoxyamines. Org. Biomol. Chem. 2017, 15, 8425–8439. [Google Scholar] [CrossRef] [PubMed]
- Audran, G.; Bagryanskaya, E.; Edeleva, M.; Marque, S.R.; Parkhomenko, D.; Tretyakov, E.; Zhivetyeva, S. Coordination-Initiated nitroxide-mediated polymerization (CI-NMP). Aust. J. Chem. 2018, 71, 334–340. [Google Scholar] [CrossRef]
- Siegwart, D.J.; Oh, J.K.; Matyjaszewski, K. ATRP in the design of functional materials for biomedical applications. Prog. Polym. Sci. 2012, 37, 18–37. [Google Scholar] [CrossRef]
- Neugebauer, D. Two decades of molecular brushes by ATRP. Polymer 2015, 72, 413–421. [Google Scholar] [CrossRef]
- Krol, P.; Chmielarz, P. Recent advances in ATRP methods in relation to the synthesis of copolymer coating materials. Prog. Org. Coat. 2014, 77, 913–948. [Google Scholar] [CrossRef]
- Gillies, M.B.; Matyjaszewski, K.; Norrby, P.-O.; Pintauer, T.; Poli, R.; Richard, P. A DFT Study of R−X Bond Dissociation Enthalpies of Relevance to the Initiation Process of Atom Transfer Radical Polymerization. Macromolecules 2003, 36, 8551–8559. [Google Scholar] [CrossRef]
- Lin, C.Y.; Coote, M.L.; Gennaro, A.; Matyjaszewski, K. Ab initio evaluation of the thermodynamic and electrochemical properties of alkyl halides and radicals and their mechanistic implications for atom transfer radical polymerization. J. Am. Chem. Soc. 2008, 130, 12762–12774. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Kwak, Y.; Braunecker, W.; Tsarevsky, N.V.; Coote, M.L.; Matyjaszewski, K. Understanding atom transfer radical polymerization: Effect of ligand and initiator structures on the equilibrium constants. J. Am. Chem. Soc. 2008, 130, 10702–10713. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Fantin, M.; Pan, X.; de Fiebre, K.; Coote, M.L.; Matyjaszewski, K.; Liu, P. Mechanistically guided predictive models for ligand and initiator effects in copper-catalyzed atom transfer radical polymerization (Cu-ATRP). J. Am. Chem. Soc. 2019, 141, 7486–7497. [Google Scholar] [CrossRef]
- Woodruff, S.R.; Davis, B.J.; Tsarevsky, N.V. Selecting the Optimal Reaction Conditions for Copper-Mediated Atom Transfer Radical Polymerization at Low Catalyst Concentration. In Progress in Controlled Radical Polymerization: Mechanisms and Techniques; ACS Publications: Washington, DC, USA, 2012; pp. 99–113. [Google Scholar]
- Ehm, C.; Zaccaria, F.; Cipullo, R. From mechanistic investigation to quantitative prediction: Kinetics of homogeneous transition metal-catalyzed α-olefin polymerization predicted by computational chemistry. In Computational Quantum Chemistry; Elsevier: Amsterdam, The Netherlands, 2019; pp. 287–326. [Google Scholar]
- Kang, X.; Luo, Y.; Hou, Z. Theoretical Insights into Olefin Polymerization Catalyzed by Cationic Organo Rare-Earth Metal Complexes. Comput. Quantum Chem. 2019, 327–356. [Google Scholar]
- Zaccaria, F.; Budzelaar, P.H.; Zuccaccia, C.; Cipullo, R.; Macchioni, A.; Busico, V.; Ehm, C. Chain Transfer to Solvent and Monomer in Early Transition Metal Catalyzed Olefin Polymerization: Mechanisms and Implications for Catalysis. Catalysts 2021, 11, 215. [Google Scholar] [CrossRef]
- Kaufman, J.J. Quantum Chemical Investigations of the Mechanism of Cationic Polymerization; Johns Hopkins Univ Baltimore Md: Baltimore, MD, USA, 1980. [Google Scholar]
- Kaufman, J.J.; Hariharan, P.; Tobin, F.L.; Petrongolo, C. Electrostatic Molecular Potential Contour Maps from Ab-initio Calculations. 1. Biologically Significant Molecules. 2. Mechanism of Cationic Polymerization. In Chemical Applications of Atomic and Molecular Electrostatic Potentials; Springer: New York, NY, USA, 1981; pp. 335–380. [Google Scholar]
- Kaufman, J.J.; Hariharan, P.; Roszak, S.; Keegstra, P. Ab-initio electrostatic molecular potential contour maps for initiation step and Ab-Initio MRD-CI calculations for propagation step of cationic polymerization of oxetanes. In Makromolekulare Chemie. Macromolecular Symposia; Hüthig & Wepf: Basel, Switzerland, 1986; pp. 315–330. [Google Scholar]
- Hlil, A.R.; Balogh, J.; Moncho, S.; Su, H.L.; Tuba, R.; Brothers, E.N.; Al-Hashimi, M.; Bazzi, H.S. Ring opening metathesis polymerization (ROMP) of five-to eight-membered cyclic olefins: Computational, thermodynamic, and experimental approach. J. Polym. Sci. Part A: Polym. Chem. 2017, 55, 3137–3145. [Google Scholar] [CrossRef]
- Chen, J.; Motta, A.; Zhang, J.; Gao, Y.; Marks, T.J. Mechanism of organoscandium-catalyzed ethylene copolymerization with amino-olefins: A quantum chemical analysis. ACS Catal. 2019, 9, 8810–8818. [Google Scholar] [CrossRef]
- Vo, M.N.; Basdogan, Y.; Derksen, B.S.; Proust, N.; Cox, G.A.; Kowall, C.; Keith, J.A.; Johnson, J.K. Mechanism of isobutylene polymerization: Quantum chemical insight into AlCl3/H2O-catalyzed reactions. ACS Catal. 2018, 8, 8006–8013. [Google Scholar] [CrossRef]
- Vo, M.N. Quantum Chemical Studies of Metal Ion Solvation and Coordination and Elucidation of the Isobutylene Polymerization Mechanism; University of Pittsburgh: Pittsburgh, PA, USA, 2017. [Google Scholar]
- Fusco, R.; Longo, L.; Masi, F.; Garbassi, F. Ethylene polymerization with homogeneous Ziegler-Natta catalysts: Theoretical study on the role of ion pairs in the polymerization mechanism. Macromol. Rapid Commun. 1997, 18, 433–441. [Google Scholar] [CrossRef]
- Xu, Z.; Vanka, K.; Ziegler, T. Influence of the Counterion MeB (C6F5)3− and Solvent Effects on Ethylene Polymerization Catalyzed by [(CpSiMe2NR) TiMe]+: A Combined Density Functional Theory and Molecular Mechanism Study. Organometallics 2004, 23, 104–116. [Google Scholar] [CrossRef]
- Araujo, C.M.; Doherty, M.D.; Konezny, S.J.; Luca, O.R.; Usyatinsky, A.; Grade, H.; Lobkovsky, E.; Soloveichik, G.L.; Crabtree, R.H.; Batista, V.S. Tuning redox potentials of bis (imino) pyridine cobalt complexes: An experimental and theoretical study involving solvent and ligand effects. Dalton Trans. 2012, 41, 3562–3573. [Google Scholar] [CrossRef] [PubMed]
- Belelli, P.G.; Damiani, D.E.; Castellani, N.J. DFT theoretical studies of UV–Vis spectra and solvent effects in olefin polymerization catalysts. Chem. Phys. Lett. 2005, 401, 515–521. [Google Scholar] [CrossRef]
- Wang, X.; Kang, X.; Zhou, G.; Qu, J.; Hou, Z.; Luo, Y. DFT studies on cis-1,4-polymerization of dienes catalyzed by a cationic rare-earth metal complex bearing an ancillary PNP ligand. Polymers 2017, 9, 53. [Google Scholar] [CrossRef]
- Belelli, P.G.; Castellani, N.J. Solvent Effects in Olefin Polymerization Catalysts: A Dft Study; Universidad Nacional del Sur: Bahía Blanca, Argentina, 2005. [Google Scholar]
- Meelua, W.; Keawkla, N.; Oláh, J.; Jitonnom, J. DFT study of formation and properties of dinuclear zirconocene cations: Effects of ligand structure, solvent, and metal on the dimerization process. J. Organomet. Chem. 2020, 905, 121024. [Google Scholar] [CrossRef]
- Castro, L.; Kirillov, E.; Miserque, O.; Welle, A.; Haspeslagh, L.; Carpentier, J.-F.; Maron, L. Are solvent and dispersion effects crucial in olefin polymerization DFT calculations? Some insights from propylene coordination and insertion reactions with group 3 and 4 metallocenes. ACS Catal. 2015, 5, 416–425. [Google Scholar] [CrossRef]
- Cavallo, L.; Del Piero, S.; Ducéré, J.-M.; Fedele, R.; Melchior, A.; Morini, G.; Piemontesi, F.; Tolazzi, M. Key interactions in heterogeneous Ziegler− Natta catalytic systems: Structure and energetics of TiCl4−Lewis base complexes. J. Phys. Chem. C 2007, 111, 4412–4419. [Google Scholar] [CrossRef]
- Correa, A.; Credendino, R.; Pater, J.T.; Morini, G.; Cavallo, L. Theoretical Investigation of Active Sites at the Corners of MgCl2 Crystallites in Supported Ziegler–Natta Catalysts. Macromolecules 2012, 45, 3695–3701. [Google Scholar] [CrossRef]
- Harvey, J.N. DFT computation of relative spin-state energetics of transition metal compounds. Princ. Appl. Density Funct. Theory Inorg. Chem. I 2004, 151–184. [Google Scholar]
- Harvey, J.N. On the accuracy of density functional theory in transition metal chemistry. Annu. Rep. Sect. C (Phys. Chem.) 2006, 102, 203–226. [Google Scholar] [CrossRef]
- Harvey, J.N.; Poli, R.; Smith, K.M. Understanding the reactivity of transition metal complexes involving multiple spin states. Coord. Chem. Rev. 2003, 238, 347–361. [Google Scholar] [CrossRef]
- Cysewski, P.; Król, P.; Shyichuk, A. First principle simulations of ethylene glycol addition to diisocyanates. Macromol. Theory Simul. 2007, 16, 541–547. [Google Scholar] [CrossRef]
- Samuilov, A.Y.; Zenitova, L.; Samuilov, Y.D.; Konovalov, A. Quantum-chemical study on the reaction of phenyl isocyanate with linear methanol associates: II. Addition at the C=O bond. Russ. J. Org. Chem. 2009, 45, 68–73. [Google Scholar] [CrossRef]
- Samuilov, A.Y.; Balabanova, F.; Kamalov, T.; Samuilov, Y.D.; Konovalov, A. Quantum-chemical study on reactions of isocyanates with linear methanol associates: III.* Reaction of methyl isocyanate with linear methanol associates. Russ. J. Org. Chem. 2010, 46, 1452–1460. [Google Scholar] [CrossRef]
- Samuilov, A.Y.; Samuilov, Y.D. Noncatalytic and Autocatalytic Rate Constants of the Reaction of Phenyl Isocyanate with Butan-1-ol. Russ. J. Org. Chem. 2018, 54, 1749–1753. [Google Scholar] [CrossRef]
- Çoban, M.; Konuklar, F.A.S. A computational study on the mechanism and the kinetics of urethane formation. Comput. Theor. Chem. 2011, 963, 168–175. [Google Scholar] [CrossRef]
- Raspoet, G.; Nguyen, M.T.; McGarraghy, M.; Hegarty, A.F. The alcoholysis reaction of isocyanates giving urethanes: Evidence for a multimolecular mechanism. J. Org. Chem. 1998, 63, 6878–6885. [Google Scholar] [CrossRef]
- Cheikh, W.; Rózsa, Z.B.; Camacho López, C.O.; Mizsey, P.; Viskolcz, B.; Szőri, M.; Fejes, Z. Urethane formation with an excess of isocyanate or alcohol: Experimental and Ab initio study. Polymers 2019, 11, 1543. [Google Scholar] [CrossRef]
- Gertig, C.; Erdkamp, E.; Ernst, A.; Hemprich, C.; Kröger, L.C.; Langanke, J.; Bardow, A.; Leonhard, K. Reaction Mechanisms and Rate Constants of Auto-Catalytic Urethane Formation and Cleavage Reactions. ChemistryOpen 2021, 10, 534. [Google Scholar] [CrossRef] [PubMed]
- Tracy, J.; D’hooge, D.R.; Bosco, N.; Delgado, C.; Dauskardt, R. Evaluating and predicting molecular mechanisms of adhesive degradation during field and accelerated aging of photovoltaic modules. Prog. Photovolt. Res. Appl. 2018, 26, 981–993. [Google Scholar] [CrossRef]
- Reese, M.O.; Nardes, A.M.; Rupert, B.L.; Larsen, R.E.; Olson, D.C.; Lloyd, M.T.; Shaheen, S.E.; Ginley, D.S.; Rumbles, G.; Kopidakis, N. Photoinduced degradation of polymer and polymer–fullerene active layers: Experiment and theory. Adv. Funct. Mater. 2010, 20, 3476–3483. [Google Scholar] [CrossRef]
- Norrman, K.; Madsen, M.V.; Gevorgyan, S.A.; Krebs, F.C. Degradation patterns in water and oxygen of an inverted polymer solar cell. J. Am. Chem. Soc. 2010, 132, 16883–16892. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, J.; Hu, J.; Wania, F. Degradation of fluorotelomer-based polymers contributes to the global occurrence of fluorotelomer alcohol and perfluoroalkyl carboxylates: A combined dynamic substance flow and environmental fate modeling analysis. Environ. Sci. Technol. 2017, 51, 4461–4470. [Google Scholar] [CrossRef]
- Hamid, S.; Prichard, W. Mathematical modeling of weather-induced degradation of polymer properties. J. Appl. Polym. Sci. 1991, 43, 651–678. [Google Scholar] [CrossRef]
- Celina, M.C. Review of polymer oxidation and its relationship with materials performance and lifetime prediction. Polym. Degrad. Stab. 2013, 98, 2419–2429. [Google Scholar] [CrossRef]
- Smith, L.M.; Aitken, H.M.; Coote, M.L. The fate of the peroxyl radical in autoxidation: How does polymer degradation really occur? Acc. Chem. Res. 2018, 51, 2006–2013. [Google Scholar] [CrossRef] [PubMed]
- Ranzi, E.; Dente, M.; Faravelli, T.; Bozzano, G.; Fabini, S.; Nava, R.; Cozzani, V.; Tognotti, L. Kinetic modeling of polyethylene and polypropylene thermal degradation. J. Anal. Appl. Pyrolysis 1997, 40, 305–319. [Google Scholar] [CrossRef]
- Sánchez-Jiménez, P.E.; Pérez-Maqueda, L.A.; Perejón, A.; Criado, J.M. A new model for the kinetic analysis of thermal degradation of polymers driven by random scission. Polym. Degrad. Stab. 2010, 95, 733–739. [Google Scholar] [CrossRef]
- Bolland, J.; Gee, G. Kinetic studies in the chemistry of rubber and related materials. II. The kinetics of oxidation of unconjugated olefins. Trans. Faraday Soc. 1946, 42, 236–243. [Google Scholar] [CrossRef]
- Bolland, J.; Ten Have, P. Kinetic studies in the chemistry of rubber and related materials. V. The inhibitory effect of phenolic compounds on the thermal oxidation of ethyl linoleate. Discuss. Faraday Soc. 1947, 2, 252–260. [Google Scholar] [CrossRef]
- Bateman, L. Olefin oxidation. Q. Rev. Chem. Soc. 1954, 8, 147–167. [Google Scholar] [CrossRef]
- Gryn’ova, G.; Hodgson, J.L.; Coote, M.L. Revising the mechanism of polymer autooxidation. Org. Biomol. Chem. 2011, 9, 480–490. [Google Scholar] [CrossRef]
- Okanishi, T.; Tsuji, Y.; Sakiyama, Y.; Matsuno, S.; Bae, B.; Miyatake, K.; Uchida, M.; Watanabe, M. Effect of PEFC operating conditions on the durability of sulfonated poly (arylene ether sulfone ketone) multiblock membranes. Electrochim. Acta 2011, 56, 8989–8996. [Google Scholar] [CrossRef]
- Kabasawa, A.; Saito, J.; Yano, H.; Miyatake, K.; Uchida, H.; Watanabe, M. Durability of a novel sulfonated polyimide membrane in polymer electrolyte fuel cell operation. Electrochim. Acta 2009, 54, 1076–1082. [Google Scholar] [CrossRef]
- Kerres, J. Blend concepts for fuel cell membranes. In Polymer Membranes for Fuel Cells; Springer: Berlin/Heidelberg, Germany, 2008; pp. 1–37. [Google Scholar]
- Kang, M.-S.; Lee, M.-J. Anhydrous solid proton conductors based on perfluorosulfonic ionomer with polymeric solvent for polymer electrolyte fuel cell. Electrochem. Commun. 2009, 11, 457–460. [Google Scholar] [CrossRef]
- Kraka, E.; Cremer, D. Characterization of CF bonds with multiple-bond character: Bond lengths, stretching force constants, and bond dissociation energies. ChemPhysChem 2009, 10, 686–698. [Google Scholar] [CrossRef]
- Bernardes, C.E.; Minas da Piedade, M.E.; Amaral, L.M.; Ferreira, A.I.; Ribeiro da Silva, M.A.; Diogo, H.P.; Costa Cabral, B.J. Energetics of C−F, C−Cl, C−Br, and C−I bonds in 2-haloethanols. enthalpies of formation of XCH2CH2OH (X = F, Cl, Br, I) compounds and of the 2-hydroxyethyl radical. J. Phys. Chem. A 2007, 111, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Nam, P.-C.; Nguyen, M.T.; Chandra, A.K. The C−H and α (C−X) bond dissociation enthalpies of toluene, C6H5-CH2X (X = F, Cl), and their substituted derivatives: A dft study. J. Phys. Chem. A 2005, 109, 10342–10347. [Google Scholar] [CrossRef]
- Izgorodina, E.I.; Coote, M.L.; Radom, L. Trends in R−X bond dissociation energies (R = Me, Et, i-Pr, t-Bu; X = H, CH3, OCH3, OH, F): A surprising shortcoming of density functional theory. J. Phys. Chem. A 2005, 109, 7558–7566. [Google Scholar] [CrossRef] [PubMed]
- Coote, M.L.; Pross, A.; Radom, L. Variable trends in R−X bond dissociation energies (R = Me, Et, i-Pr, t-Bu). Org. Lett. 2003, 5, 4689–4692. [Google Scholar] [CrossRef]
- Blanksby, S.J.; Ellison, G.B. Bond dissociation energies of organic molecules. Acc. Chem. Res. 2003, 36, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Zavitsas, A.A. The relation between bond lengths and dissociation energies of carbon-carbon bonds. J. Phys. Chem. A 2003, 107, 897–898. [Google Scholar] [CrossRef]
- Matsunaga, N.; Rogers, D.W.; Zavitsas, A.A. Pauling’s electronegativity equation and a new corollary accurately predict bond dissociation enthalpies and enhance current understanding of the nature of the chemical bond. J. Org. Chem. 2003, 68, 3158–3172. [Google Scholar] [CrossRef] [PubMed]
- Beyer, M.K. The mechanical strength of a covalent bond calculated by density functional theory. J. Chem. Phys. 2000, 112, 7307–7312. [Google Scholar] [CrossRef]
- Pratt, D.A.; Wright, J.S.; Ingold, K. Theoretical Study of Carbon−Halogen Bond Dissociation Enthalpies of Substituted Benzyl Halides. How Important Are Polar Effects? J. Am. Chem. Soc. 1999, 121, 4877–4882. [Google Scholar] [CrossRef]
- Tokumasu, T.; Ogawa, I.; Koyama, M.; Ishimoto, T.; Miyamoto, A. A DFT study of bond dissociation trends of perfluorosulfonic acid membrane. J. Electrochem. Soc. 2010, 158, B175. [Google Scholar] [CrossRef]
- Devi, K.J.; Chandra, A.K. Theoretical investigation of the gas-phase reactions of (CF3)2CHOCH3 with OH radical. Chem. Phys. Lett. 2011, 502, 23–28. [Google Scholar] [CrossRef]
- Yu, T.H.; Sha, Y.; Liu, W.-G.; Merinov, B.V.; Shirvanian, P.; Goddard, W.A., III. Mechanism for degradation of Nafion in PEM fuel cells from quantum mechanics calculations. J. Am. Chem. Soc. 2011, 133, 19857–19863. [Google Scholar] [CrossRef]
- Xie, T.; Hayden, C.A. A kinetic model for the chemical degradation of perfluorinated sulfonic acid ionomers: Weak end groups versus side chain cleavage. Polymer 2007, 48, 5497–5506. [Google Scholar] [CrossRef]
- Panchenko, A. DFT investigation of the polymer electrolyte membrane degradation caused by OH radicals in fuel cells. J. Membr. Sci. 2006, 278, 269–278. [Google Scholar] [CrossRef]
- Sai, N.; Leung, K.; Zádor, J.; Henkelman, G. First principles study of photo-oxidation degradation mechanisms in P3HT for organic solar cells. Phys. Chem. Chem. Phys. 2014, 16, 8092–8099. [Google Scholar] [CrossRef] [PubMed]
- Ebadi, M.; Marchiori, C.; Mindemark, J.; Brandell, D.; Araujo, C.M. Assessing structure and stability of polymer/lithium-metal interfaces from first-principles calculations. J. Mater. Chem. A 2019, 7, 8394–8404. [Google Scholar] [CrossRef]
- Xing, L.; Borodin, O.; Smith, G.D.; Li, W. Density functional theory study of the role of anions on the oxidative decomposition reaction of propylene carbonate. J. Phys. Chem. A 2011, 115, 13896–13905. [Google Scholar] [CrossRef]
- Ma, Q.; Shuler, P.J.; Aften, C.W.; Tang, Y. Theoretical studies of hydrolysis and stability of polyacrylamide polymers. Polym. Degrad. Stab. 2015, 121, 69–77. [Google Scholar] [CrossRef]
- Duan, L.; D’hooge, D.R.; Cardon, L. Recent progress on flexible and stretchable piezoresistive strain sensors: From design to application. Prog. Mater. Sci. 2020, 114, 100617. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Sheldon, B.W. Deformation and stress in electrode materials for Li-ion batteries. Prog. Mater. Sci. 2014, 63, 58–116. [Google Scholar] [CrossRef]
- He, Q.; Yu, B.; Li, Z.; Zhao, Y. Density functional theory for battery materials. Energy Environ. Mater. 2019, 2, 264–279. [Google Scholar] [CrossRef]
- Allam, O.; Cho, B.W.; Kim, K.C.; Jang, S.S. Application of DFT-based machine learning for developing molecular electrode materials in Li-ion batteries. RSC Adv. 2018, 8, 39414–39420. [Google Scholar] [CrossRef]
- Yan, L.-M.; Su, J.-M.; Sun, C.; Yue, B.-H. Review of the first principles calculations and the design of cathode materials for Li-ion batteries. Adv. Manuf. 2014, 2, 358–368. [Google Scholar] [CrossRef]
- Wang, A.; Kadam, S.; Li, H.; Shi, S.; Qi, Y. Review on modeling of the anode solid electrolyte interphase (SEI) for lithium-ion batteries. NPJ Comput. Mater. 2018, 4, 1–26. [Google Scholar] [CrossRef]
- Yang, X.; Luo, J.; Sun, X. Towards high-performance solid-state Li–S batteries: From fundamental understanding to engineering design. Chem. Soc. Rev. 2020, 49, 2140–2195. [Google Scholar] [CrossRef]
- Adekoya, D.; Qian, S.; Gu, X.; Wen, W.; Li, D.; Ma, J.; Zhang, S. DFT-guided design and fabrication of carbon-nitride-based materials for energy storage devices: A review. Nano-Micro Lett. 2021, 13, 1–44. [Google Scholar] [CrossRef]
- Leung, K. DFT modelling of explicit solid–solid interfaces in batteries: Methods and challenges. Phys. Chem. Chem. Phys. 2020, 22, 10412–10425. [Google Scholar] [CrossRef]
- Moens, J.; Geerlings, P.; Roos, G. A conceptual DFT approach for the evaluation and interpretation of redox potentials. Chem. A Eur. J. 2007, 13, 8174–8184. [Google Scholar] [CrossRef]
- Tanaka, A.; Nakashima, K. DFT studies of ESR parameters for N–O centered radicals, N-alkoxyaminyl and aminoxyl radicals. Magn. Reson. Chem. 2011, 49, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, N.; Blase, X.; Hautier, G.; Charlier, J.-C.; Rignanese, G.-M. Ab initio calculations of open-cell voltage in Li-ion organic radical batteries. J. Phys. Chem. C 2015, 119, 23373–23378. [Google Scholar] [CrossRef]
- de la Cruz, C.; Molina, A.; Patil, N.; Ventosa, E.; Marcilla, R.; Mavrandonakis, A. New insights into phenazine-based organic redox flow batteries by using high-throughput DFT modelling. Sustain. Energy Fuels 2020, 4, 5513–5521. [Google Scholar] [CrossRef]
- Ma, R.; Sharma, V.; Baldwin, A.F.; Tefferi, M.; Offenbach, I.; Cakmak, M.; Weiss, R.; Cao, Y.; Ramprasad, R.; Sotzing, G.A. Rational design and synthesis of polythioureas as capacitor dielectrics. J. Mater. Chem. A 2015, 3, 14845–14852. [Google Scholar] [CrossRef]
- Cui, F.-Z.; Liu, Z.; Ma, D.-L.; Liu, L.; Huang, T.; Zhang, P.; Tan, D.; Wang, F.; Jiang, G.-F.; Wu, Y. Polyarylimide and porphyrin based polymer microspheres for zinc ion hybrid capacitors. Chem. Eng. J. 2021, 405, 127038. [Google Scholar] [CrossRef]
- Wang, C.; Pilania, G.; Boggs, S.; Kumar, S.; Breneman, C.; Ramprasad, R. Computational strategies for polymer dielectrics design. Polymer 2014, 55, 979–988. [Google Scholar] [CrossRef]
- Wang, C.; Pilania, G.; Ramprasad, R.; Agarwal, M.; Misra, M.; Kumar, S.; Yuan, X.; Mike Chung, T. Dielectric permittivity enhancement in hydroxyl functionalized polyolefins via cooperative interactions with water. Appl. Phys. Lett. 2013, 102, 152901. [Google Scholar] [CrossRef]
- Gonze, X.; Lee, C. Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory. Phys. Rev. B 1997, 55, 10355. [Google Scholar] [CrossRef]
- Gonze, X. First-principles responses of solids to atomic displacements and homogeneous electric fields: Implementation of a conjugate-gradient algorithm. Phys. Rev. B 1997, 55, 10337. [Google Scholar] [CrossRef]
- Pilania, G.; Wang, C.; Jiang, X.; Rajasekaran, S.; Ramprasad, R. Accelerating materials property predictions using machine learning. Sci. Rep. 2013, 3, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pilania, G.; Wang, C.; Wu, K.; Sukumar, N.; Breneman, C.; Sotzing, G.; Ramprasad, R. New group IV chemical motifs for improved dielectric permittivity of polyethylene. J. Chem. Inf. Modeling 2013, 53, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Hehre, W.J. A Guide to Molecular Mechanics and Quantum Chemical Calculations; Wavefunction: Irvine, CA, USA, 2003; Volume 2. [Google Scholar]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]









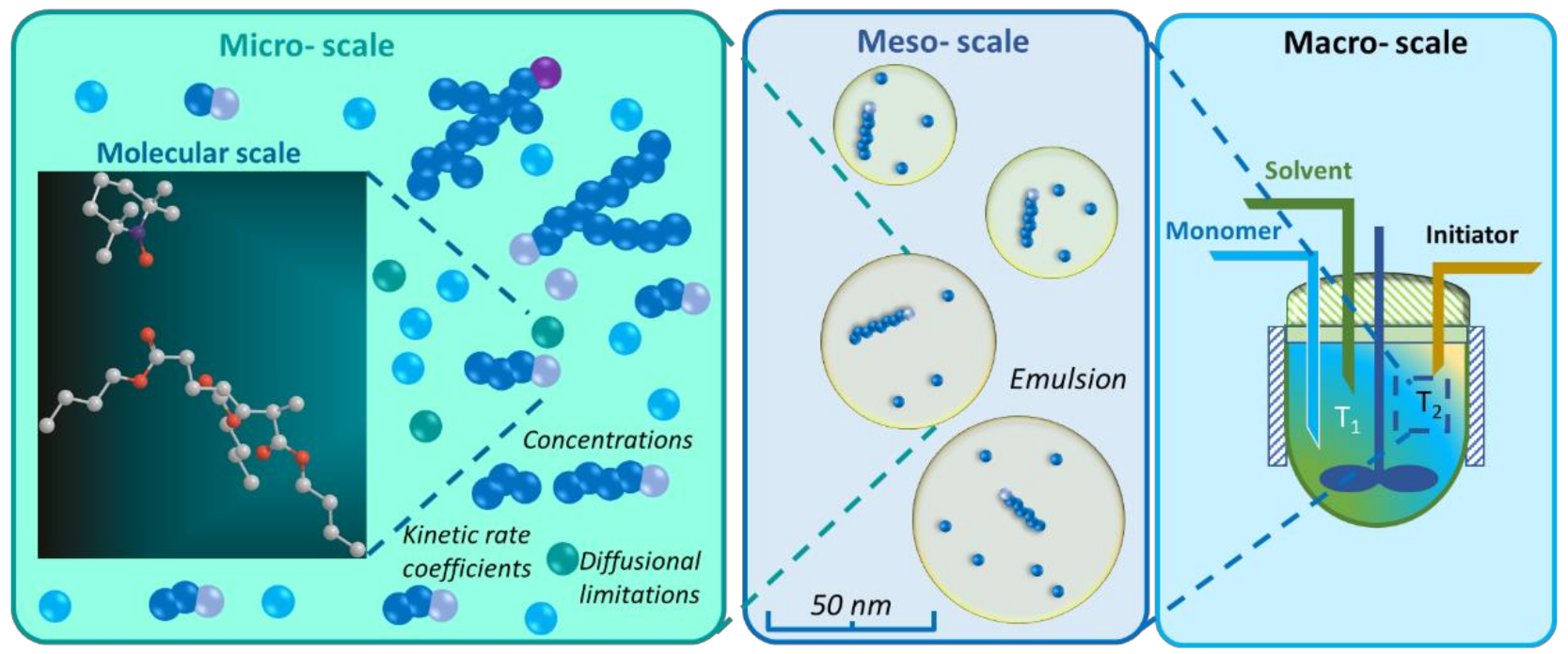{kind=link}
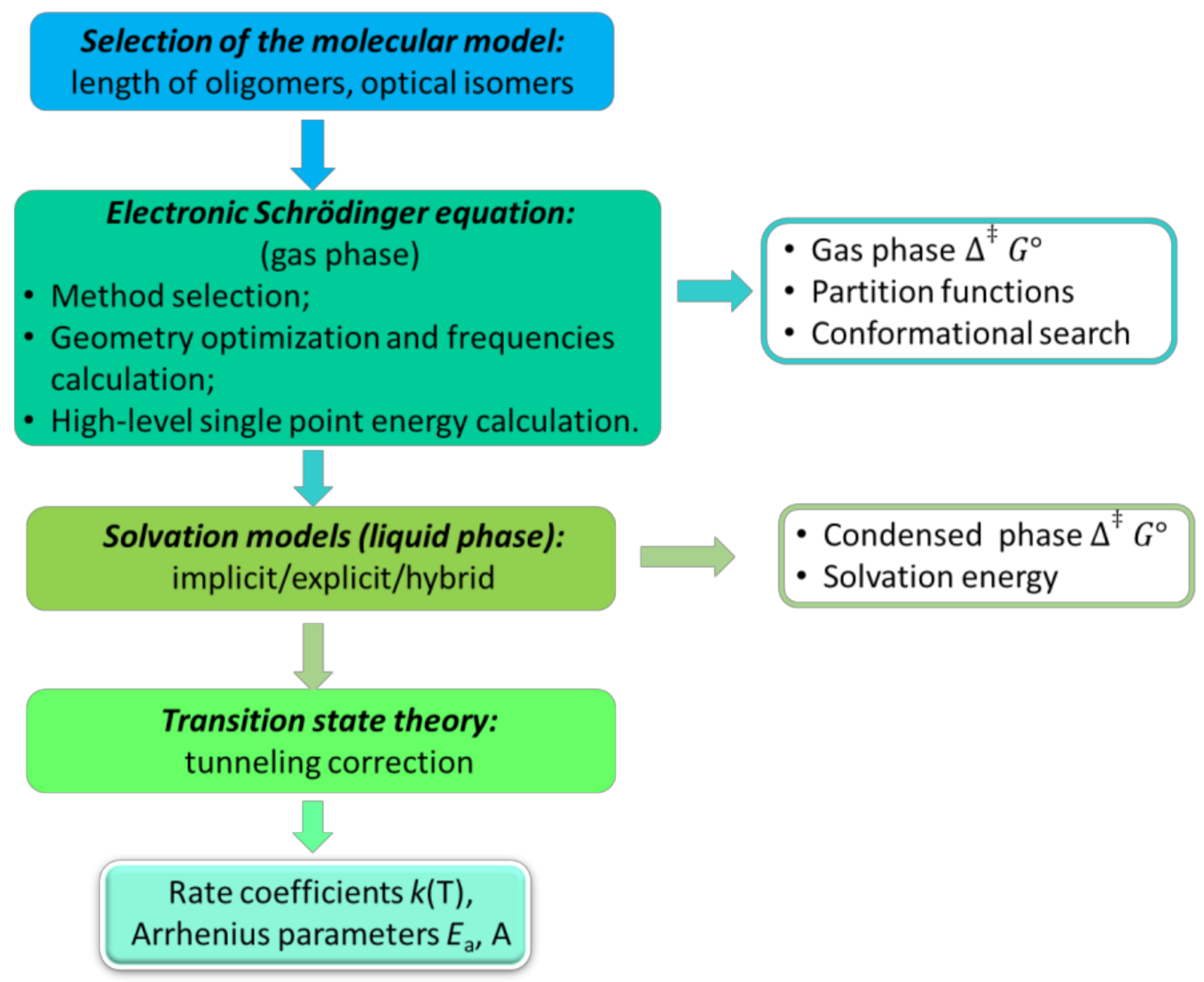{kind=link}
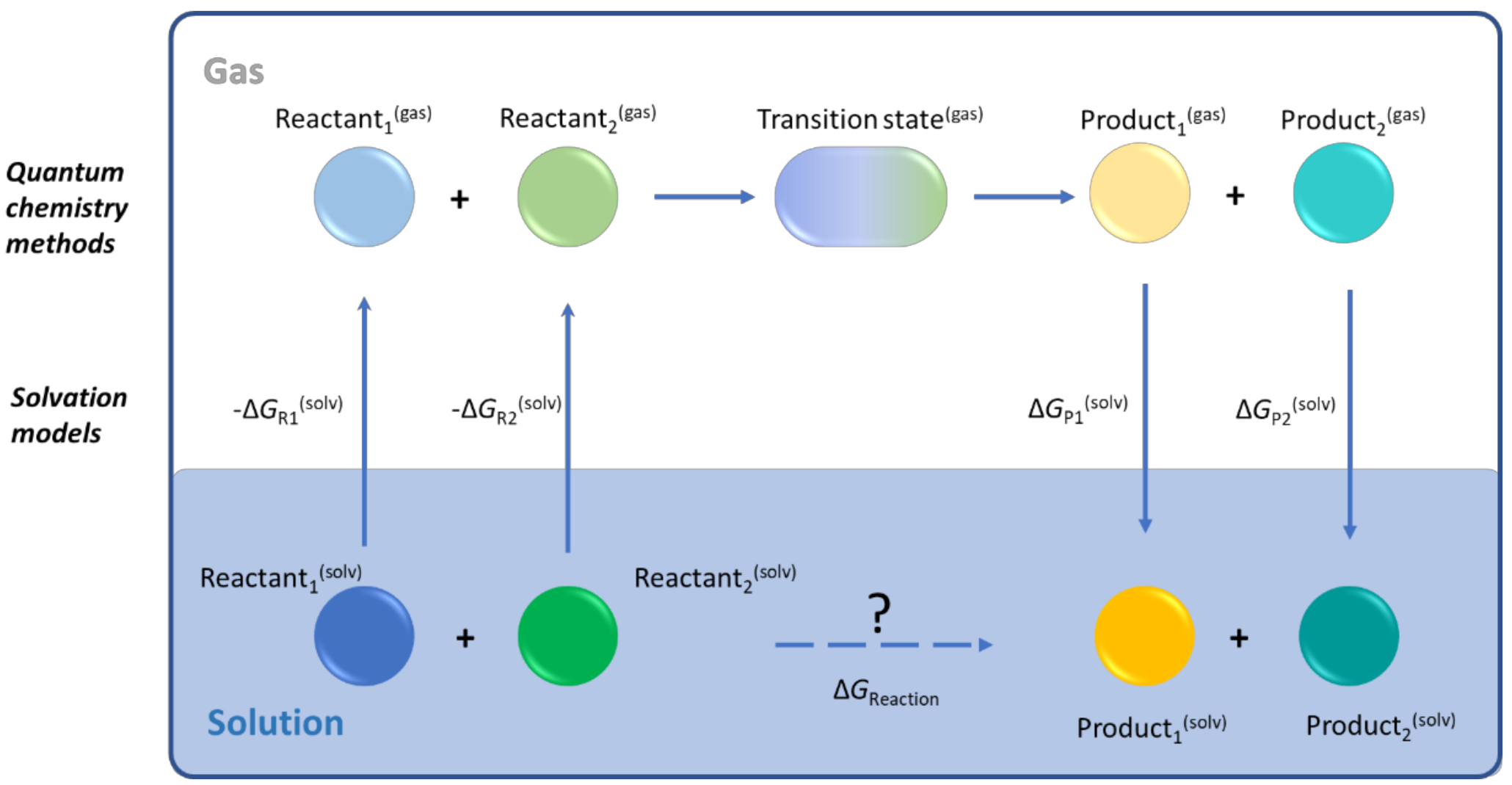{kind=link}
{kind=link}
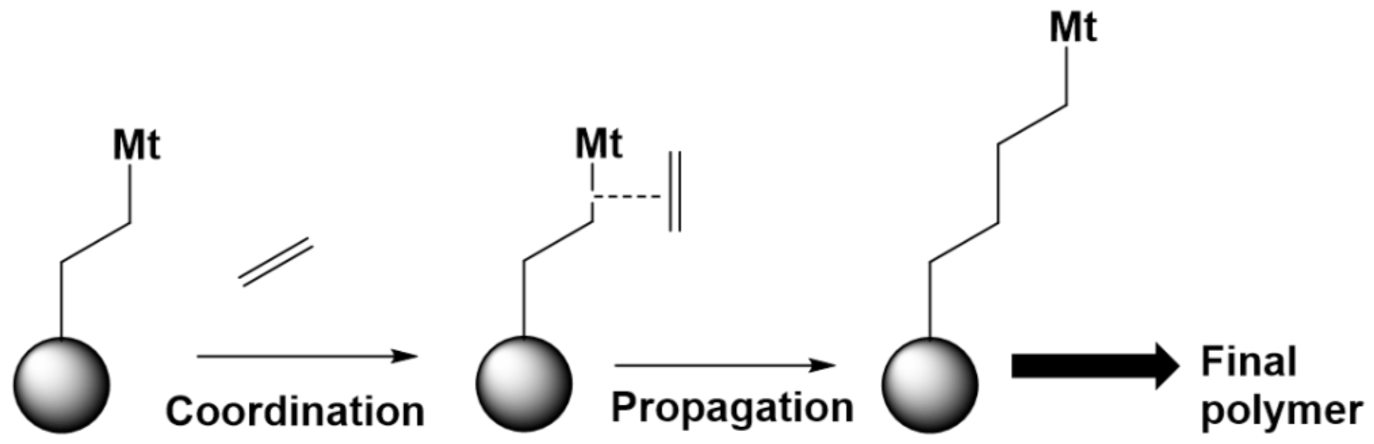{kind=link}
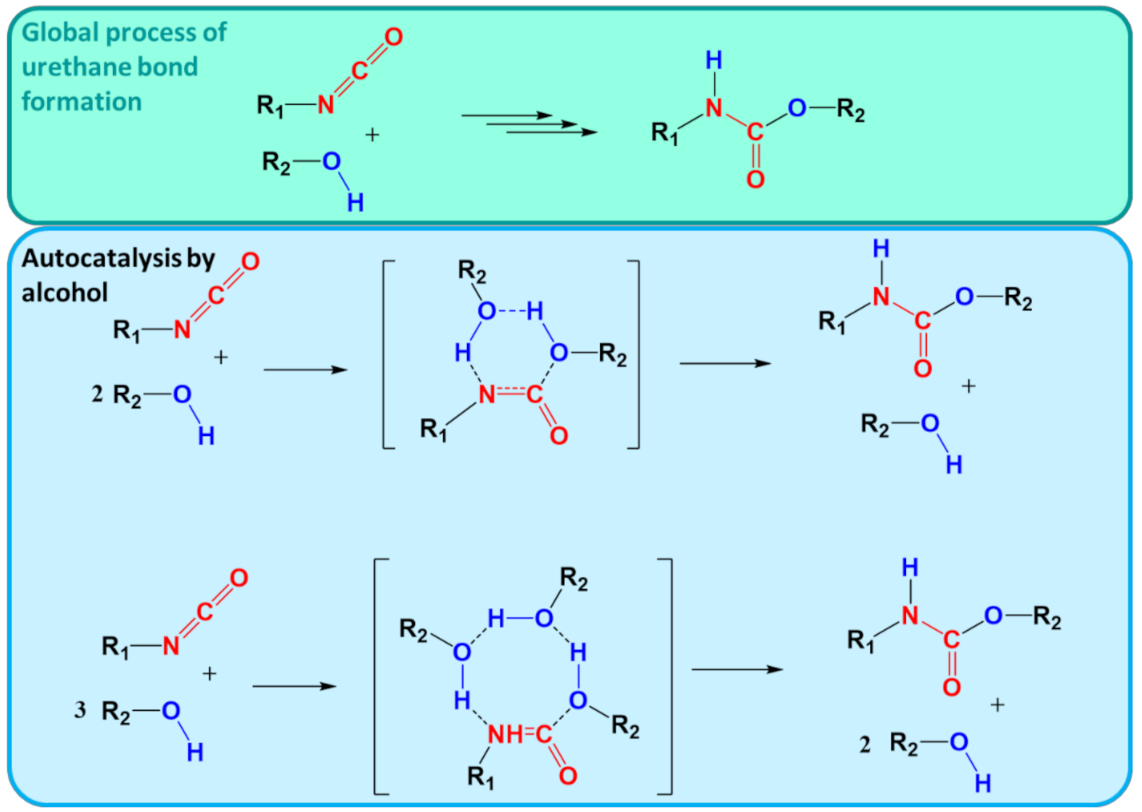{kind=link}
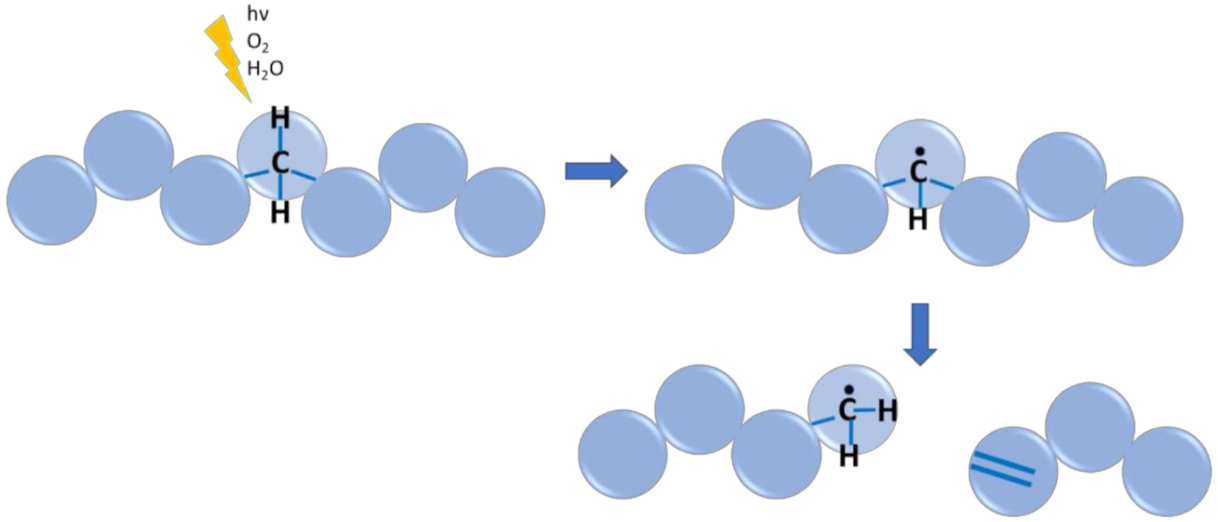{kind=link}
{kind=link}
| Explicit Models | Implicit Models | |
|---|---|---|
| Features | All solvent molecules are explicitly represented. | Representation of solvent as a continuum. |
| Advantages | Realistic physical system description. Detailed interaction modeling is provided. High accuracy of calculation. Full details on the molecular geometry. | Simple. Low computational costs No explicit solvent atoms. High level of theory is possible due to low computational costs. |
| Disadvantages | Expensive for computation. Long iterative cycle required to equilibrate solvent to solute. Often solvent and solute are not polarizable. Large fluctuations in calculation results due to use of small system size. | Ignoring of specific short-range effects. Less accurate. Need to define an artificial boundary between the solute and solvent. No “good” model for treating short-range effects (dispersion and cavity). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edeleva, M.; Van Steenberge, P.H.M.; Sabbe, M.K.; D’hooge, D.R. Connecting Gas-Phase Computational Chemistry to Condensed Phase Kinetic Modeling: The State-of-the-Art. Polymers 2021, 13, 3027. https://doi.org/10.3390/polym13183027
Edeleva M, Van Steenberge PHM, Sabbe MK, D’hooge DR. Connecting Gas-Phase Computational Chemistry to Condensed Phase Kinetic Modeling: The State-of-the-Art. Polymers. 2021; 13(18):3027. https://doi.org/10.3390/polym13183027
Chicago/Turabian StyleEdeleva, Mariya, Paul H.M. Van Steenberge, Maarten K. Sabbe, and Dagmar R. D’hooge. 2021. "Connecting Gas-Phase Computational Chemistry to Condensed Phase Kinetic Modeling: The State-of-the-Art" Polymers 13, no. 18: 3027. https://doi.org/10.3390/polym13183027
APA StyleEdeleva, M., Van Steenberge, P. H. M., Sabbe, M. K., & D’hooge, D. R. (2021). Connecting Gas-Phase Computational Chemistry to Condensed Phase Kinetic Modeling: The State-of-the-Art. Polymers, 13(18), 3027. https://doi.org/10.3390/polym13183027