
Crystallization of Polymers under the Influence of an External Force Field



Abstract
:1. Introduction
2. A Thermodynamic Analysis of the Influence of Stretching on the Crystallization of Polymers
3. Simulation Results
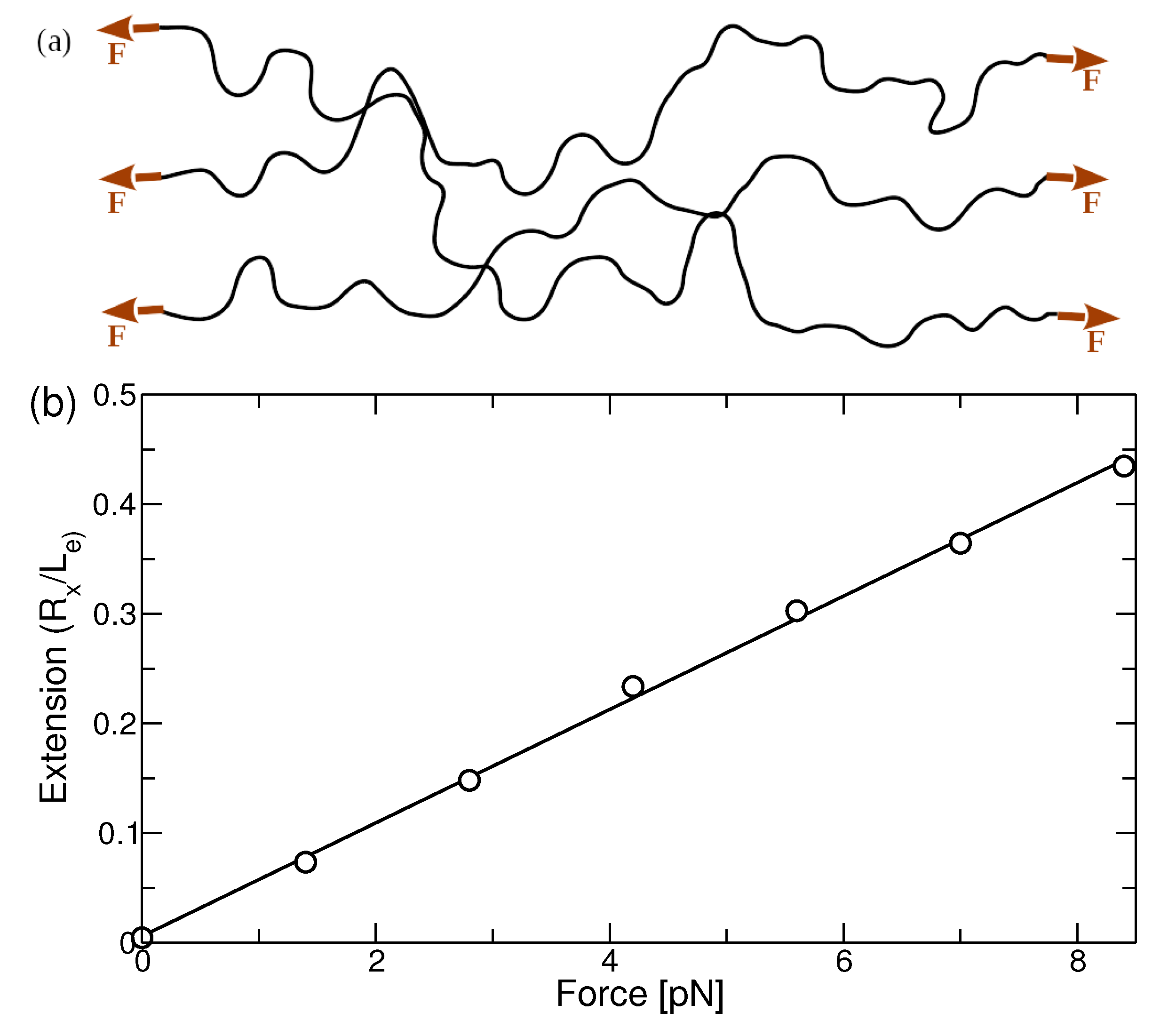
3.1. Application of Force Dipoles to Melt State
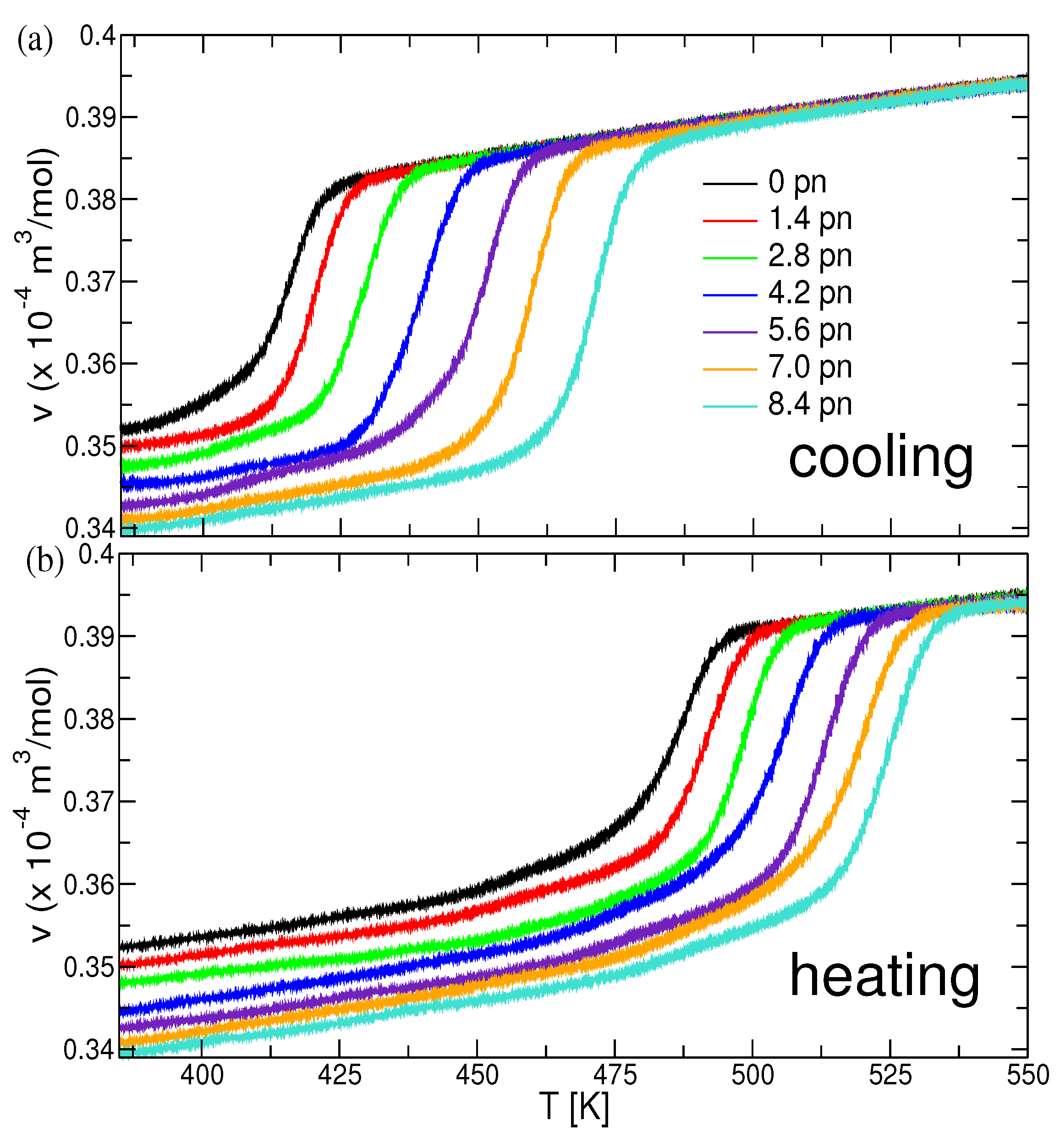
3.2. The Crystallization of Polymer Melts under Force
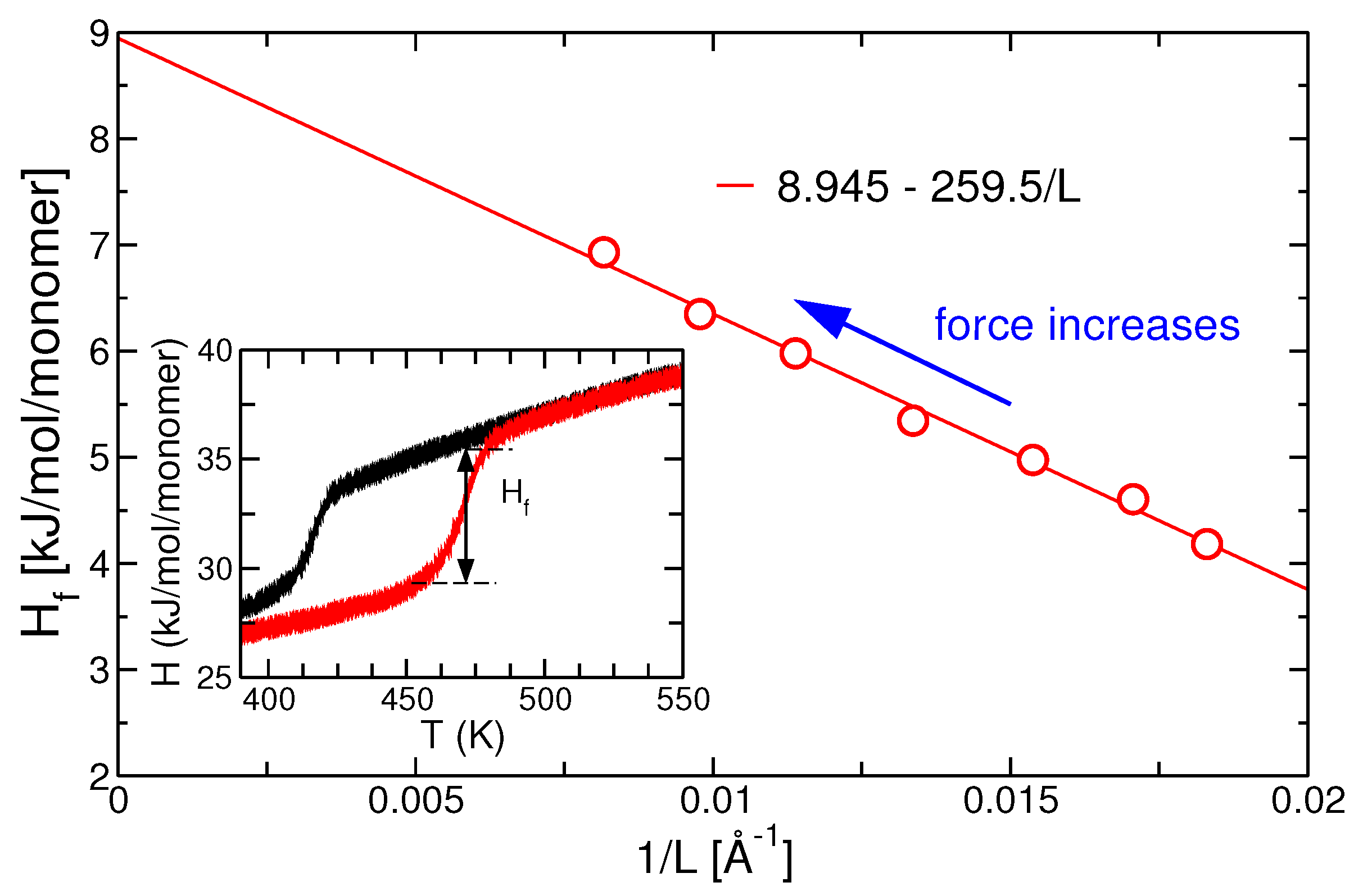
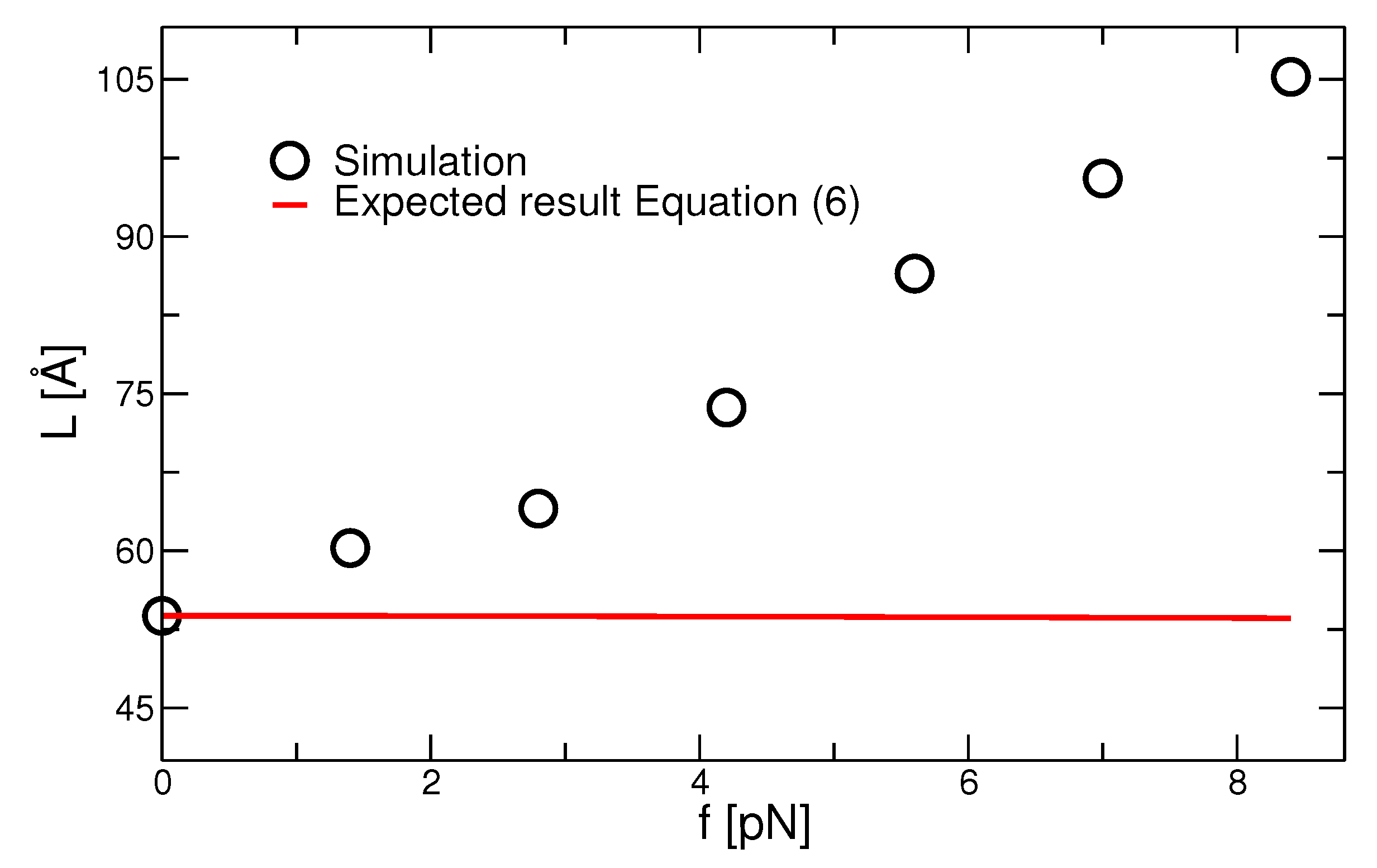
3.3. Thermodynamic Analysis and Instantaneous Quenching Simulations
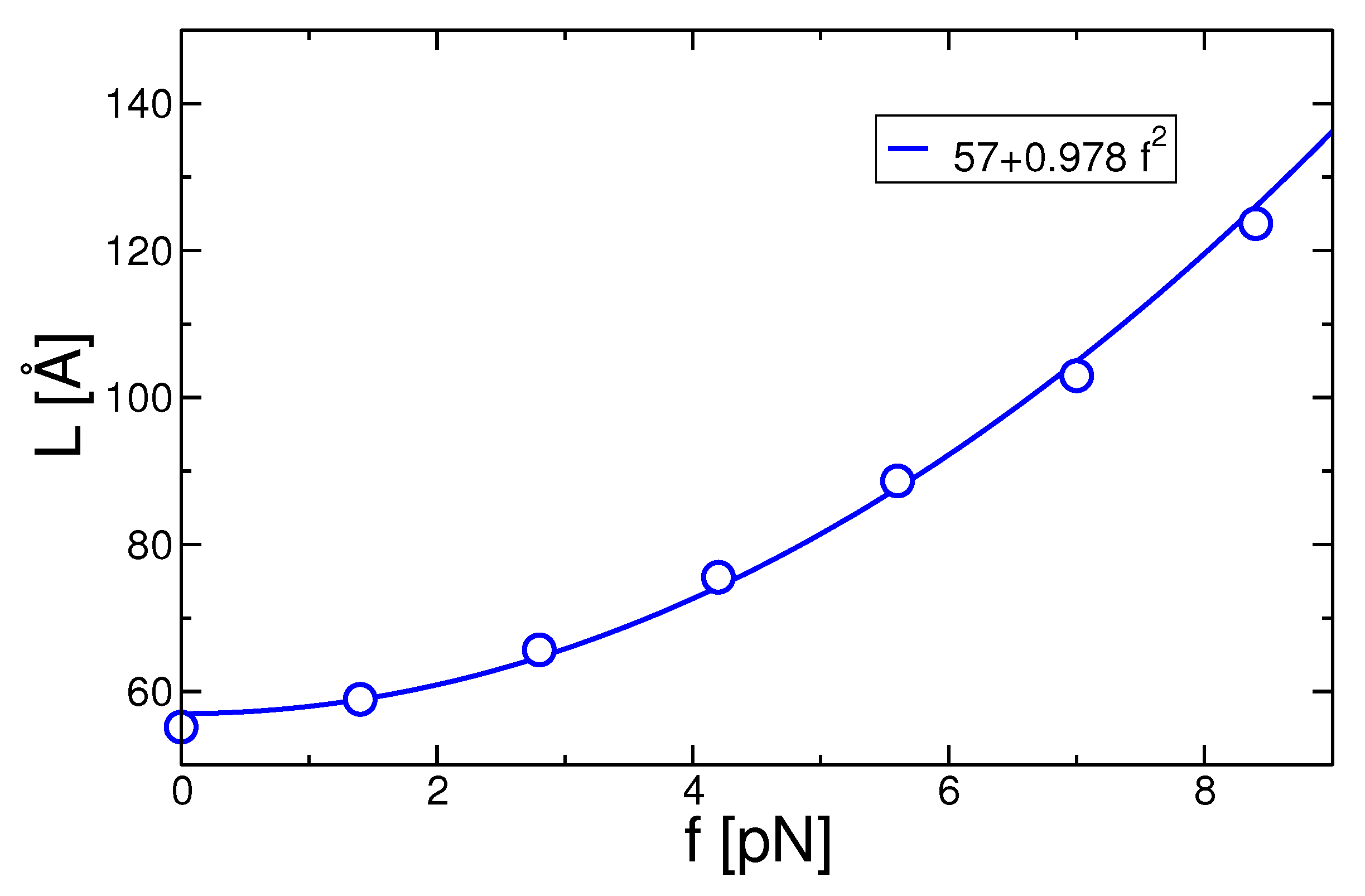
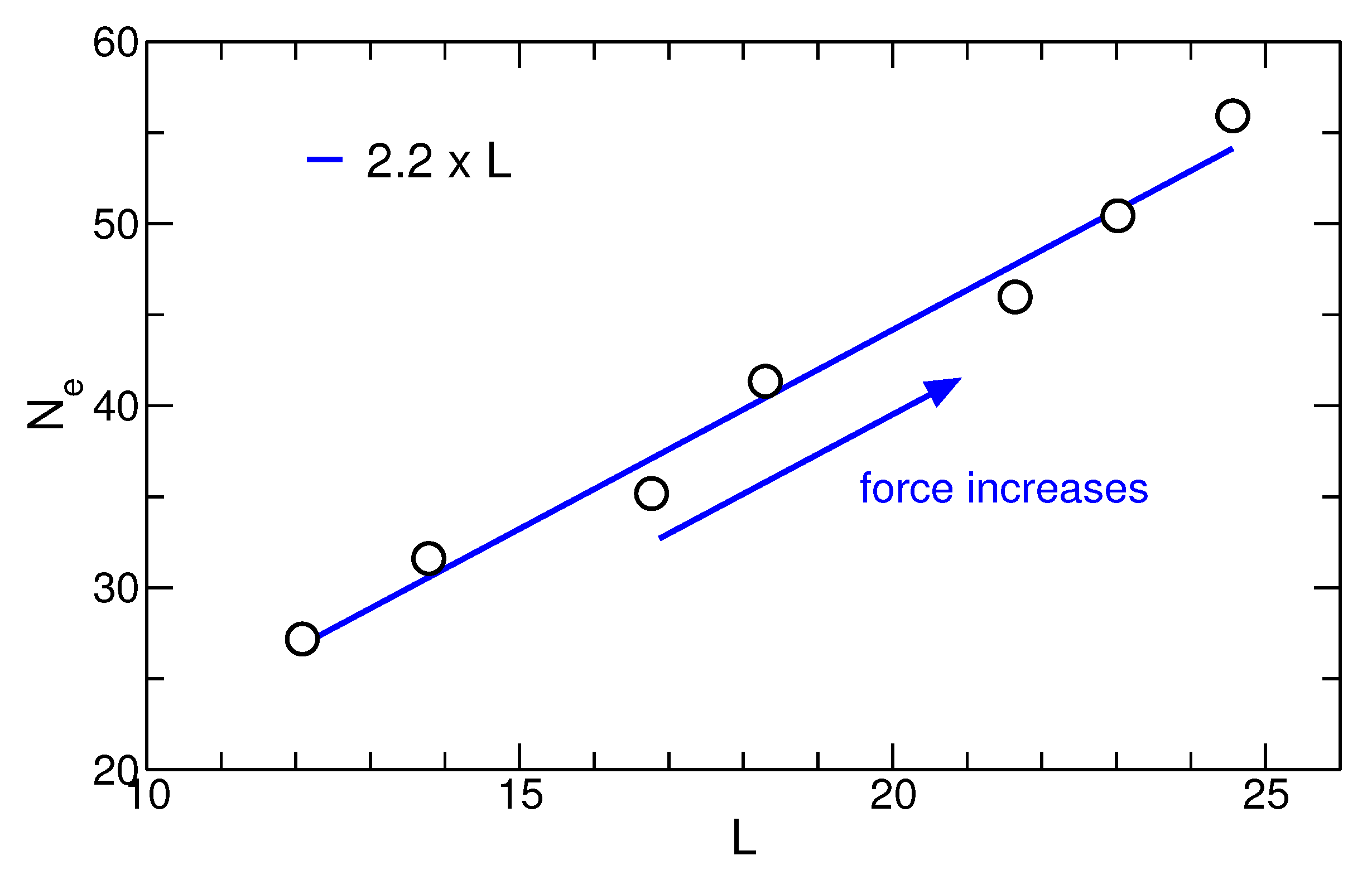
3.4. Entanglements and Stem Length Selection under Force
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Keller, A. A Note on single crystals in polymers—Evidence for a folded chain configuration. Phil. Mag. 1957, 2, 1171. [Google Scholar] [CrossRef]
- Fischer, E.W. Stufenförmiges und Spiralförmiges Wachstum bei Hochpolymeren. Z. Naturf. A 1957, 12, 753–754. [Google Scholar] [CrossRef] [Green Version]
- Strobl, G. Colloquium: Laws controlling crystallization and melting in bulk polymers. Rev. Mod. Phys. 2009, 81, 1287–1300. [Google Scholar] [CrossRef]
- Nakamura, K.; Watanabe, T.; Katayama, K.; Amano, T. Some aspects of nonisothermal crystallization of polymers. I. Relationship between crystallization temperature, crystallinity, and cooling conditions. J. Appl. Polym. Sci. 1972, 16, 1077–1091. [Google Scholar] [CrossRef]
- Lee, W.I.; Talbott, M.F.; Springer, G.S.; Berglund, L.A. Effects of cooling rate on the crystallinity and mechanical properties of thermoplastic composites. J. Reinf. Plast. Compos. 1987, 6, 2–12. [Google Scholar] [CrossRef]
- Mackley, M.; Keller, A. Flow induced crystallization of polyethylene melts. Polymer 1973, 14, 16–20. [Google Scholar] [CrossRef]
- Rao, I.J.; Rajagopal, K.R. A study of strain-induced crystallization of polymers. Int. J. Solids Struct. 2001, 38, 1149–1167. [Google Scholar] [CrossRef]
- Flory, P. Thermodynamics of crystallisation in High Polymers. I. crystallisation induced by Stretching. J. Chem. Phys. 1947, 15, 397. [Google Scholar] [CrossRef]
- Hamad, F.; Colby, R.; Milner, S. Lifetime of Flow-Induced Precursors in Isotactic Polypropylene. Macromolecules 2015, 48, 7286. [Google Scholar] [CrossRef]
- Ma, Z.; Balzano, L.; Peters, G. Dissolution and Re-emergence of Flow-Induced Shish in Polyethylene with a Broad Molecular Weight Distribution. Macromolecules 2016, 49, 2724. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, Z.; Li, L. Flow-Induced Crystallization of Polymers: Molecular and Thermodynamic Considerations. Macromolecules 2016, 49, 1505. [Google Scholar] [CrossRef]
- Nicolson, D.; Rutledge, G. An assessment of models for flow-enhanced nucleation in an n-alkane melt by molecular simulation. J. Rheol. 2019, 63, 465. [Google Scholar] [CrossRef]
- Kimata, S.; Sakurai, T.; Nozue, Y.; Kasahara, T.; Yamaguchi, N.; Karino, T.; Shibayama, M.; Kornfield, J.A. Molecular basis of the shish-kebab morphology in polymer crystallization. Science 2007, 316, 1014–1017. [Google Scholar] [CrossRef] [Green Version]
- Balzano, L.; Ma, Z.; Cavallo, D.; van Erp, T.; Fernandez-Ballester, L.; Peters, G. Molecular Aspects of the Formation of Shish-Kebab in Isotactic Polypropylene. Macromolecules 2013, 49, 3799. [Google Scholar] [CrossRef]
- Katayama, K.; Amano, T.; Nakakura, N. Structural formation during melt spinning process. Kolloid Z. Z. Polym. 1968, 226, 125. [Google Scholar] [CrossRef]
- Spuriell, J.; White, J. Structure development during polymer processing: Studies of the melt spinning of polyethylene and polypropylene fibers. Polym. Eng. Sci. 1975, 15, 660. [Google Scholar] [CrossRef]
- White, J.; Cakmak, M. Orientation development and crystallization in melt spinning of fibers. Adv. Polym. Technol. 1986, 6, 295. [Google Scholar] [CrossRef]
- Janeschitz-Kriegl, H. How to understand nucleation in crystallizing polymer melts under real processing conditions. Colloid Polym. Sci. 2003, 281, 1157. [Google Scholar] [CrossRef]
- Yamamoto, T. Molecular dynamics in fiber formation of polyethylene and large deformation of the fiber. Polymer 2013, 54, 3086. [Google Scholar] [CrossRef]
- Liu, X.; Yu, W. Weak Shear-Induced Slowdown in Crystallization of Less-Entangled Poly(ϵ-caprolactone). Macromolecules 2021, 54, 3347–3357. [Google Scholar] [CrossRef]
- Sliozberg, Y.R.; Yeh, I.-C.; Kröger, M.; Masser, K.A.; Lenhart, J.L.; Andzelm, J.W. Ordering and Crystallization of Entangled Polyethylene Melts under Uniaxial Tension: A Molecular Dynamics Study. Macromolecules 2018, 51, 9635–9648. [Google Scholar] [CrossRef]
- Reith, D.; Meyer, H.; Müller-Plathe, F. Mapping atomistic to coarse-grained polymer models using automatic simplex optimization to fit structural properties. Macromolecules 2001, 34, 2335–2345. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.-F.; Sommer, J.-U. Coding coarse grained polymer model for LAMMPS and its application to polymer crystallization. Comput. Phys. Commun. 2009, 180, 1382. [Google Scholar] [CrossRef]
- Luo, C.-F.; Sommer, J.-U. Coexistence of Melting and Growth during Heating of a Semicrystalline Polymers. Phys. Rev. Lett. 2009, 102, 147801. [Google Scholar] [CrossRef] [PubMed]
- Everaers, R.; Sukumaran, S.K.; Grest, G.S.; Svaneborg, C.; Sivasubramanian, A.; Kremer, K. Rheology and microscopic topology of entangled polymeric liquids. Science 2004, 303, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.-F.; Sommer, J.-U. Disentanglement of Linear Polymer Chains Towards Unentangled Crystals. ACS Macro Lett. 2013, 2, 31. [Google Scholar] [CrossRef]
- Luo, C.; Sommer, J.-U. Frozen topology: Entanglements control nucleation and crystallization in polymers. Phys. Rev. Lett. 2014, 112, 195702. [Google Scholar] [CrossRef]
- Luo, C.-F.; Kröger, M.; Sommer, J.-U. Entanglements and crystallization of concentrated polymer solutions: Molecular dynamics simulations. Macromolecules 2016, 49, 9017. [Google Scholar] [CrossRef]
- Luo, C.-F.; Sommer, J.-U. The Role of Thermal History and Entanglement Related Thickness Selection in Polymer Crystallization. ACS Macro Lett. 2016, 5, 30. [Google Scholar] [CrossRef]
- Donets, S.; Sommer, J.-U. Molecular Dynamics Simulations of Strain-Induced Phase Transition of Poly(ethyleneoxide) in Water. J. Phys. Chem. B 2018, 122, 392. [Google Scholar] [CrossRef] [Green Version]
- Mkandawire, W.D.; Milner, S.T. Simulated Osmotic Equation of State for Poly(ethylene Oxide) Solutions Predicts Tension-Induced Phase Separation. Macromolecules 2021, 54, 3613–3619. [Google Scholar] [CrossRef]
- Dunderdale, G.; Davidson, S.; Ryan, A.; Mykhaylyk, O. Flow-induced crystallisation of polymers from aqueous solution. Nat. Comm. 2020, 11, 3372. [Google Scholar] [CrossRef]
- Hoffmann, J.D.; Davis, G.T.; Lauritzen, J.I. The Rate of Crystallization of Linear Polymers with Chain Folding, Treatise in Solid State Chemistry ed.; Hannay, N.B., Ed.; Plenum Press: New York, NY, USA, 1976; Volume 3, pp. 497–614. [Google Scholar]
- Mandelkern, L. Crystallization of Polymers: Equilibrium Concepts; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Sommer, J.-U. The role of the amorphous fraction for the equilibrium shape of polymer single crystals. Eur. Phys. J. E 2006, 19, 413–422. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Miller, R.; Marand, H.; Roitman, D. Relationhip between the lateral surface free energy σ and the chain structure of melt-crystallized polymers. Macromolecules 1992, 25, 2221–2229. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Jabbari-Farouji, S. Static and dynamic scaling behavior of a polymer melt model with triple-well bending potential. J. Polym. Sci. B: Polym. Phys. 2018, 56, 1376–1392. [Google Scholar] [CrossRef] [Green Version]
- Kröger, M. Shortest multiple disconnected path for the analysis of entanglements in two-and three-dimensional polymeric systems. Comput. Phys. Commun. 2005, 168, 209–232. [Google Scholar] [CrossRef]
- Keller, A.; Kolnaar, H.W. Flow-Induced Orientation and Structure Formation; Wiley Online Library: Hoboken, NJ, USA, 2006. [Google Scholar]
- Luo, C.; Sommer, J.-U. Growth pathway and precursor states in single lamellar crystallization: MD simulations. Macromolecules 2011, 44, 1523–1529. [Google Scholar] [CrossRef]
- Hikosaka, M.; Watanabe, K.; Okada, K.; Yamazaki, S. Interphases and Mesophases in Polymer Crystallization III; Springer: Berlin/Heidelberg, Germany, 2005; pp. 137–186. [Google Scholar]
- Liu, K.; de Boer, E.; Yao, Y.; Romano, D.; Ronca, S.; Rastogi, S. Heterogeneous Distribution of Entanglements in a Nonequilibrium Polymer Melt of UHMWPE: Influence on Crystallization without and with Graphene Oxide. Macromolecues 2016, 49, 7497. [Google Scholar] [CrossRef] [Green Version]
- Muthukumar, M. Molecular modelling of nucleation in polymers. Phil. Trans. R. Soc. Lond. A 2003, 361, 539–556. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Force | |||||
|---|---|---|---|---|---|
| (pN) | (kJ/mol/Monomer) | (kJ/mol/Monomer) | (%) | (%) | K |
| 0.0 | 4.183 | 0.000 | 0.0 | 0.0 | 523.2 |
| 1.4 | 4.605 | 0.017 | 0.3 | 0.2 | 524.2 |
| 2.8 | 4.975 | 0.069 | 1.4 | 0.8 | 527.4 |
| 4.2 | 5.345 | 0.159 | 3.0 | 1.8 | 532.6 |
| 5.6 | 5.979 | 0.286 | 4.8 | 3.2 | 539.9 |
| 7.0 | 6.349 | 0.443 | 7.0 | 4.9 | 548.8 |
| 8.4 | 6.929 | 0.632 | 9.1 | 7.0 | 559.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Payal, R.S.; Sommer, J.-U. Crystallization of Polymers under the Influence of an External Force Field. Polymers 2021, 13, 2078. https://doi.org/10.3390/polym13132078
Payal RS, Sommer J-U. Crystallization of Polymers under the Influence of an External Force Field. Polymers. 2021; 13(13):2078. https://doi.org/10.3390/polym13132078
Chicago/Turabian StylePayal, Rajdeep Singh, and Jens-Uwe Sommer. 2021. "Crystallization of Polymers under the Influence of an External Force Field" Polymers 13, no. 13: 2078. https://doi.org/10.3390/polym13132078
APA StylePayal, R. S., & Sommer, J.-U. (2021). Crystallization of Polymers under the Influence of an External Force Field. Polymers, 13(13), 2078. https://doi.org/10.3390/polym13132078