
Synthesis of Novel Dental Nanocomposite Resins by Incorporating Polymerizable, Quaternary Ammonium Silane-Modified Silica Nanoparticles

 ,
,  ,
, 
Abstract
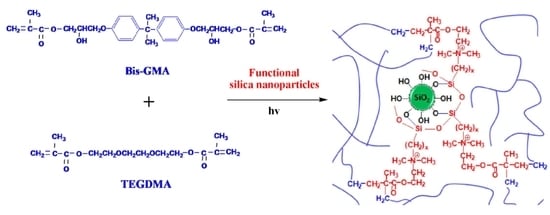
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Nanosilica Organomodifiers (QASiC)
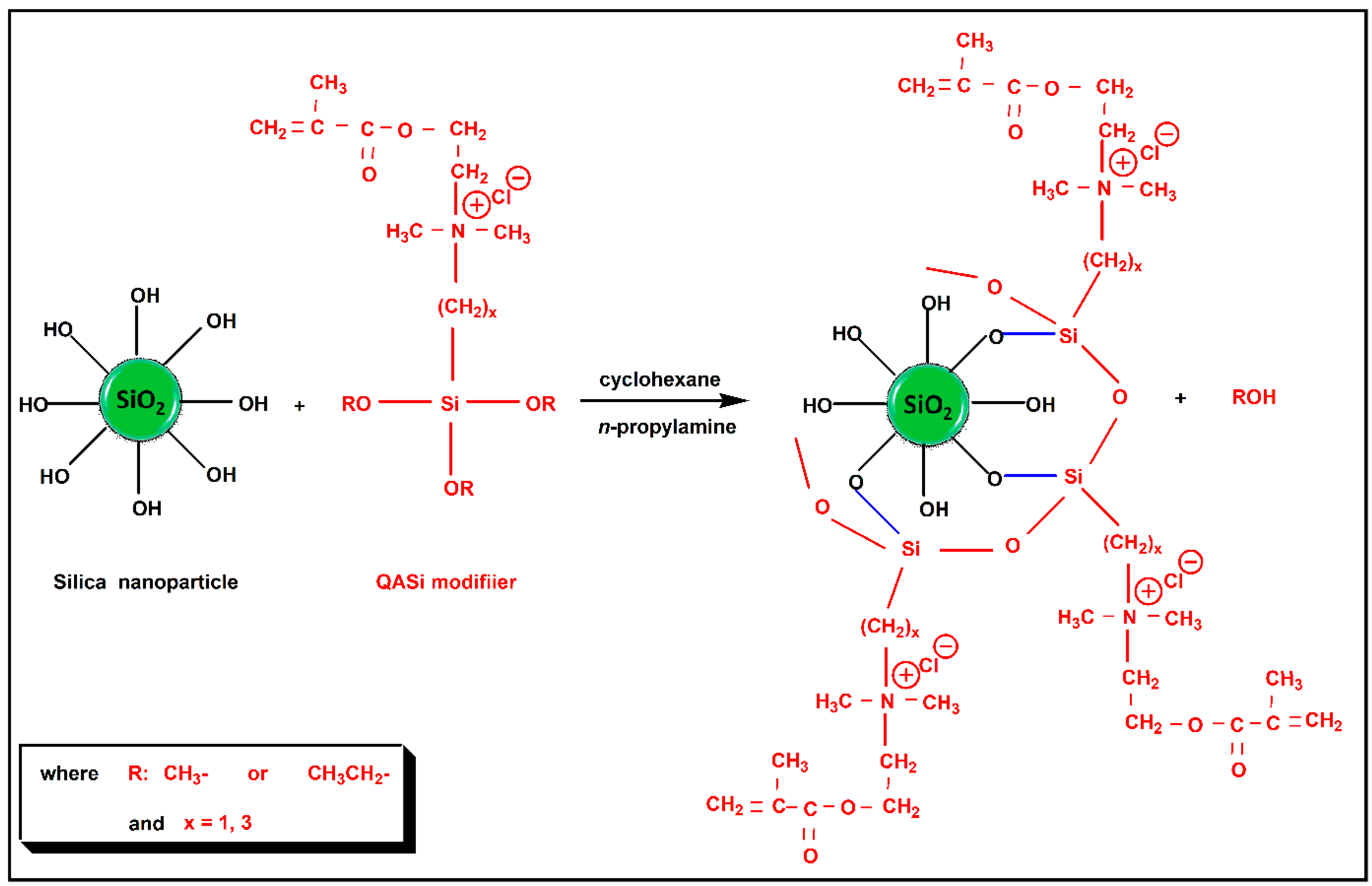
2.3. Surface Modification of Nanosilica with Quaternary Ammonium Silane Coupling Agents
2.4. Preparation of the Uncured Dental Composite Pastes
2.5. Measuremens
2.5.1. Structural Characterization of QASiC
2.5.2. Characterization of the Organically Modified Nanosilica
2.5.3. Surface Morphology Measurements of Nanocomposite Resins
2.5.4. Physicochemical Properties
2.5.5. Mechanical Properties
2.5.6. Statistical Analysis
3. Results and Discussion
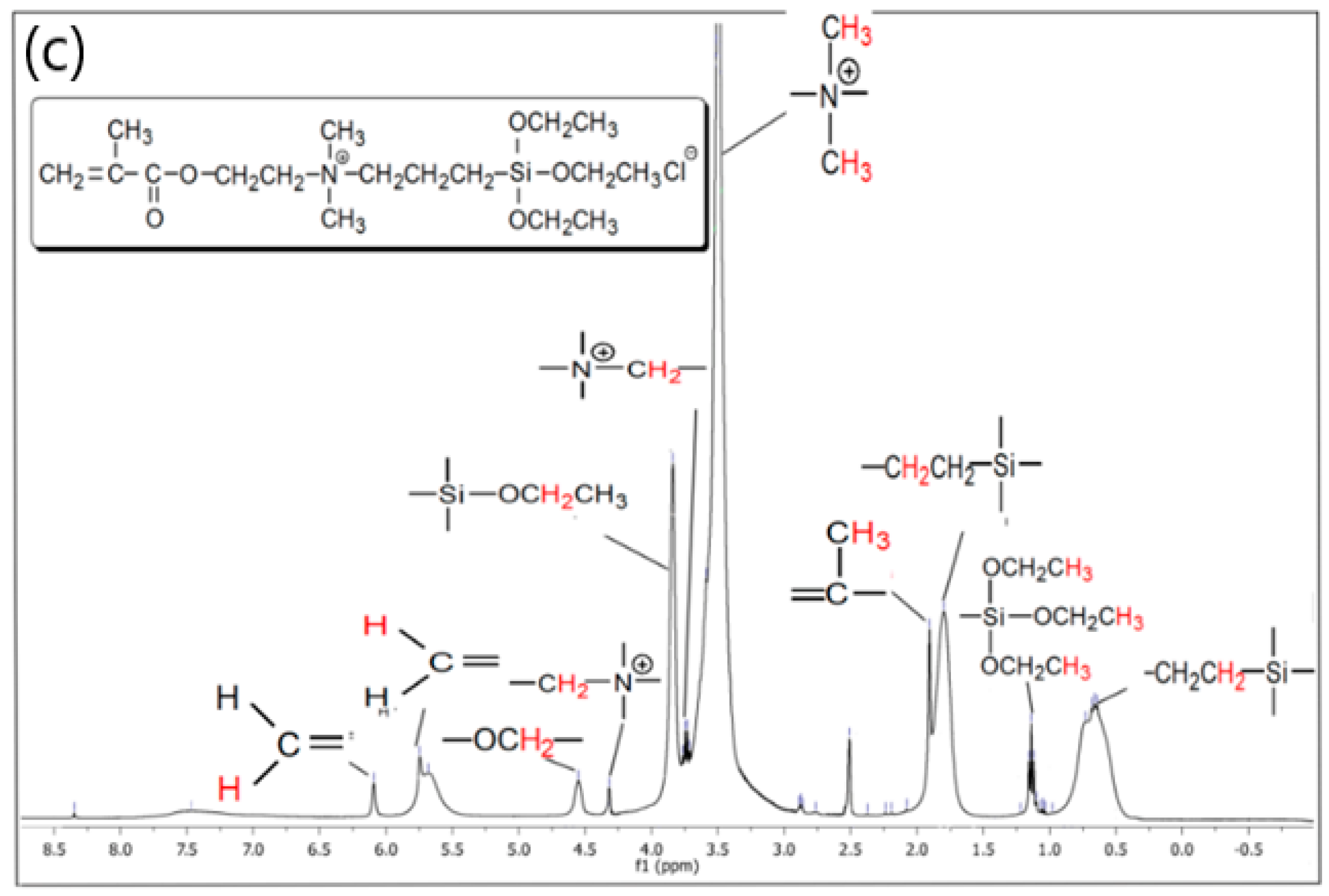
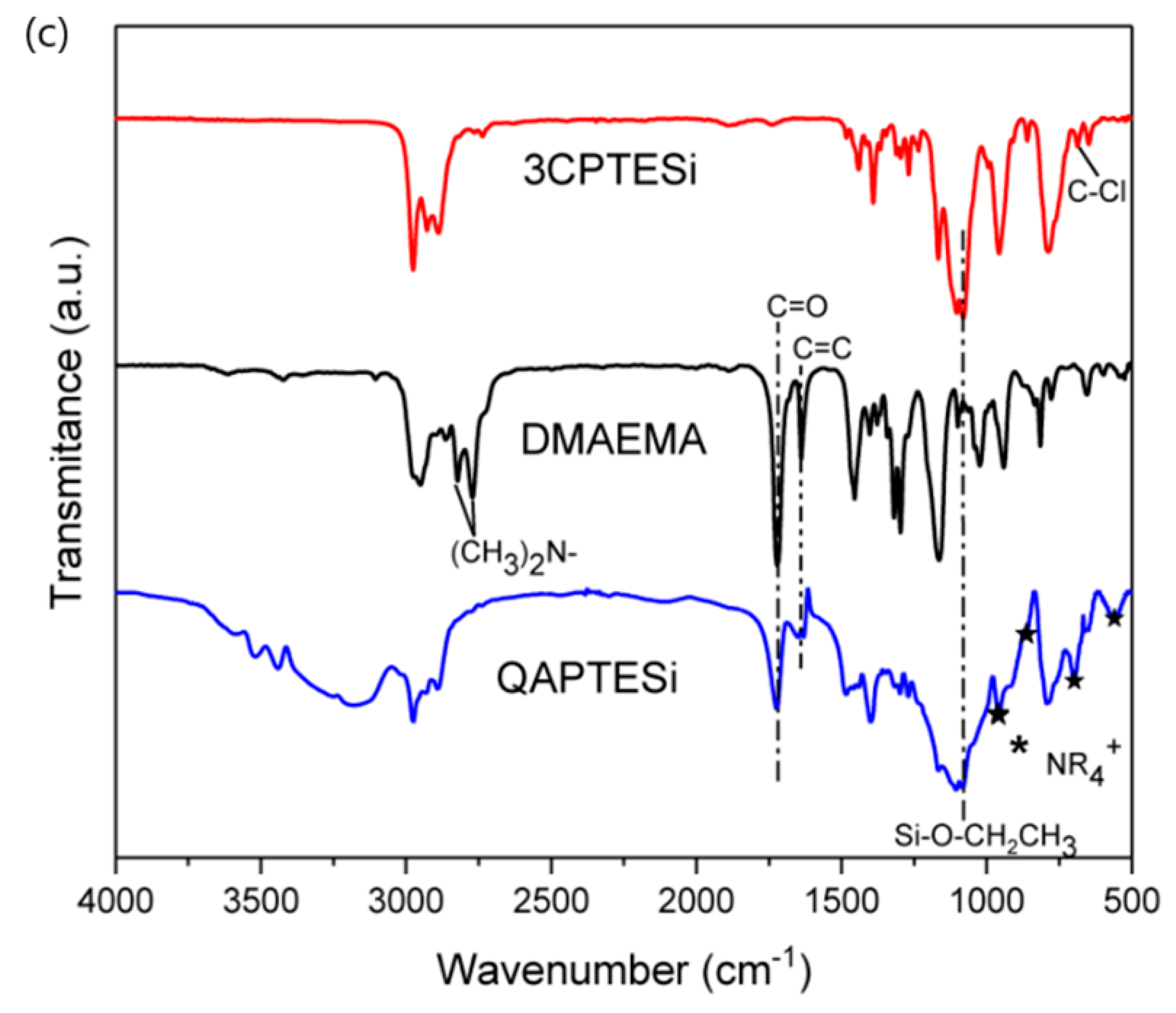
3.1. 1H-NMR and FTIR Data of the Synthesized QASiC
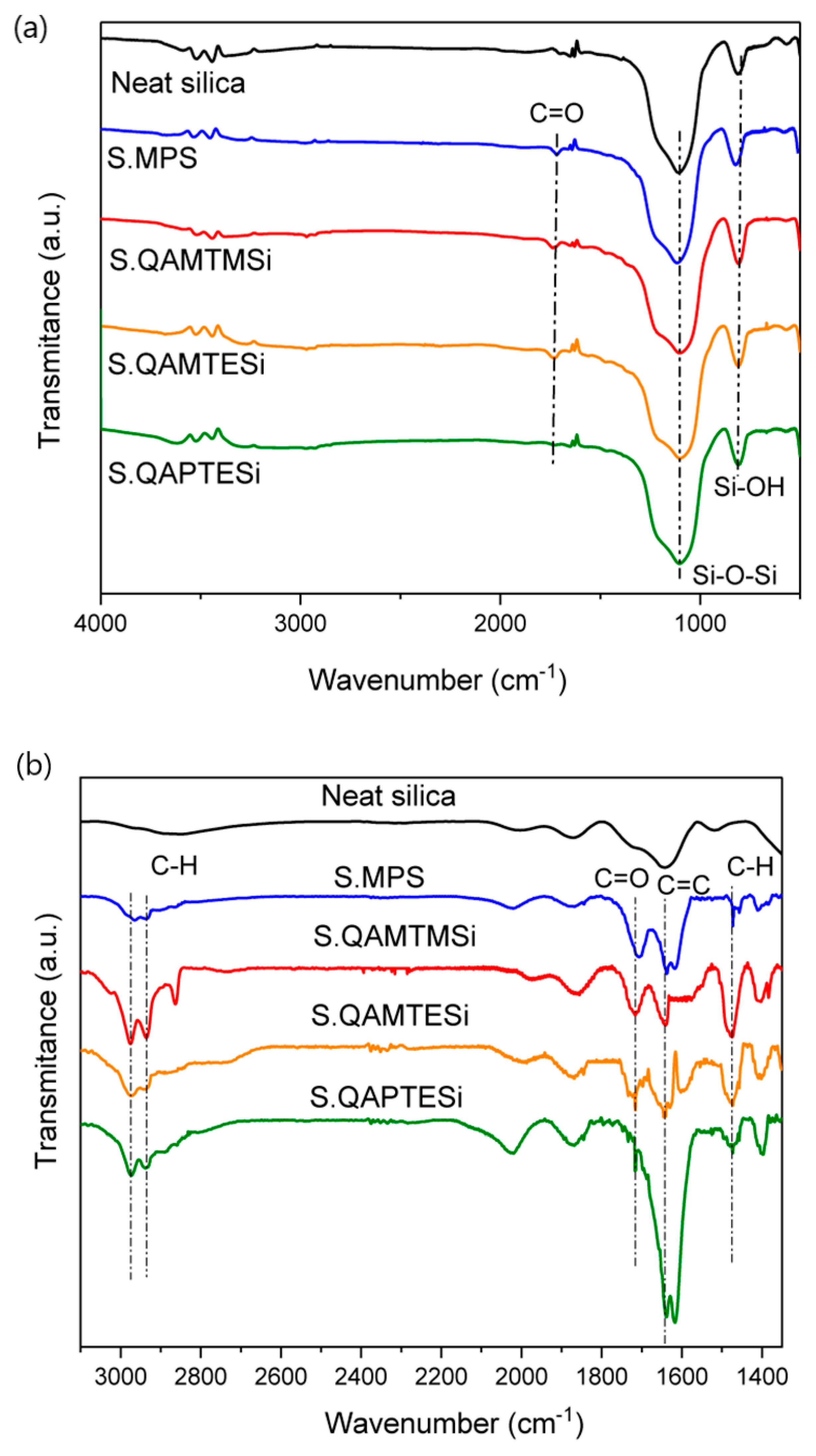
3.2. FTIR Analysis of Silica Nanoparticles
3.3. Thermogravimetric Analysis (TGA) of Silica Nanoparticles
3.4. SEM Images of Nanocomposites’ Surface
3.5. FTIR Analysis-Degree of Conversion (DC%)
3.6. Polymerization Shrinkage Kinetics
3.7. Flexural Properties
3.8. Compression Tests
4. Conclusions
Author Contributions
Funding

Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Staninec, M.; Holt, M. Bonding of amalgam to tooth structure: Tensile adhesion and microleakage tests. J. Prosthet. Dent. 1988, 59, 397–402. [Google Scholar] [CrossRef]
- Kim, J.W.; Kim, A.L.U.; Kim, C.K. Size Control of Silica Nanoparticles and Their Surface Treatment for Fabrication of Dental Nanocomposites. Biomacromolecules 2007, 8, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Kydd, W.L.; Daly, C.H. The biologic and mechanical effects of stress on oral mucosa. J. Prosthet. Dent. 1982, 47, 317–329. [Google Scholar] [CrossRef]
- Baliga, S.; Muglikar, S.; Kale, R. Salivary pH: A diagnostic biomarker. J. Indian Soc. Periodontol. 2013, 17, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.D.; Bishara, S.E.; Quinn, J.K. Biodegradation of orthodontic appliances. Part I. Biodegradation of nickel and chromium in vitro. Am. J. Orthod. Dentofac. Orthop. 1993, 103, 8–14. [Google Scholar] [CrossRef]
- Rosan, B.; Lamont, R.J. Dental plaque formation. Microbes Infect. 2000, 2, 1599–1607. [Google Scholar] [CrossRef]
- Eliades, T.; Athanasiou, A.E. In vivo aging of orthodontic alloys: Implications for corrosion potential, nickel release, and biocompatibility. Angle Orthod. 2002, 72, 222–237. [Google Scholar] [PubMed]
- Oliveira, K.M.C.; Lancellotti, A.C.R.A.; Ccahuana-Vásquez, R.A.; Consani, S. Influence of filling techniques on shrinkage stress in dental composite restorations. J. Dent. Sci. 2013, 8, 53–60. [Google Scholar] [CrossRef]
- Biradar, B.; Biradar, S.; Ms, A. Evaluation of the Effect of Water on Three Different Light Cured Composite Restorative Materials Stored in Water: An In Vitro Study. Int. J. Dent. 2012, 2012, 640942. [Google Scholar] [CrossRef]
- Zhao, X.; Zanetti, F.; Wang, L.; Pan, J.; Majeed, S.; Malmstrom, H.; Peitsch, M.C.; Hoeng, J.; Ren, Y. Effects of different discoloration challenges and whitening treatments on dental hard tissues and composite resin restorations. J. Dent. 2019, 89, 103182. [Google Scholar] [CrossRef]
- Marashdeh, M.Q.; Gitalis, R.; Levesque, C.; Finer, Y. Enterococcus faecalis Hydrolyzes Dental Resin Composites and Adhesives. J. Endod. 2018, 44, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Chaughule, R.S. Dental Applications of Nanotechnology, 1st ed.; Springer Nature AG: Cham, Switzerland, 2018; pp. 72–77, 247–268. [Google Scholar]
- Slowing, I.I.; Trewyn, B.G.; Giri, S.; Lin, V.S.-Y. Mesoporous Silica Nanoparticles for Drug Delivery and Biosensing Applications. Adv. Funct. Mater. 2007, 17, 1225–1236. [Google Scholar] [CrossRef]
- Lino, M.M.; Paulo, C.S.; Vale, C.; Vaz, M.F.; Ferreira, L.S. Antifungal activity of dental resins containing amphotericin B-conjugated nanoparticles. Dent. Mater. 2013, 29, e252–e262. [Google Scholar] [CrossRef] [PubMed]
- Habib, E.; Wang, R.; Zhu, X. Monodisperse silica-filled composite restoratives mechanical and light transmission properties. Dent. Mater. 2017, 33, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Hosseinalipour, M.; Javadpour, J.; Rezaie, H.; Dadras, T.; Hayati, A.N. Investigation of Mechanical Properties of Experimental Bis-GMA/TEGDMA Dental Composite Resins Containing Various Mass Fractions of Silica Nanoparticles. J. Prosthodont. 2010, 19, 112–117. [Google Scholar] [CrossRef]
- Rezvani, M.B.; Atai, M.; Hamze, F.; Hajrezai, R. The effect of silica nanoparticles on the mechanical properties of fiber-reinforced composite resins. J. Dent. Res. Dent. Clin. Dent. Prospect. 2016, 10, 112–117. [Google Scholar] [CrossRef]
- Rodríguez, H.A.; Kriven, W.M.; Casanova, H. Development of mechanical properties in dental resin composite: Effect of filler size and filler aggregation state. Mater. Sci. Eng. C 2019, 101, 274–282. [Google Scholar] [CrossRef]
- Chen, M.-H.; Chen, C.-R.; Hsu, S.-H.; Sun, S.-P.; Su, W.-F. Low shrinkage light curable nanocomposite for dental restorative material. Dent. Mater. 2006, 22, 138–145. [Google Scholar] [CrossRef]
- Karabela, M.M.; Sideridou, I.D. Effect of the structure of silane coupling agent on sorption characteristics of solvents by dental resin-nanocomposites. Dent. Mater. 2008, 24, 1631–1639. [Google Scholar] [CrossRef]
- Karabela, M.M.; Sideridou, I.D. Synthesis and study of properties of dental resin composites with different nanosilica particles size. Dent. Mater. 2011, 27, 825–835. [Google Scholar] [CrossRef]
- Rodríguez, H.A.; Giraldo, L.F.; Casanova, H. Formation of functionalized nanoclusters by solvent evaporation and their effect on the physicochemical properties of dental composite resins. Dent. Mater. 2015, 31, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Sideridou, I.D.; Karabela, M.M. Effect of the amount of 3-methacyloxypropyltrimethoxysilane coupling agent on physical properties of dental resin nanocomposites. Dent. Mater. 2009, 25, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Salgado, V.E.; Cavalcante, L.M.; Silikas, N.; Schneider, L.F.J. The influence of nanoscale inorganic content over optical and surface properties of model composites. J. Dent. 2013, 41, e45–e53. [Google Scholar] [CrossRef] [PubMed]
- Salgado, V.E.; Cavalcante, L.M.; Moraes, R.R.; Davis, H.B.; Ferracane, J.L.; Schneider, L.F. Degradation of optical and surface properties of resin-based composites with distinct nanoparticle sizes but equivalent surface area. J. Dent. 2017, 59, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Karabela, M.M.; Sideridou, I.D. Synthesis and study of physical properties of dental light-cured nanocomposites using different amounts of a urethane dimethacrylatetrialkoxysilane coupling agent. Dent. Mater. 2011, 27, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Imazato, S.; Toriil, M.; Tsuchitani, Y.; McCabe, J.; Russell, R. Incorporation of Bacterial Inhibitor into Resin Composite. J. Dent. Res. 1994, 73, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, J.M.; Zeiger, D.N.; Tang, K.; Lin-Gibson, S.; Fowler, B.O.; Lin, N.J. Synthesis and characterization of dimethacrylates containing quaternary ammonium functionalities for dental applications. Dent. Mater. 2012, 28, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, R.; Izutani, N.; Hirose, N.; Hayashi, M.; Imazato, S.; Kitagawa, H. Development of an Antibacterial Root Canal Filling System Containing MDPB. J. Dent. Res. 2014, 93, 1277–1282. [Google Scholar] [CrossRef]
- Melo, M.A.; Wu, J.; Weir, M.D.; Xu, H.H. Novel antibacterial orthodontic cement containing quaternary ammonium monomer dimethylaminododecyl methacrylate. J. Dent. 2014, 42, 1193–1201. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, K.; Zhou, X.; Xu, N.; Xu, H.H.K.; Weir, M.D.; Ge, Y.; Wang, S.; Li, M.; Li, Y.; et al. Antibacterial Effect of Dental Adhesive Containing Dimethylaminododecyl Methacrylate on the Development of Streptococcus mutans Biofilm. Int. J. Mol. Sci. 2014, 15, 12791–12806. [Google Scholar] [CrossRef]
- Feng, J.; Cheng, L.; Zhou, X.; Xu, H.H.; Weir, M.D.; Meyer, M.; Maurer, H.; Li, Q.; Hannig, M.; Rupf, S. In situ antibiofilm effect of glass-ionomer cement containing dimethylaminododecyl methacrylate. Dent. Mater. 2015, 31, 992–1002. [Google Scholar] [CrossRef]
- Yu, F.; Yu, H.; Lin, P.; Dong, Y.; Zhang, L.; Sun, X.; Liu, Z.; Guo, H.; Huang, L.; Chen, J. Effect of an Antibacterial Monomer on the Antibacterial Activity of a Pit-and-Fissure Sealant. PLoS ONE 2016, 11, e0162281. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.A.S.; Weir, M.D.; Passos, V.F.; Rolim, J.P.M.; Lynch, C.D.; Rodrigues, L.K.A.; Xu, H.H.K. Human in situ study of the effect of bis(2-methacryloyloxyethyl) dimethylammonium bromide immobilized in dental composite on controlling mature cariogenic biofilm. Int. J. Mol. Sci. 2018, 19, 3443. [Google Scholar] [CrossRef] [PubMed]
- Paunovska, M.L.; Coleman, N.J.; Stevanovic, M.M.; Dimkov, A.G.; Gabric, D.; Gjorgievska, E.S. Effects of Addition of Quaternary Ammonium Antimicrobial Compounds into Root Canal Sealers. Eur. J. Dent. 2019, 13, 243–247. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Campos, K.D.P.L.; Viana, G.M.; Cabral, L.M.; Portela, M.B.; Junior, R.H.; Cavalcante, L.M.; Lourenço, E.J.V.; Telles, D.D.M. Self-cured resin modified by quaternary ammonium methacrylates and chlorhexidine: Cytotoxicity, antimicrobial, physical, and mechanical properties. Dent. Mater. 2020, 36, 68–75. [Google Scholar] [CrossRef]
- Garcia, I.M.; Rodrigues, S.B.; Balbinot, G.D.S.; Visioli, F.; Leitune, V.C.B.; Collares, F.M. Quaternary ammonium compound as antimicrobial agent in resin-based sealants. Clin. Oral Investig. 2019, 24, 777–784. [Google Scholar] [CrossRef]
- Cheng, L.; Weir, M.D.; Xu, H.H.; Antonucci, J.M.; Kraigsley, A.M.; Lin, N.J.; Lin-Gibson, S.; Zhou, X. Antibacterial amorphous calcium phosphate nanocomposites with a quaternary ammonium dimethacrylate and silver nanoparticles. Dent. Mater. 2012, 28, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Weir, M.D.; Zhang, K.; Xu, S.M.; Chen, Q.; Zhou, X.; Xu, H.H.K. Antibacterial Nanocomposite with Calcium Phosphate and Quaternary Ammonium. J. Dent. Res. 2012, 91, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Zhang, K.; Zhou, C.-C.; Weir, M.D.; Zhou, X.-D.; Xu, H.H.K. One-year water-ageing of calcium phosphate composite containing nano-silver and quaternary ammonium to inhibit biofilms. Int. J. Oral Sci. 2016, 8, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Weir, M.D.; Zhang, K.; Deng, D.; Cheng, L.; Xu, H.H.K. Synthesis of new antibacterial quaternary ammonium monomer for incorporation into CaP nanocomposite. Dent. Mater. 2013, 29, 859–870. [Google Scholar] [CrossRef]
- Zhang, K.; Cheng, L.; Weir, M.D.; Bai, Y.-X.; Xu, H.H. Effects of quaternary ammonium chain length on the antibacterial and remineralizing effects of a calcium phosphate nanocomposite. Int. J. Oral Sci. 2016, 8, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhou, H.; Weir, M.D.; Melo, M.A.S.; Levine, E.D.; Xu, H.H. Effect of dimethylaminohexadecyl methacrylate mass fraction on fracture toughness and antibacterial properties of CaP nanocomposite. J. Dent. 2015, 43, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Yudovin-Farber, I.; Beyth, N.; Nyska, A.; Weiss, E.I.; Golenser, J.; Domb, A.J. Surface Characterization and Biocompatibility of Restorative Resin Containing Nanoparticles. Biomacromolecules 2008, 9, 3044–3050. [Google Scholar] [CrossRef] [PubMed]
- Yudovin-Farber, I.; Beyth, N.; Weiss, E.I.; Domb, A.J. Antibacterial effect of composite resins containing quaternary ammonium polyethyleneimine nanoparticles. J. Nanopart. Res. 2010, 12, 591–603. [Google Scholar] [CrossRef]
- Ebi, N.; Imazato, S.; Noiri, Y.; Ebisu, S. Inhibitory effects of resin composite containing bactericide-immobilized filler on plaque accumulation. Dent. Mater. 2001, 17, 485–491. [Google Scholar] [CrossRef]
- Imazato, S. Antibacterial activity of bactericide-immobilized filler for resin-based restoratives. Biomaterials 2003, 24, 3605–3609. [Google Scholar] [CrossRef]
- Makvandi, P.; Ghaemy, M.; Ghadiri, A.; Mohseni, M. Photocurable, Antimicrobial Quaternary Ammonium–modified Nanosilica. J. Dent. Res. 2015, 94, 1401–1407. [Google Scholar] [CrossRef]
- Zaltsman, N.; Weiss, E.I. Compositions and Medical Devices Comprising Anti-Microbial Particles. Int. Pub. No.: WO 2019/043713 A1. 2019. Available online: https://patents.google.com/patent/WO2019043713A1/en (accessed on 26 April 2021).
- Wang, L.; Xie, X.; Imazato, S.; Weir, M.D.; Reynolds, M.A.; Xu, H.H. A protein-repellent and antibacterial nanocomposite for Class-V restorations to inhibit periodontitis-related pathogens. Mater. Sci. Eng. C 2016, 67, 702–710. [Google Scholar] [CrossRef]
- Liu, Q.; Ding, J.; Chambers, D.E.; Debnath, S.; Wunder, S.L. Filler-coupling agent-matrix interactions in silica/polymethylmethacrylate composites. J. Biom. Mater. Res. 2001, 57, 384–393. [Google Scholar] [CrossRef]
- Wilson, K.S.; Zhang, K.; Antonucci, J.M. Systematic variation of interfacial phase reactivity in dental nanocomposites. Biomaterials 2005, 26, 5095–5103. [Google Scholar] [CrossRef]
- Söderholm, K.-J.; Shang, S.-W. Molecular Orientation of Silane at the Surface of Colloidal Silica. J. Dent. Res. 1993, 72, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Brauer, G. Solvent Effects on Bonding Organo-silane to Silica Surfaces. J. Dent. Res. 1982, 61, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Watts, D.C.; Marouf, A.S. Optimal specimen geometry in bonded-disk shrinkage-strain measurements on light-cured biomaterials. Dent. Mater. 2000, 16, 447–451. [Google Scholar] [CrossRef]
- Watts, D.; Marouf, A.; Al-Hindi, A. Photo-polymerization shrinkage-stress kinetics in resin-composites: Methods development. Dent. Mater. 2003, 19, 1–11. [Google Scholar] [CrossRef]
- Al Sunbul, H.; Silikas, N.; Watts, D.C. Polymerization shrinkage kinetics and shrinkage-stress in dental resin-composites. Dent. Mater. 2016, 32, 998–1006. [Google Scholar] [CrossRef]
- Rueggeberg, F.; Hashinger, D.; Fairhurst, C. Calibration of FTIR conversion analysis of contemporary dental resin composites. Dent. Mater. 1990, 6, 241–249. [Google Scholar] [CrossRef]
- Wang, Y.; Li, P.; Kong, L. Chitosan-Modified PLGA Nanoparticles with Versatile Surface for Improved Drug Delivery. AapsPharmscitech 2013, 14, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Dierkes, W.K.; Noordermeer, J.W.M.; Rinker, M.; Kelting, K.-U.; Van de Pol, C. Increasing the silanisation efficiency of silica compounds: Upscaling. Kautsch. Gummi Kunstst. 2003, 56, 338–344. [Google Scholar]
- Achilias, D.S. A Review of Modeling of Diffusion Controlled Polymerization Reactions. Macromol. Theory Simul. 2007, 16, 319–347. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Z.; Zhao, C.; Bu, W.; Na, H. Preparation and characterization of silane-modified SiO2 particles reinforced resin composites with fluorinated acrylate polymer. J. Mech. Behav. Biomed. Mater. 2018, 80, 11–19. [Google Scholar] [CrossRef]
- Ruyter, I.E.; Øysaed, H. Composites for use in posterior teeth: Composition and conversion. J. Biomed. Mater. Res. 1987, 21, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, M.; Liu, F.; Bao, S.; Wu, T.; Jiang, X.; Zhang, Q.; Zhu, M. Investigation on the physical–mechanical properties of dental resin composites reinforced with novel bimodal silica nanostructures. Mater. Sci. Eng. C 2015, 50, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Suárez, C.E.; Pimentel-García, B.; Rivera-Gonzaga, A.; Álvarez-Gayosso, C.; Ancona-Meza, A.L.; Grazioli, G.; Zamarripa-Calderón, E. Examining the Effect of Radiant Exposure on Commercial Photopolimerizable Dental Resin Composites. Dent. J. 2018, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Achilias, D.S.; Verros, G.D. Modeling of diffusion-controlled reactions in free radical solution and bulk polymerization: Model validation by DSC experiments. J. Appl. Polym. Sci. 2010, 116, 1842–1856. [Google Scholar] [CrossRef]
- Verros, G.D.; Achilias, D.S. Modeling gel effect in branched polymer systems: Free-radical solution homopolymerization of vinyl acetate. J. Appl. Polym. Sci. 2009, 111, 2171–2185. [Google Scholar] [CrossRef]
- Verros, G.; Latsos, T.; Achilias, D. Development of a unified framework for calculating molecular weight distribution in diffusion controlled free radical bulk homo-polymerization. Polymer 2005, 46, 539–552. [Google Scholar] [CrossRef]
- Miletic, V. Dental Composite Materials for Direct Restorations, 1st ed.; Springer Nature AG: Cham, Switzerland, 2018; p. 238. [Google Scholar]
- Atai, M.; Watts, D.C.; Atai, Z. Shrinkage strain-rates of dental resin-monomer and composite systems. Biomaterials 2005, 26, 5015–5020. [Google Scholar] [CrossRef] [PubMed]
- Satterthwaite, J.D.; Vogel, K.; Watts, D.C. Effect of resin-composite filler particle size and shape on shrinkage–strain. Dent. Mater. 2009, 25, 1612–1615. [Google Scholar] [CrossRef]
- Peutzfeldt, A. Resin composites in dentistry: The monomer systems. Eur. J. Oral Sci. 1997, 105, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Ferracane, J.L. Resin composite—State of the art. Dent. Mater. 2011, 27, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Liu, F.; He, J. Preparation of low shrinkage stress dental composite with synthesized dimethacrylate oligomers. J. Mech. Behav. Biomed. Mater. 2019, 94, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Campos, L.M.D.P.; Boaro, L.C.; Ferreira, H.P.; Dos Santos, L.K.G.; dos Santos, T.R.; Parra, D.F. Evaluation of polymerization shrinkage in dental restorative experimental composites based: BisGMA/TEGDMA, filled with MMT. J. Appl. Polym. Sci. 2016, 133, 43543. [Google Scholar] [CrossRef]
- Antonucci, J.M.; Dickens, S.H.; Fowler, B.O.; Xu, H.H.K.; McDonough, W.G. Chemistry of silanes: Interfaces in dental polymers and composites. J. Res. Natl. Inst. Stand. Technol. 2005, 110, 541–558. [Google Scholar] [CrossRef] [PubMed]
- Condon, J.; Ferracane, J. In vitro Wear of Composite with Varied Cure, Filler Level, and Filler Treatment. J. Dent. Res. 1997, 76, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, Y.; Nishiwaki, T.; Nemoto, K.; Ben, G. Effect of filler content on bending properties of dental composites: Numerical simulation with the use of the finite-element method. J. Biomed. Mater. Res. 2004, 71, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Alkhudhairy, F.I. The effect of curing intensity on mechanical properties of different bulk-fill composite resins. Clin. Cosmet. Investig. Dent. 2017, 9, 1–6. [Google Scholar] [CrossRef] [PubMed]
- ISO 4049. Dentistry-Polymer-Based Restorative Materials, 5th ed.; International Standard Organization: Geneva, Switzerland, 2019; Available online: https://www.iso.org/standard/67596.html (accessed on 26 April 2021).
- Bindu, M.; Satapathy, B.K.; Jaggi, H.S.; Ray, A.R. Size-scale effects of silica on bis-GMA/TEGDMA based nanohybrid dental restorative composites. Compos. Part B Eng. 2013, 53, 92–102. [Google Scholar] [CrossRef]
- Miao, X.; Li, Y.; Zhang, Q.; Zhu, M.; Wang, H. Low shrinkage light curable dental nanocomposites using SiO2 microspheres as fillers. Mater. Sci. Eng. C 2012, 32, 2115–2121. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Specifications | Supplier |
|---|---|---|
| 2,2-Bis[p-(2′-hydroxy-3′-methacryloxypropoxy)phenylene]propane (Bis-GMA) | SIGMA-ALDRICH CHEMIE GmbH (Steinheim, Germany) | |
| Triethylene glycol dimethacrylate (TEGDMA) | 95% | |
| Cyclohexane | ≥99.5% | |
| Silica nanopowder | 99.8%, average diameter: 12 nm, specific surface area (BET): 200 ± 25 m2·g−1 | |
| (3-chloropropyl)triethoxysilane (3CPTESi) | 95% | |
| Camphorquinone | 98% | J&K Scientific GmbH (Pforzheim, Germany) |
| 2-(dimethylamino)ethylmethacrylate (DMAEMA) | 99% | |
| 3-(trimethoxysilyl)propyl methacrylate (γ-MPS) | 98% | |
| (chloromethyl)trimethoxysilane (CMTMSi) | 96% | |
| (chloromethyl)triethoxysilane (CMTESi) | 97% | abcr GmbH (Karlsruhe, Germany) |
| Propylamine | ≥99.0% | Merck KGaA (Darmstadt, Germany) |
| Methanol | ≥99.8% | Honeywell/Riedel-de-Haën (Seelze, Germany) |
| Organomodified Nanosilica | Total (wt%) | Physically Absorbed | Chemically Absorbed | Effective Functional Ratio | ||||
|---|---|---|---|---|---|---|---|---|
| wt% | μmol·m−2 | Molecules·nm−2 | wt% | mmol·m−2 | Molecules·nm−2 | |||
| S.MPS | 5.21 | 1.10 | 0.22 | 0.13 | 4.11 | 0.83 | 0.50 | 3.74 |
| S.QAMTMSi | 8.98 | 3.55 | 0.54 | 0.33 | 5.43 | 0.83 | 0.50 | 1.53 |
| S.QAMTESi | 9.77 | 4.50 | 0.61 | 0.37 | 5.27 | 0.71 | 0.43 | 1.17 |
| S.QAPTESi | 6.94 | 3.50 | 0.44 | 0.27 | 3.44 | 0.43 | 0.26 | 0.98 |
| Material | Final DC (%) | Total Strain (%) |
|---|---|---|
| Organic matrix | 54.20 | 8.84 ± 0.26 |
| Neat silica nanocomposite | 54.98 | 4.67 ± 0.70 |
| S.MPS nanocomposite | 59.51 | 7.26 ± 0.46 |
| S.QAMTMSi nanocomposite | 59.16 | 6.30 ± 0.70 |
| S.QAMTESi nanocomposite | 55.67 | 4.44 ± 0.98 |
| S.QAPTESi nanocomposite | 36.17 | 4.82 ± 0.44 |
| FiltekTMZ350 XT (3M ESPE, St. Paul, MN, USA) [65] | 50.96 | 1.66 ± 0.15 |
| Tetric® N-Ceram Bulk Fill (Ivoclar-Vivadent, Schaan, Liechtenstein) [65] | 49.50 | 1.36 ± 0.08 |
| Material | Flexural Modulus (GPa) 1 | Flexural Strength (MPa) 1 | Compressive Strength (MPa) |
|---|---|---|---|
| Organic matrix | 0.96 ± 0.06 a,b,c,e,g | 50.37 ± 3.16 j,k,l,m | 221.48 ± 43.95 |
| Neat silica nanocomposite | 1.91 ± 0.13 a,c,e,g | 63.54 ± 10.56 j,l,n,o | 222.86 ± 133.86 |
| S.MPS nanocomposite | 1.79 ± 0.09 b,d,f,h | 70.86 ± 4.94 k,m,p,q,r | 276.72 ± 87.82 |
| S.QAMTMSi nanocomposite | 1.67 ± 0.10 c,d,g | 51.58 ± 5.51 n,p,r | 231.81 ± 78.41 |
| S.QAMTESi nanocomposite | 1.62 ± 0.10 e,f,i | 56.36 ± 13.35 q,s | 264.17 ± 60.03 |
| S.QAPTESi nanocomposite | 0.02 ± 0.00 g,h,i | 1.57 ± 0.40 l,m,o,r,s | 184.87 ± 30.17 |
| FiltekTMZ350 XT (3M ESPE, St. Paul, MN, USA) [65,79] | 9.13 ± 0.66 | 80.52 ± 15.88 | 226.92 ± 6.64 |
| Tetric® N-Ceram Bulk Fill (Ivoclar-Vivadent, Schaan, Liechtenstein) [65,79] | 7.05 ± 0.60 | 60.37 ± 11.05 | 214.28 ± 3.67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolaidis, A.K.; Koulaouzidou, E.A.; Gogos, C.; Achilias, D.S. Synthesis of Novel Dental Nanocomposite Resins by Incorporating Polymerizable, Quaternary Ammonium Silane-Modified Silica Nanoparticles. Polymers 2021, 13, 1682. https://doi.org/10.3390/polym13111682
Nikolaidis AK, Koulaouzidou EA, Gogos C, Achilias DS. Synthesis of Novel Dental Nanocomposite Resins by Incorporating Polymerizable, Quaternary Ammonium Silane-Modified Silica Nanoparticles. Polymers. 2021; 13(11):1682. https://doi.org/10.3390/polym13111682
Chicago/Turabian StyleNikolaidis, Alexandros K., Elisabeth A. Koulaouzidou, Christos Gogos, and Dimitris S. Achilias. 2021. "Synthesis of Novel Dental Nanocomposite Resins by Incorporating Polymerizable, Quaternary Ammonium Silane-Modified Silica Nanoparticles" Polymers 13, no. 11: 1682. https://doi.org/10.3390/polym13111682
APA StyleNikolaidis, A. K., Koulaouzidou, E. A., Gogos, C., & Achilias, D. S. (2021). Synthesis of Novel Dental Nanocomposite Resins by Incorporating Polymerizable, Quaternary Ammonium Silane-Modified Silica Nanoparticles. Polymers, 13(11), 1682. https://doi.org/10.3390/polym13111682