
UV Polymerization of Methacrylates—Preparation and Properties of Novel Copolymers

Abstract
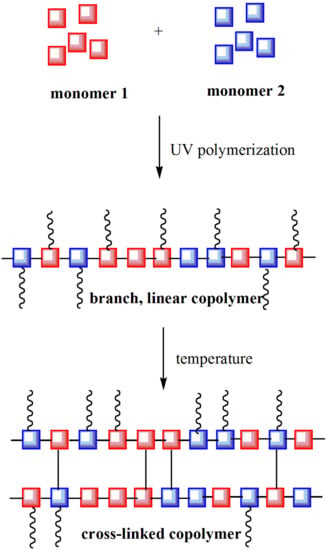
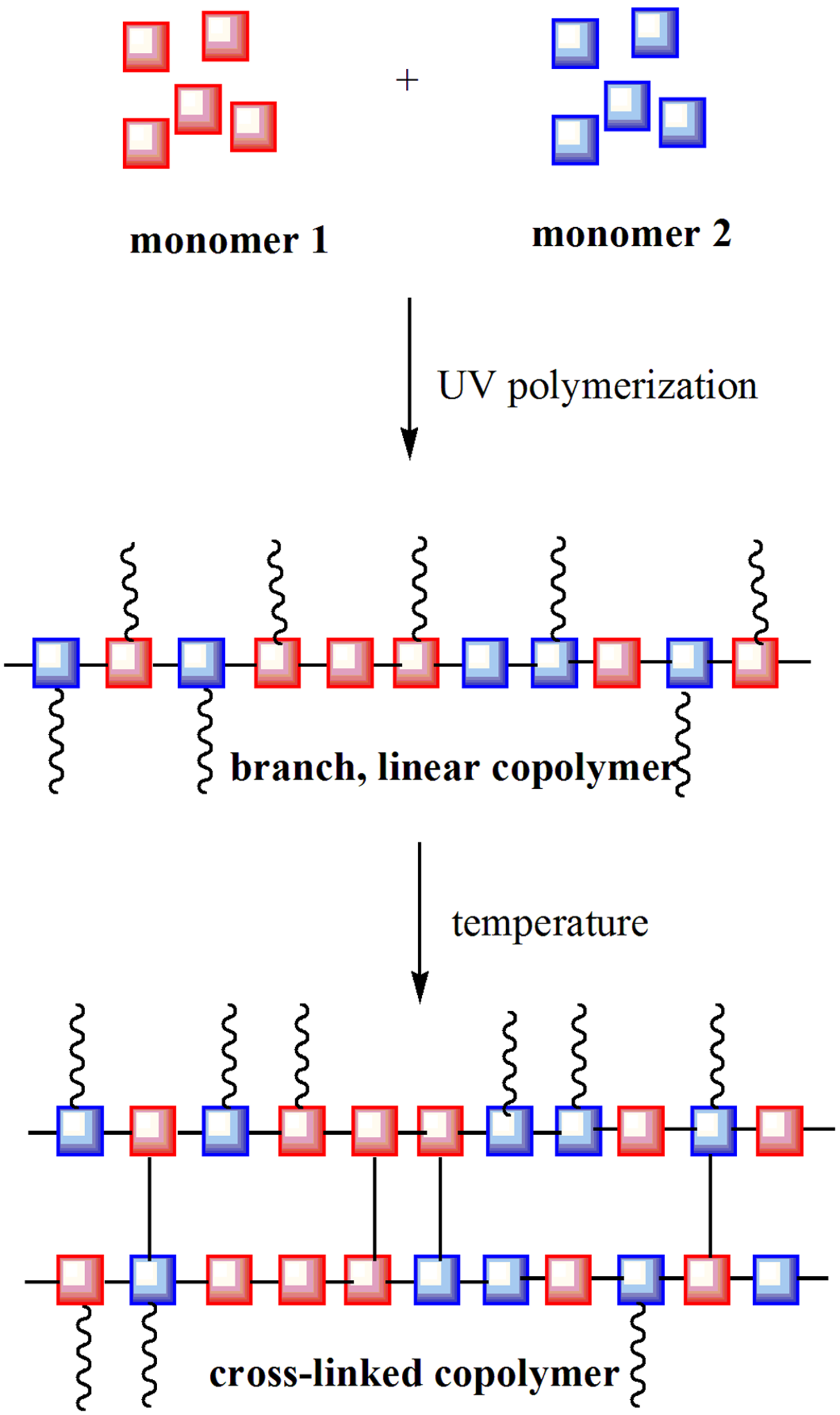
1. Introduction
2. Materials and Methods
2.1. Materials
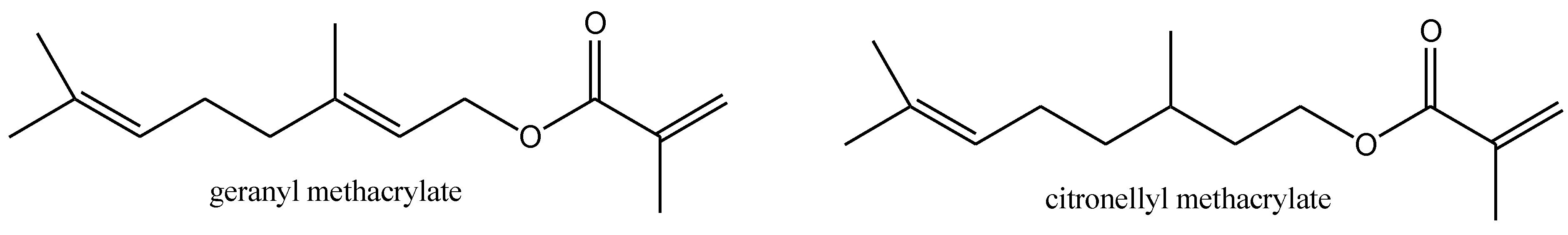
2.2. Synthesis of Methacrylate Monomers
2.3. UV-Polymerization
2.4. Characterization of Copolymers
2.4.1. ATR-FTIR
2.4.2. Conversion of Double Bonds
2.4.3. Solubility Tests
2.4.4. Chemical Resistance
2.4.5. Glass Transition Temperature
2.4.6. Thermal Properties
2.4.7. Simultaneous TG–FTIR Analysis
3. Results and Discussion
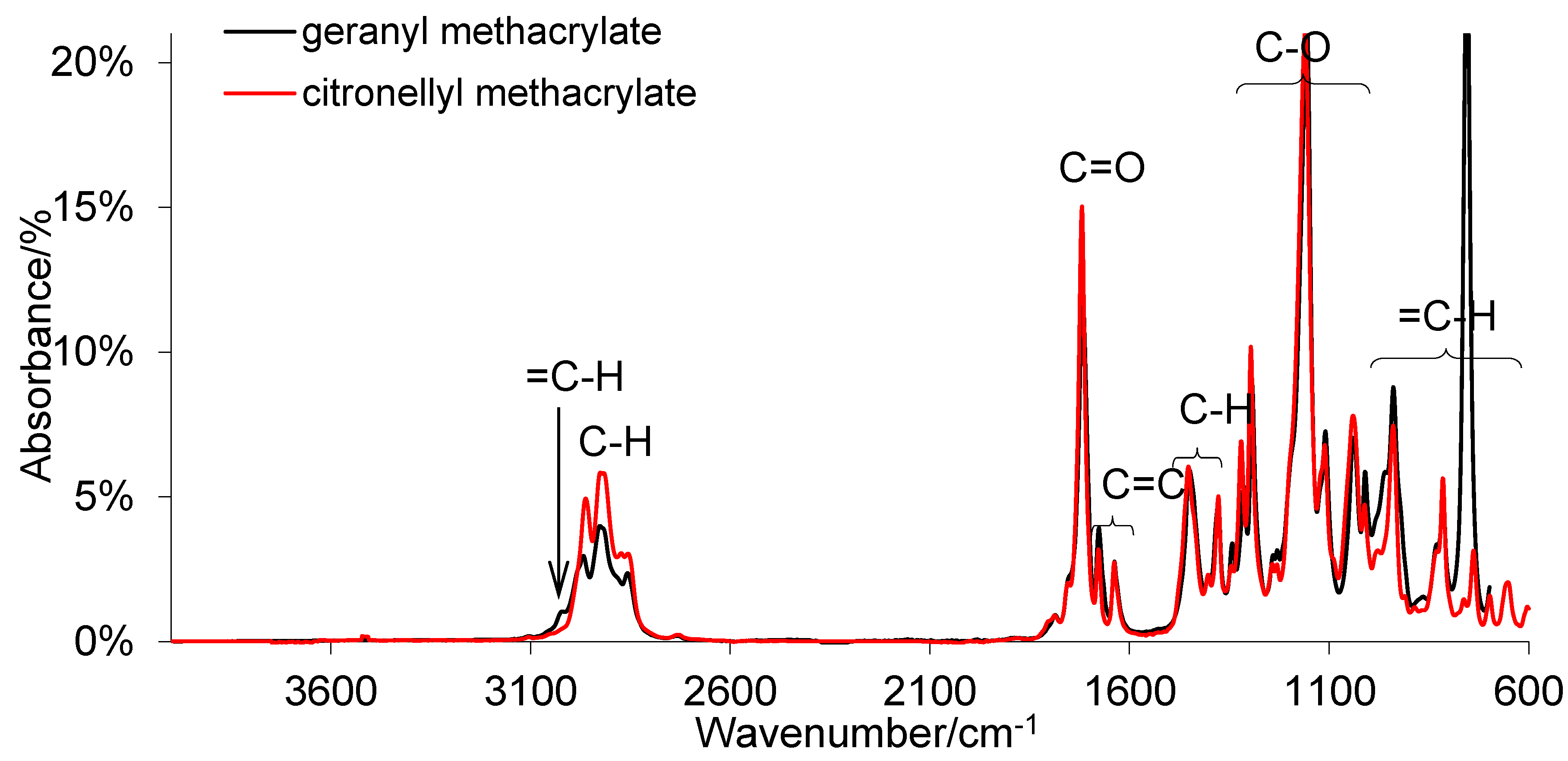
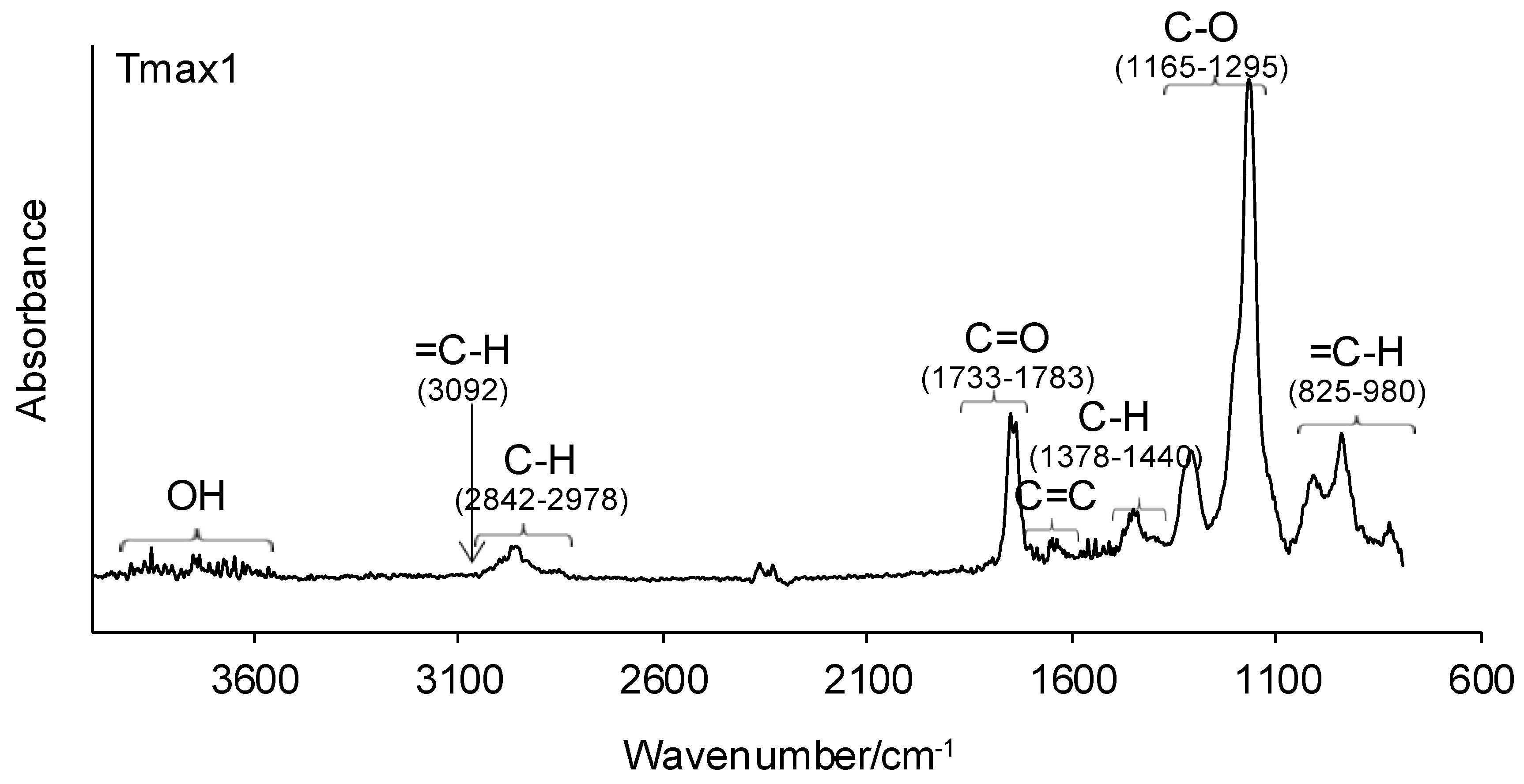
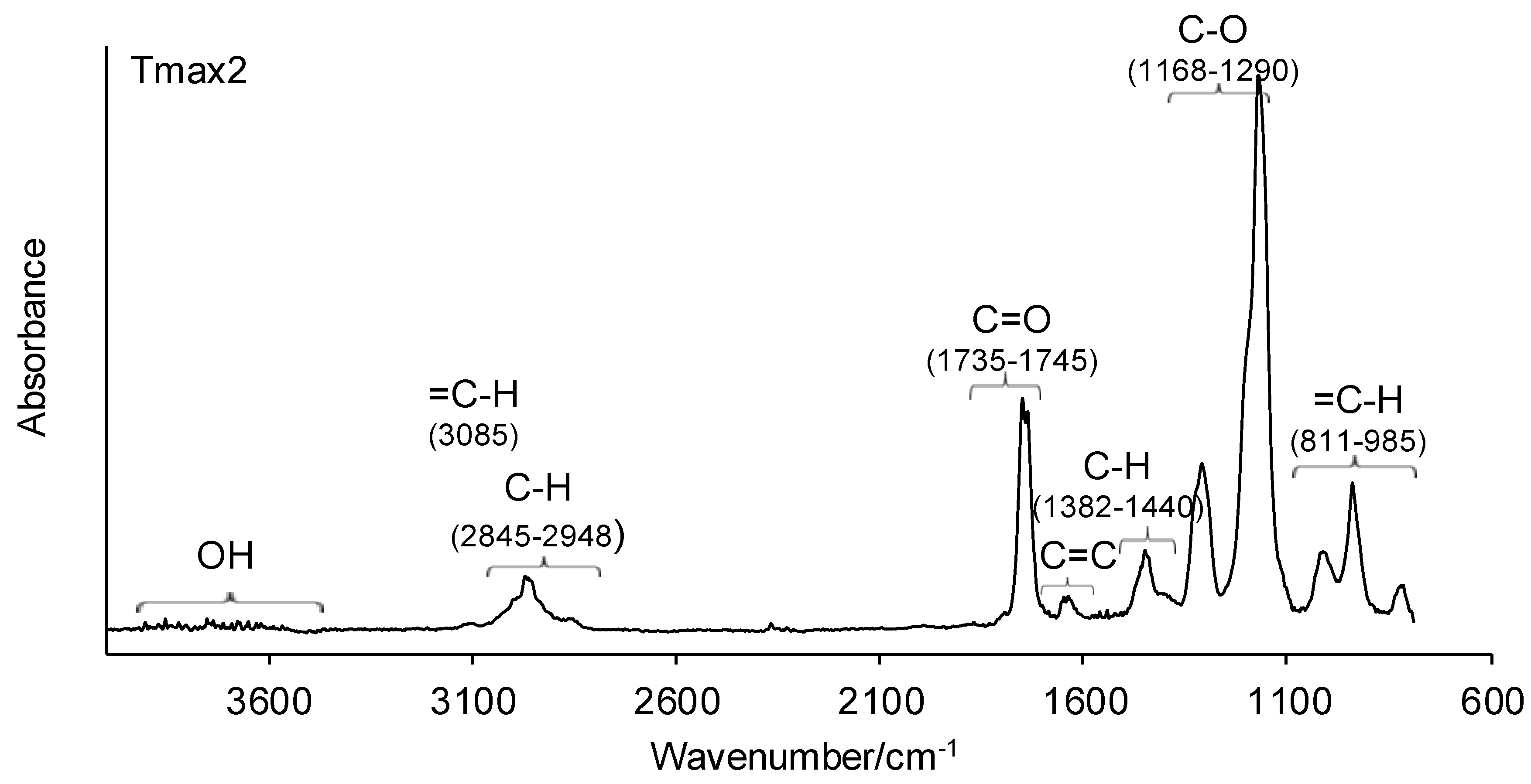
3.1. ATR-FTIR of Monomers
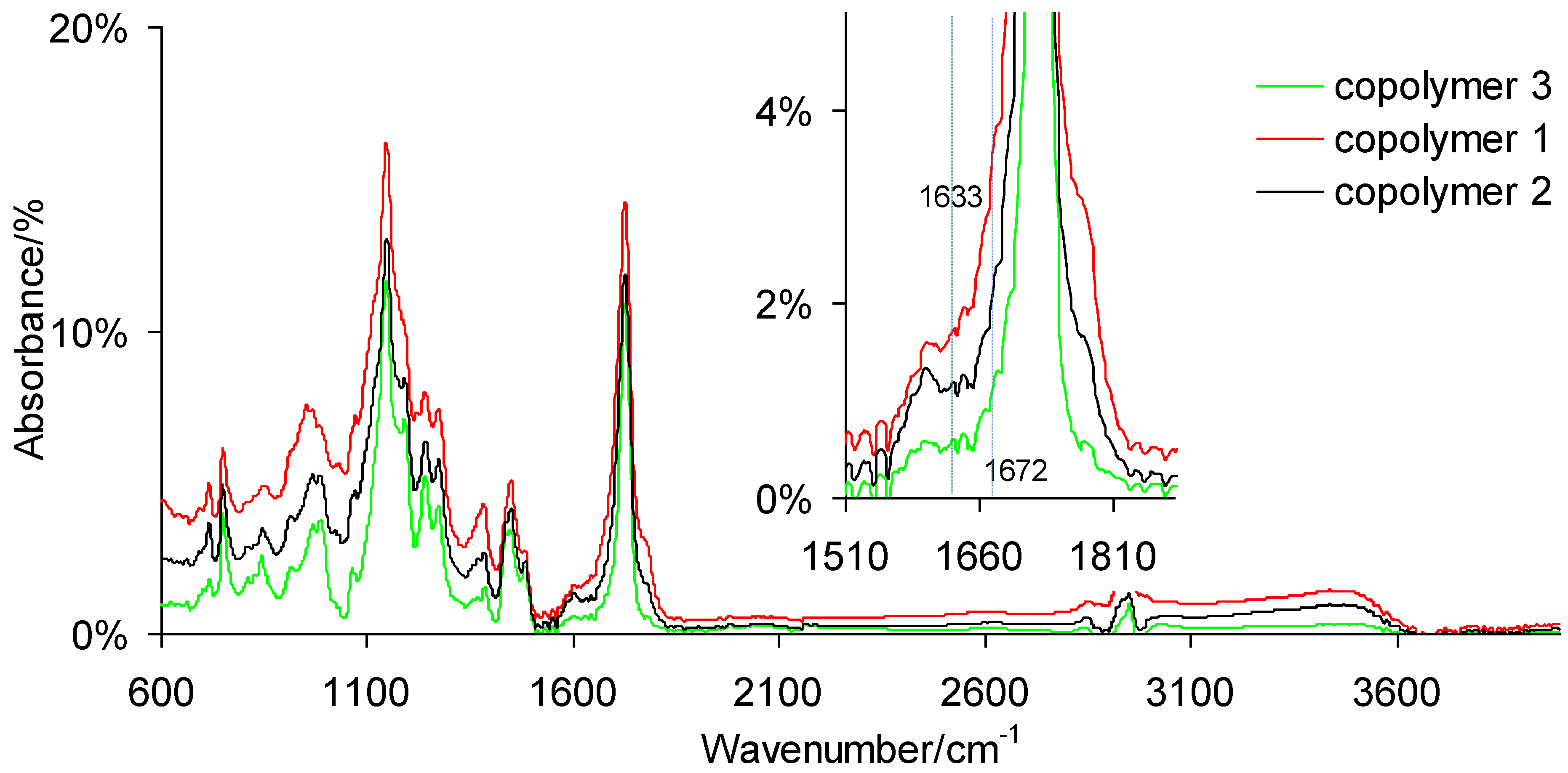
3.2. Conversion of Double Bonds
3.3. Solubility Tests
3.4. Chemical Resistance
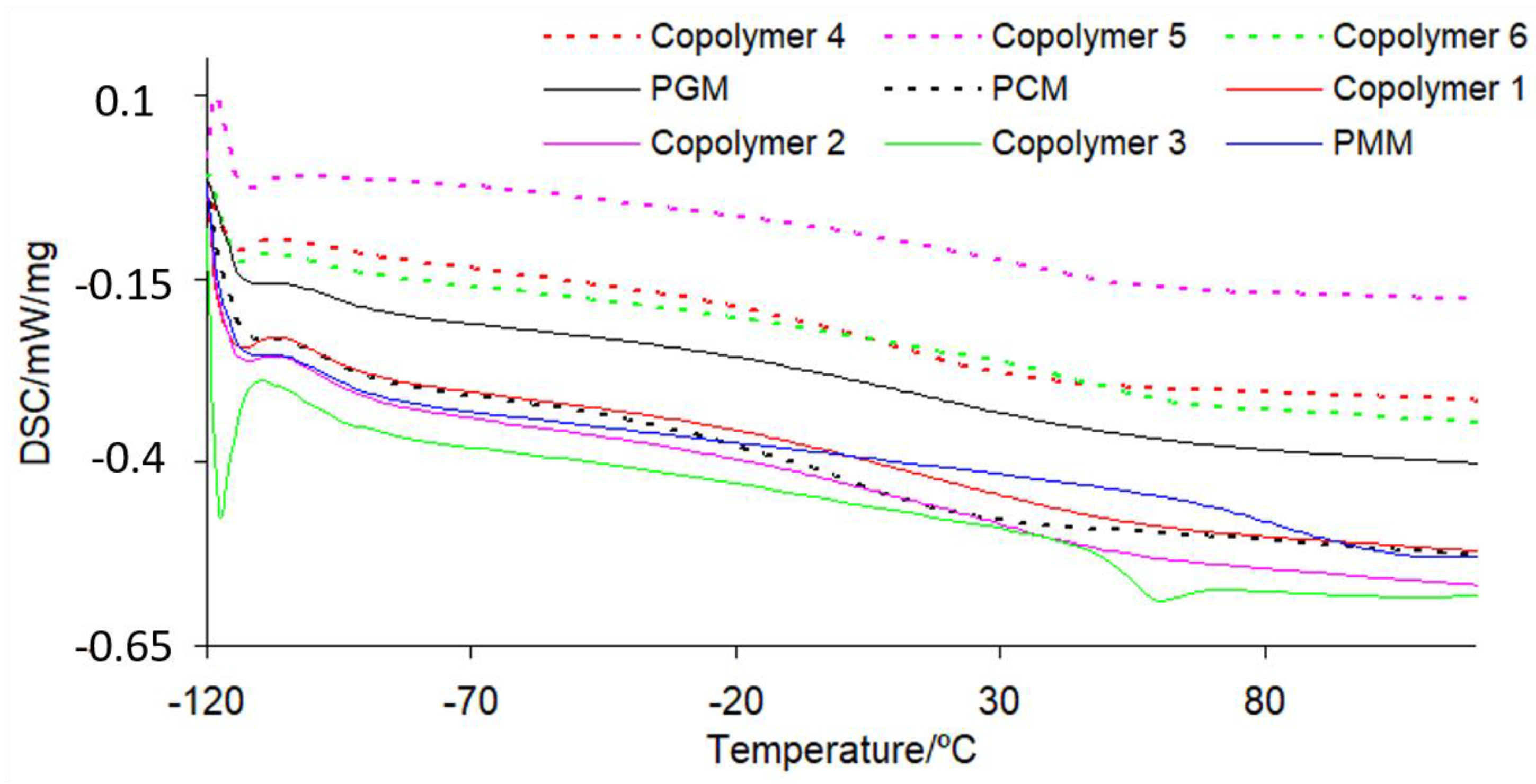
3.5. Glass Transition Temperature
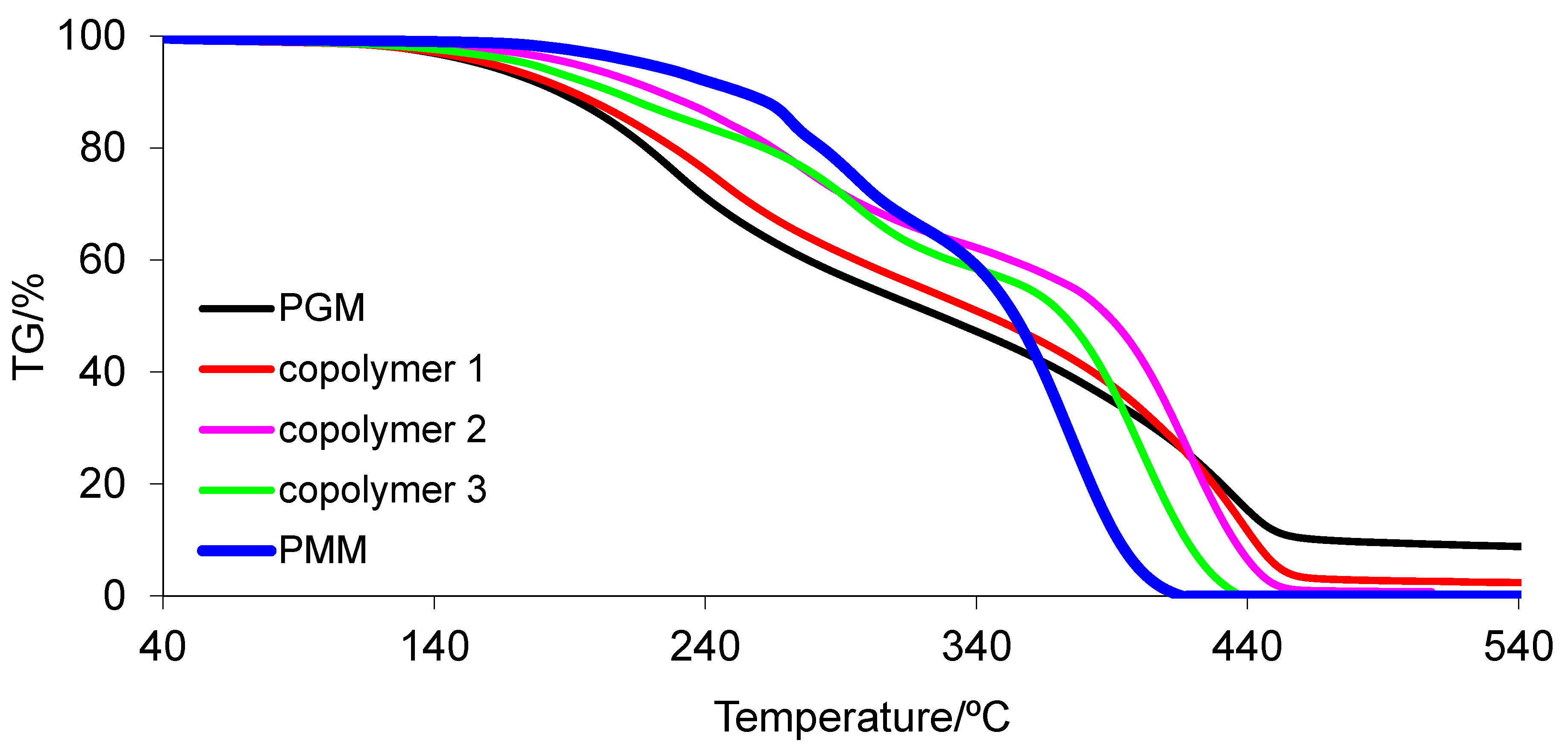
3.6. Thermal Properties
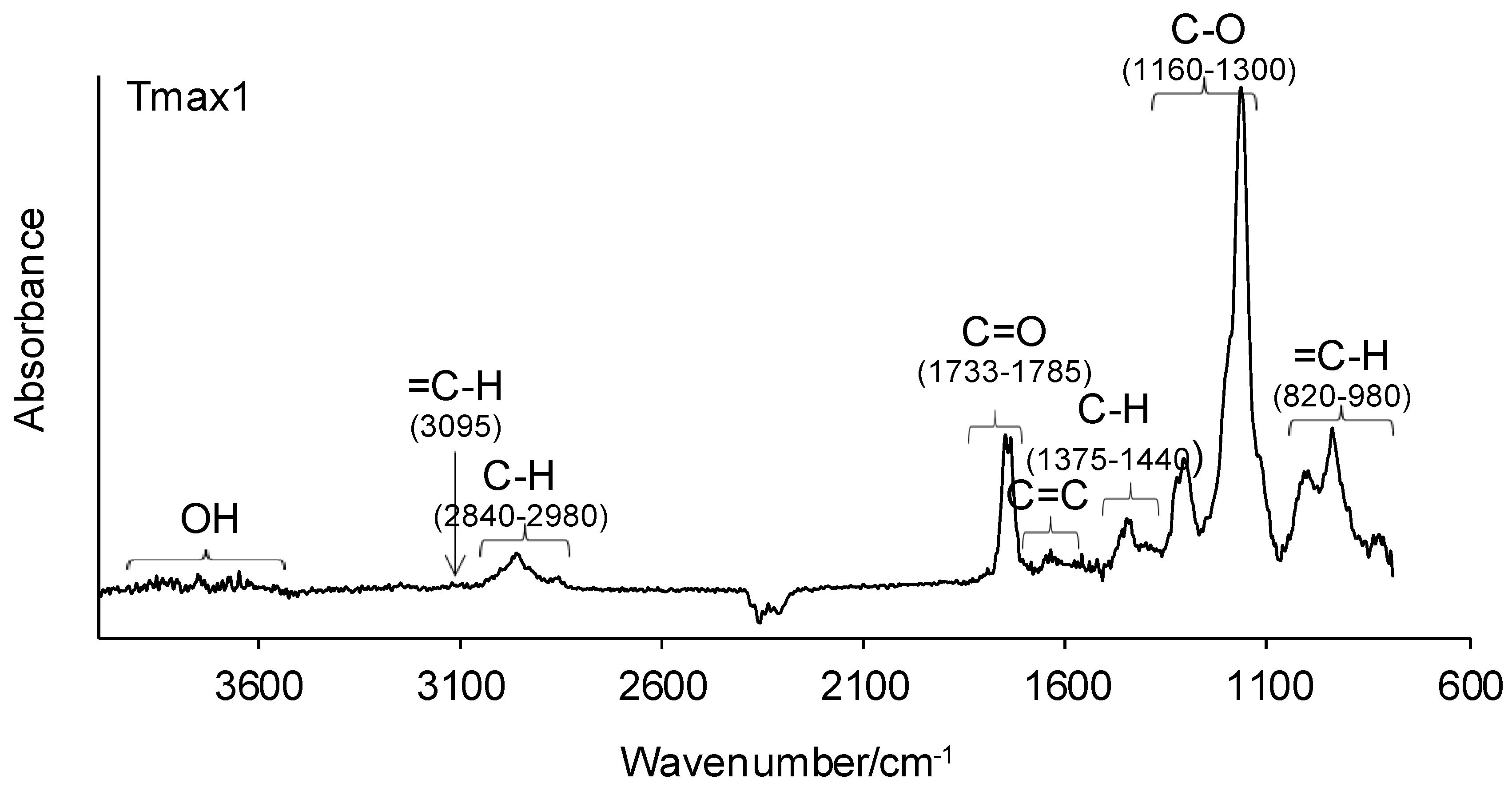
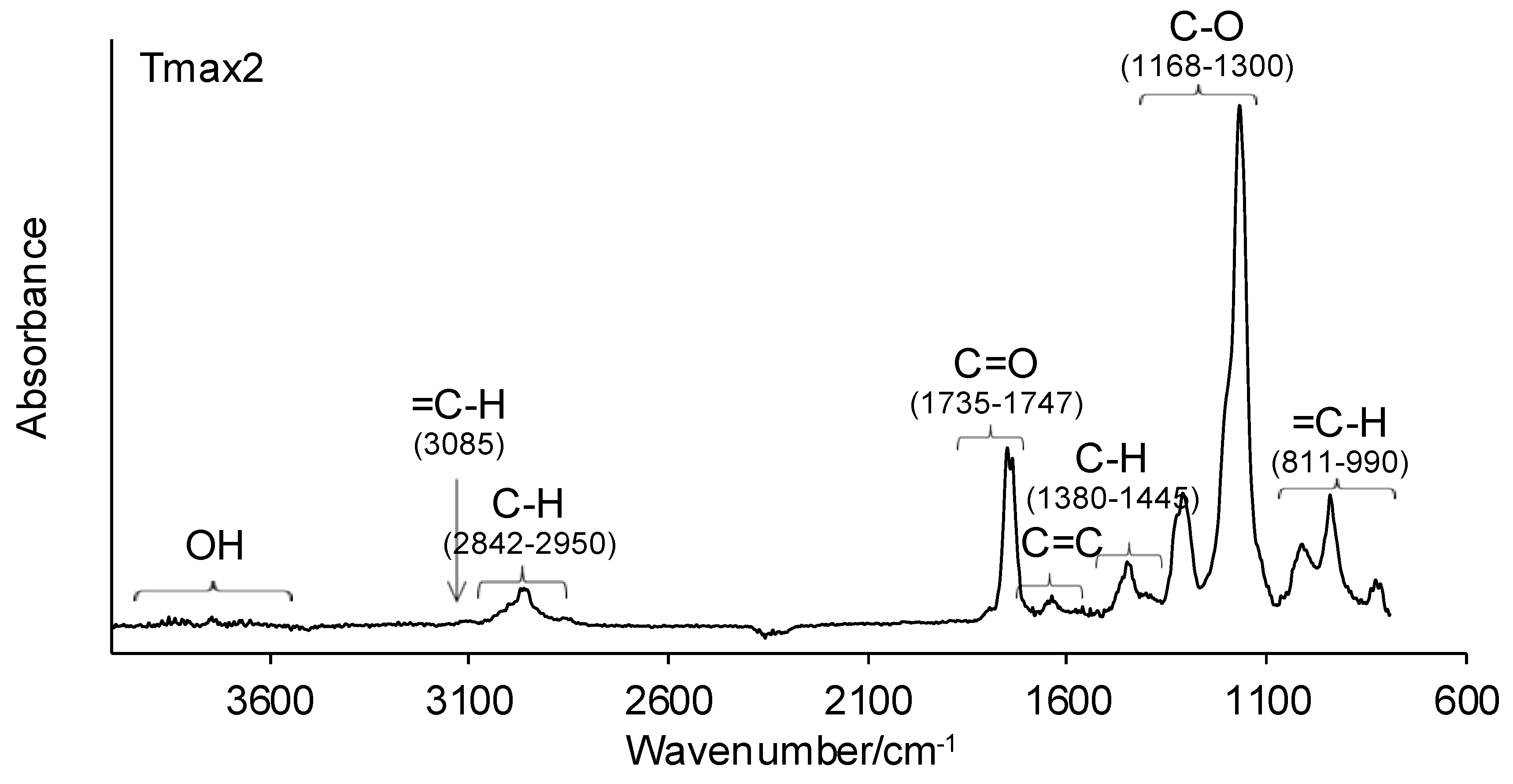
3.7. Simultaneous TG–FTIR Analysis
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gibson, I.; Rosen, D.; Stucker, B. Additive Manufacturing Technologies; Springer: Boston, MA, USA, 2010; pp. 78–119. [Google Scholar]
- Shao, J.; Huang, Y.; Fan, Q. Visible light initiating systems for photopolymerization: Status, development and challenges. Polym. Chem. 2014, 14, 4195–4210. [Google Scholar] [CrossRef]
- Fertig, J.; Goldberg, A.I.; Skoultchi, M. Ultraviolet stabilizing monomers and polymers. I. Synthesis and polymerization of phenyl 5-methacryloxymethylsalicylate. J. Appl. Polym. Sci. 1965, 9, 903–910. [Google Scholar] [CrossRef]
- Park, J.W.; Shim, G.S.; Lee, J.G.; Jang, S.W.; Kim, H.J.; Choi, J.N. Evaluation of UV curing properties of mixture systems with differently sized monomers. Materials 2018, 11, 509. [Google Scholar] [CrossRef] [PubMed]
- Decker, C.; Moussa, K. Radiation Curing of Polymeric Materials; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1990; Chapter 31; pp. 439–456. [Google Scholar]
- McKeen, L.W. The Effect of Creep and Other Time Related Factors on Plastics and Elastomers; Elsevier: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Decker, C. Handbook of Adhesives and Sealants; Elsevier: Amsterdam, The Netherlands, 2011; pp. 303–340. [Google Scholar]
- Licari, J.J.; Swanson, D.W. Adhesives Technology for Electronic Applications. Materials, Processing, Reliability; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Anastasio, R.; Peerbooms, W.; Cardinaels, R.; Breemen, L.C.A. Characterization of ultraviolet-cured methacrylate networks: From photopolymerization to ultimate mechanical properties. Macromolecules 2019, 52, 9220–9231. [Google Scholar] [CrossRef]
- Chylińska, M.; Kaczmarek, H.; Moszyński, D.; Królikowski, B.; Kowalonek, J. Surface studies of UV irradiated polypropylene films modified with mineral fillers designed as piezoelectric materials. Polymers 2020, 12, 562. [Google Scholar] [CrossRef] [PubMed]
- Sutton, P.; Airoldi, M.; Porcarelli, L.; Olmedo-Martínez, J.L.; Mugemana, C.; Bruns, N.; Mecerreyes, D.; Steiner, U.; Gunkel, I. Tuning the properties of a UV-polymerized, cross-linked solid polymer electrolyte for lithium batteries. Polymers 2020, 12, 595. [Google Scholar] [CrossRef]
- Sanai, Y.; Kubota, K. Effect of UV-curing conditions on the polymer structures: A comparison between coating and adhesive. Polym. J. 2020, 52, 1153–1163. [Google Scholar] [CrossRef]
- Li, Y.; Sawut, A.; Hou, G.; He, M.; Yimit, M. UV polymerization and property analysis of maleacylated methyl cellulose acrylic acid absorbent resin. Pol. J. Chem. Technol. 2020, 22, 34–41. [Google Scholar] [CrossRef]
- Bagheri, A.; Jin, J. Photopolymerization in 3D Printing. ACS Appl. Polym. Mater. 2019, 1, 593–611. [Google Scholar] [CrossRef]
- Fouassier, J.P.; Allonas, X.; Burget, D. Photopolymerization reactions under visible lights: Principle, mechanism and examples of applications. Prog. Org. Coat. 2003, 47, 16–36. [Google Scholar] [CrossRef]
- Garra, P.; Dietlin, C.; Morlet-Savary, F.; Dumur, F.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Photopolymerization processes of thick films and in shadow areas: A review for the access to composites. Polym. Chem. 2017, 8, 7088–7101. [Google Scholar] [CrossRef]
- Baroli, B. Photopolymerization of biomaterials: Issues and potentialities in drug delivery, tissue engineering, and cell encapsulation applications. J. Chem. Technol. Biotechnol. 2006, 81, 491–499. [Google Scholar] [CrossRef]
- Kaur, M.; Srivastava, A.K. Photopolymerization: A review. J. Macromol. Sci. Polym. Rev. Part C 2002, 42, 481–512. [Google Scholar] [CrossRef]
- Lin, J.T.; Cheng, D.C.; Chen, K.T.; Liu, H.W. Dual-wavelength (UV and blue) controlled photopolymerization confinement for 3D-Printing: Modeling and analysis of measurements. Polymers 2019, 11, 1819. [Google Scholar] [CrossRef] [PubMed]
- Elias, H.G. Macromolecules: Synthesis, Materials and Technology; Springer Science: New York, NY, USA, 1984. [Google Scholar]
- Lu, J.; Kamigaito, M.; Higashimura, T.; Deng, Y.X. Living cationic isomerization polymerization of β-pinene. 1. Initiation with HCl-2-chloroethyl vinyl ether adduct/TiCl3(OiPr) in conjunction with nBu4NCl. Macromoelules 1997, 30, 22–26. [Google Scholar] [CrossRef]
- Satoh, K.; Sugiyama, H.; Kamigaito, M. Biomass-derived heat-resistant alicyclic hydrocarbon polymers: Poly(terpenes) and hydrogenated derivatives. Green Chem. 2006, 8, 878–882. [Google Scholar] [CrossRef]
- Yu, P.; Li, A.; Liang, H.; Lu, J. Polymerization of β-pinene with Schiff-base nickel complexes catalyst: Synthesis of relatively high molecular weight poly(β-pinene) at high temperature with high productivity. J. Polym. Res. Part A Polym. Chem. 2007, 45, 3739–3746. [Google Scholar] [CrossRef]
- Kukhta, N.A.; Vasilenko, I.V.; Kostujk, S.V. Room temperature cationic polymerization of β-pinene using modified AlCl3 catalyst: Toward sustainable plastics from renewable biomass resources. Green Chem. 2011, 13, 2362–2364. [Google Scholar] [CrossRef]
- Wang, Y.; Li, A.; Liang, H.; Lu, J. Reversible addition-fragmentation chain transfer radical copolymerization of β-pinene and methyl acrylate. Eur. Polym. J. 2006, 42, 2695–2702. [Google Scholar] [CrossRef]
- Firdaus, M.; Espinosa, L.M.; Meier, M.A.R. Terpene-based renewable monomers and polymers via thiol-ene addition. Macromolecules 2011, 44, 7253–7262. [Google Scholar] [CrossRef]
- Nair, D.P.; Podgórski, M.; Chatani, S.; Gong, T.; Xi, W.; Fenoli, C.R.; Bowman, C.N. The thiol-Michael addition click reaction: A powerful and widely used tool in materials chemistry. Chem. Mater. 2014, 26, 724–744. [Google Scholar] [CrossRef]
- Lu, J.; Liang, H.; Li, A.; Cheng, Q. Synthesis of block and graft copolymers of β-pinene and styrene by transformation of living cationic polymerization to atom transfer radical polymerization. Eur. Polym. J. 2004, 40, 397–402. [Google Scholar] [CrossRef]
- Worzakowska, M. The preparation, physicochemical and thermal properties of the high moisture, solvent and chemical resistant starch-g-poly(geranyl methacrylate) copolymers. J. Therm. Anal. Calorim. 2020, 140, 189–198. [Google Scholar] [CrossRef]
- Worzakowska, M. Novel starch-g-copolymers obtained using acrylate monomers prepared from two geometric isomers of terpene alcohol. Eur. Polym. J. 2019, 110, 265–275. [Google Scholar] [CrossRef]
- Worzakowska, M. Synthesis and some physico-chemical properties of novel starch-g-poly(citronellyl acrylate) copolymers. Starch 2018, 70, 1700330. [Google Scholar] [CrossRef]
- Worzakowska, M. Chemical modification of potato starch by graft copolymerization with citronellyl methacrylate. J. Polym. Environ. 2018, 26, 1613–1624. [Google Scholar] [CrossRef]
- Kaith, B.S.; Singha, A.S.; Grupa, S.K. Graft copolymerization of flax fibres with binary vinyl monomer mixtures and evaluation of swelling, moisture absorbance and thermal behavior of the grafted fibres. J. Polym. Mater. 2003, 20, 195–199. [Google Scholar]
- Worzakowska, M. High chemical and solvent resistant, branched, terpene methacrylate polymers-Preparation, thermal properties and decomposition mechanism. Polym. Adv. Technol. 2018, 29, 1414–1425. [Google Scholar] [CrossRef]
- Available online: https://www.sigmaaldrich.com/catalog/product/aldrich/m55909?lang=pl®ion=PL&gclid=EAIaIQobChMIsu3bot6r8AIVideyCh2sPgUGEAAYASAAEgJSrPD_BwE (accessed on 15 April 2021).
- Albeladi, H.K.; Al-Romaizan, A.N.; Hussein, M.A. Role of cross-linking process on the performance of PMMA. Int. J. Biosens. Bioelectron. 2017, 3, 279–284. [Google Scholar]
- Gziut, K.; Kowalczyk, A.; Schmidt, B. Free-radical bulk-photopolymerization process as a method of obtaining thermally curable structural self-adhesive tapes and effect of used type I Photoinitiators. Polymers 2020, 12, 2191. [Google Scholar] [CrossRef] [PubMed]
- Godiya, C.B.; Gabrielli, S.; Materazzi, S.; Pianesi, M.S.; Stefanini, N.; Marcantoni, N. Depolymerization of waste poly(methyl methacrylate) scraps and purification of depolymerized products. J. Environ. Manag. 2019, 231, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.L.; Minh, L.; Blanchard, N.P. Thermal decomposition and glass transition temperature of poly(methyl methacrylate) and poly(isobutyl methacrylate). J. Macromol. Sci. Part A Chem. 1983, 19, 579–600. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer Name | Geranyl Methacrylate (GM)/g | Citronellyl Methacrylate (CM)/g | Methyl Methacrylate (MM)/g | Mass Ratio of Monomers (GM, CM to MM)/% |
|---|---|---|---|---|
| Poly(geranyl methacrylate) (PGM) | 3.0 | - | - | 100:0 |
| Poly(citronellyl methacrylate) (PCM) | - | 3.0 | - | 100:0 |
| Poly(methyl methacrylate) (PMM) | - | - | 3.0 | 0:100 |
| Copolymer 1 | 2.4 | - | 0.6 | 80:20 |
| Copolymer 2 | 1.5 | - | 1.5 | 50:50 |
| Copolymer 3 | 0.6 | - | 2.4 | 20:80 |
| Copolymer 4 | - | 2.4 | 0.6 | 80:20 |
| Copolymer 5 | - | 1.5 | 1.5 | 50:50 |
| Copolymer 6 | - | 0.6 | 2.4 | 20:80 |
| Monomer | Molecular Formulae | Molecular Mass/g/mol | Density/g/mL (25 °C) | Refractive Index n20/D | Purity/% | State |
|---|---|---|---|---|---|---|
| Geranyl methacrylate | C14H22O2 | 222.32 | 0.976 | 1.468 | ≥99% | liquid |
| Cironellyl methacrylate | C14H24O2 | 224.34 | 0.955 | 1.443 | ≥99% | liquid |
| Methyl methacrylate * | C5H8O2 | 100.12 | 0.936 | 1.414 | ≥99% | liquid |
| Polymer Name | DC/% | ||
|---|---|---|---|
| Irradiation | 50 °C | 120 °C | |
| PGM | 78 | 85 | 92 |
| PCM | 68 | 80 | 90 |
| PMM | 80 | 88 | 95 |
| Copolymer 1 | 75 | 85 | 90 |
| Copolymer 2 | 78 | 88 | 92 |
| Copolymer 3 | 78 | 88 | 94 |
| Copolymer 4 | 70 | 80 | 90 |
| Copolymer 5 | 72 | 83 | 93 |
| Copolymer 6 | 75 | 87 | 95 |
| Polymer Name | ΔmS/% | ||||||
|---|---|---|---|---|---|---|---|
| Water | Methanol | Butanol | Hexane | Toluene | Chloroform | CCl4 | |
| PGM | 19.0 | 4.8 | 3.2 | 10.8 | 18.2 | 15.0 | 16.4 |
| PCM | 10 | 3.7 | 2.1 | 10.2 | 17.6 | 11.0 | 13.2 |
| PMM | 0 | 0 | 0 | 5.0 | 100 | 100 | 4.0 |
| Copolymer 1 | 6.3 | 4.2 | 2.4 | 5.2 | 8.7 | 7.6 | 8.2 |
| Copolymer 2 | 8.4 | 6.7 | 4.8 | 6.6 | 10.2 | 9.3 | 11.3 |
| Copolymer 3 | 10 | 8.0 | 5.3 | 8.2 | 15.6 | 12.3 | 14.7 |
| Copolymer 4 | 4.8 | 2.5 | 1.5 | 5.2 | 7.3 | 6.5 | 8.0 |
| Copolymer 5 | 5.5 | 3.1 | 2.0 | 6.5 | 9.1 | 9.3 | 11.0 |
| Copolymer 6 | 8.3 | 4.2 | 2.4 | 8.0 | 10.4 | 11.2 | 12.8 |
| Polymer Name | ΔmS/% | ||||||
|---|---|---|---|---|---|---|---|
| Water | Methanol | Butanol | Hexane | Toluene | Chloroform | CCl4 | |
| PGM | 0.3 | 0.2 | 0.1 | 0.5 | 0.6 | 0.4 | 0.3 |
| PCM | 0.4 | 0.3 | 0.3 | 0.8 | 0.7 | 0.6 | 0.6 |
| PMM | 0 | 0 | 0 | 0 | 100 | 100 | 0 |
| Copolymer 1 | 0.2 | 0.1 | 0.1 | 0.3 | 0.4 | 0.4 | 0.2 |
| Copolymer 2 | 0.1 | 0.1 | 0.0 | 0.3 | 0.4 | 0.4 | 0.1 |
| Copolymer 3 | 0.1 | 0.1 | 0.0 | 0.3 | 0.3 | 0.4 | 0.2 |
| Copolymer 4 | 0.2 | 0.1 | 0.1 | 0.4 | 0.5 | 0.6 | 0.7 |
| Copolymer 5 | 0.3 | 0.1 | 0.1 | 0.3 | 0.5 | 0.4 | 0.3 |
| Copolymer 6 | 0.1 | 0.1 | 0.0 | 0.1 | 0.4 | 0.3 | 0.2 |
| Polymer Name | ΔmR/% | ||||
|---|---|---|---|---|---|
| 1M NaOH | 1M HCl | Buffer pH = 5 | Buffer pH = 7 | Buffer pH = 9 | |
| PGM | 10.2 | 8.3 | 7.5 | 6.4 | 9.3 |
| PCM | 6.7 | 5.5 | 4.8 | 4.6 | 6.5 |
| PMM | 5.0 | 2.0 | 1.0 | 0 | 4.0 |
| Copolymer 1 | 6.5 | 6.0 | 6.0 | 5.5 | 6.3 |
| Copolymer 2 | 7.8 | 6.5 | 6.3 | 5.8 | 8.0 |
| Copolymer 3 | 9.8 | 8.0 | 7.5 | 6.1 | 9.2 |
| Copolymer 4 | 5.5 | 5.3 | 5.0 | 5.0 | 5.5 |
| Copolymer 5 | 7.4 | 7.0 | 6.9 | 6.3 | 7.5 |
| Copolymer 6 | 7.6 | 7.4 | 7.0 | 6.9 | 7.7 |
| Polymer Name | ΔmR/% | ||||
|---|---|---|---|---|---|
| 1M NaOH | 1M HCl | Buffer pH = 4 | Buffer pH = 6 | Buffer pH = 10 | |
| PGM | 0.5 | 0.0 | 0.0 | 0.0 | 0.2 |
| PCM | 0.4 | 0.0 | 0.0 | 0.1 | 0.1 |
| PMM | 0 | 0 | 0 | 0 | 0 |
| Copolymer 1 | 0.3 | 0.0 | 0.0 | 0.1 | 0.2 |
| Copolymer 2 | 0.2 | 0.0 | 0.0 | 0.1 | 0.1 |
| Copolymer 3 | 0.4 | 0.0 | 0.0 | 0.1 | 0.2 |
| Copolymer 4 | 0.2 | 0.0 | 0.0 | 0.0 | 0.2 |
| Copolymer 5 | 0.2 | 0.0 | 0.0 | 0.1 | 0.1 |
| Copolymer 6 | 0.3 | 0.0 | 0.0 | 0.0 | 0.1 |
| Polymer Name | Tg/°C |
|---|---|
| PGM | 24.1 |
| PCM | 3.1 |
| PMM | 91.7 |
| Copolymer 1 | 24.9 |
| Copolymer 2 | 32.6 |
| Copolymer 3 | 53.0 |
| Copolymer 4 | 15.7 |
| Copolymer 5 | 38.6 |
| Copolymer 6 | 48.6 |
| Polymer Name | T5%/°C | Tmax1/°C | Δm1/% | Tmax2/°C | Δm2/% | rm/% |
|---|---|---|---|---|---|---|
| PGM | 158 | 228 | 48.5 | 434 | 42.7 | 8.8 |
| PCM | 153 | 252 | 28.8 | 407 | 71.2 | 0.0 |
| PMM | 218 | 235/272/295 | 34.1 | 374 | 65.9 | 0.0 |
| Copolymer 1 | 161 | 245 | 45.2 | 437 | 52.4 | 2.4 |
| Copolymer 2 | 192 | 276 | 36.7 | 418 | 62.5 | 0.8 |
| Copolymer 3 | 175 | 183/213/294 | 40.2 | 401 | 59.8 | 0.0 |
| Copolymer 4 | 195 | 211 | 24.6 | 404 | 75.4 | 0.0 |
| Copolymer 5 | 224 | 219 | 9.0 | 395 | 91.0 | 0.0 |
| Copolymer 6 | 221 | 213/297 | 28.0 | 386 | 72.0 | 0.0 |
| Polymer Name | T5%/°C | Tmax1/°C | Δm1/% | Tmax2/°C | Δm2/% | rm/% |
|---|---|---|---|---|---|---|
| PGM | 189 | 235 | 54.0 | 434 | 46.0 | 0.0 |
| PCM | 251 | 396 | 99.0 | 577 | 1.0 | 0.0 |
| PMM | 230 | 378 | 100 | - | - | 0.0 |
| Copolymer 1 | 195 | 231 | 50.0 | 435 | 50.0 | 0.0 |
| Copolymer 2 | 220 | 236 | 43.0 | 430 | 57.0 | 0.0 |
| Copolymer 3 | 219 | 231 | 29.5 | 411 | 70.5 | 0.0 |
| Copolymer 4 | 243 | 265 | 78.8 | 408 | 21.2 | 0.0 |
| Copolymer 5 | 250 | 261 | 52.3 | 401 | 47.7 | 0.0 |
| Copolymer 6 | 228 | 255 | 32.6 | 400 | 67.4 | 0.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Worzakowska, M. UV Polymerization of Methacrylates—Preparation and Properties of Novel Copolymers. Polymers 2021, 13, 1659. https://doi.org/10.3390/polym13101659
Worzakowska M. UV Polymerization of Methacrylates—Preparation and Properties of Novel Copolymers. Polymers. 2021; 13(10):1659. https://doi.org/10.3390/polym13101659
Chicago/Turabian StyleWorzakowska, Marta. 2021. "UV Polymerization of Methacrylates—Preparation and Properties of Novel Copolymers" Polymers 13, no. 10: 1659. https://doi.org/10.3390/polym13101659
APA StyleWorzakowska, M. (2021). UV Polymerization of Methacrylates—Preparation and Properties of Novel Copolymers. Polymers, 13(10), 1659. https://doi.org/10.3390/polym13101659