
Influence of the Active Layer Structure on the Photovoltaic Performance of Water-Soluble Polythiophene-Based Solar Cells

 , , ,
, , ,
Abstract
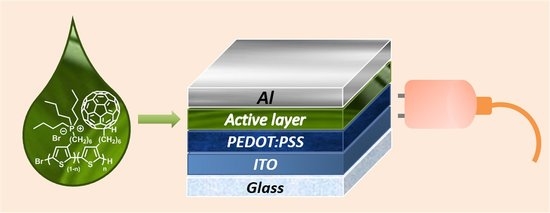
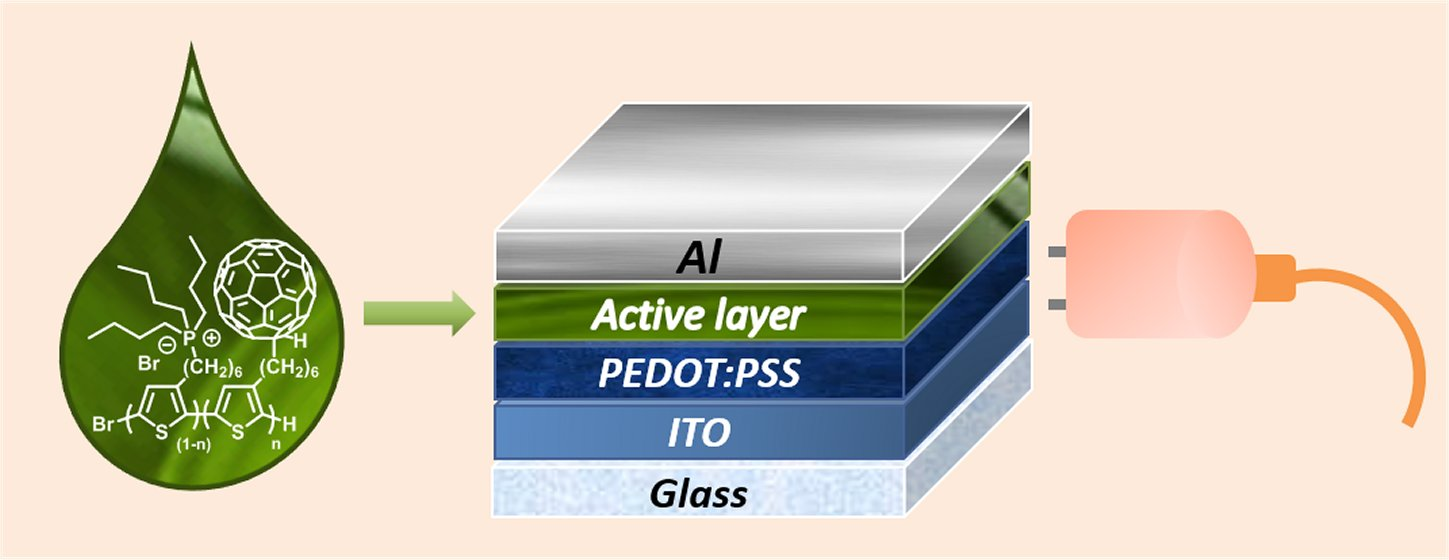
1. Introduction
2. Materials and Methods
2.1. Materials
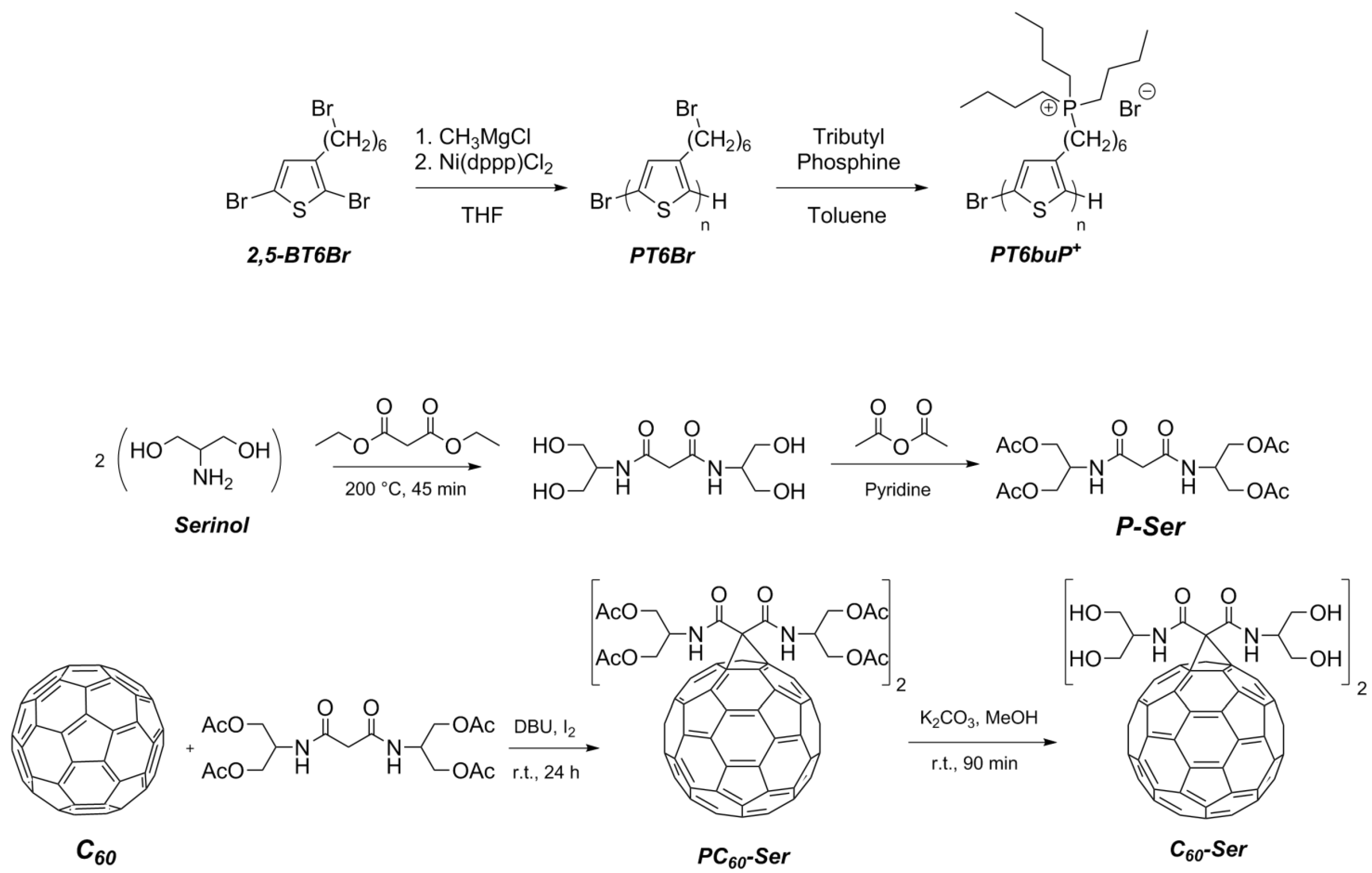
2.2. Synthesis of Homopolymers
2.2.1. Poly[3-(6-bromohexyl)thiophene] (PT6Br) by Grignard Metathesis Polymerization (GRIM)
2.2.2. Synthesis of Poly{3-[6-(tributylphosphonium)-hexyl]-thiophene-2,5-diyl Bromide} (PT6buP+) by Post-Functionalization of PT6Br
2.3. Synthesis of Water-Soluble Fullerene Derivative
2.3.1. Synthesis of N,N’-Bis{2-(acetyloxy)-1-[(acetyloxy)methyl]ethyl}-malonamide (P-Ser)
2.3.2. Synthesis of Protected Malonodiserinolamide Fullerene (PC60-Ser)
2.3.3. Synthesis of Malonodiserinolamide Fullerene (C60-Ser)
2.4. Synthesis of Water-Soluble Double-Cable Copolymer
2.4.1. Synthesis of Poly [3-(6-Bromohexyl)thiophene-co-3-(6-fullerenylhexyl)thiophene] (P[(T6Br)-co-(T6F)]) by Post-Functionalization of PT6Br
2.4.2. Synthesis of Poly [3-(6-Tributylphosphonium)thiophene-co-3-(6-fullerenylhexyl)thiophene] (P[(T6buP+)-co-(T6F)]) by Post-Functionalization of P[(T6Br)-co-(T6F)]) with Tributylphosphine
2.5. Measurements
2.6. Solar Cells
3. Results and Discussion
3.1. Synthesis
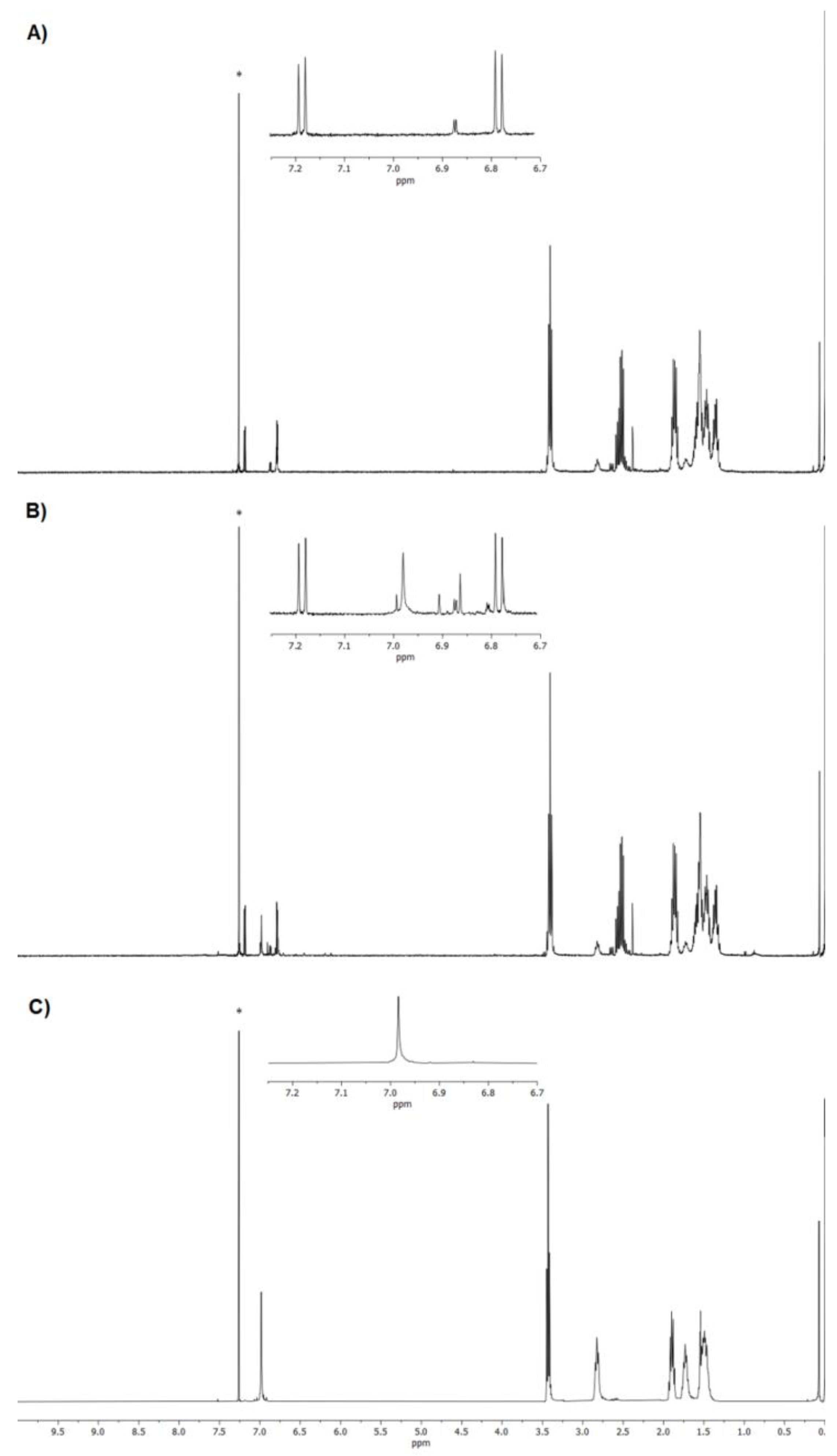
3.2. NMR Characterization
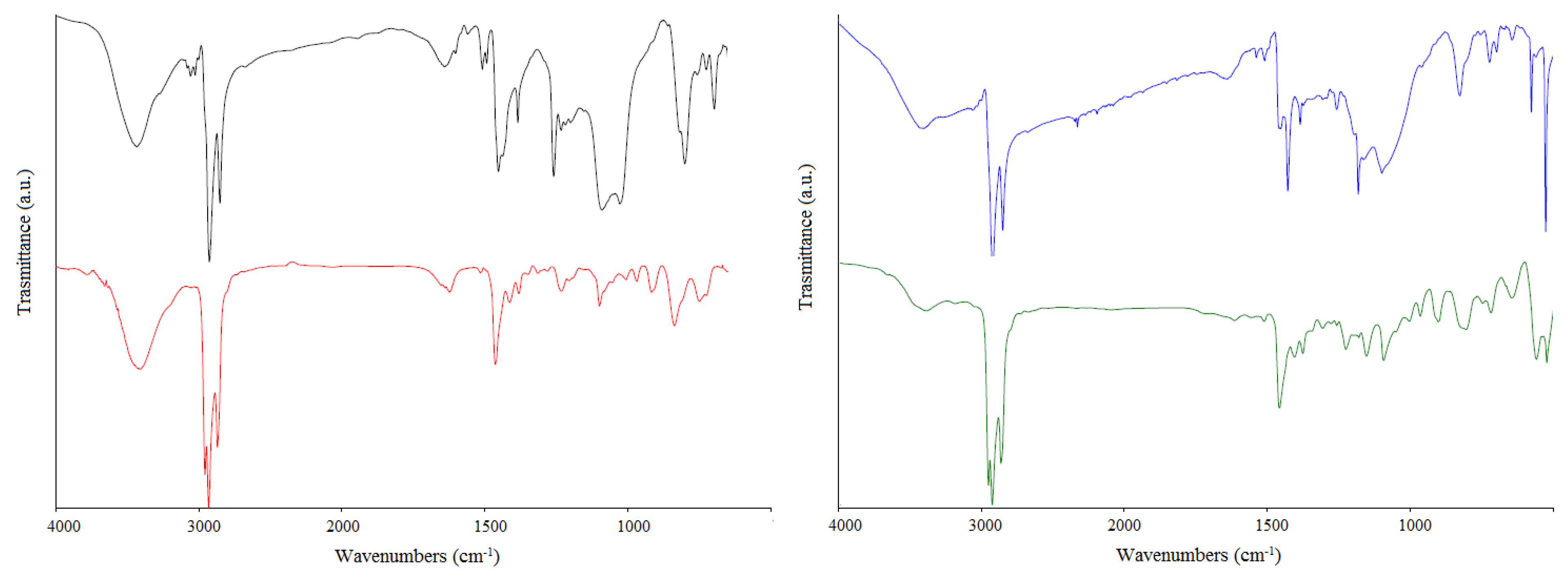
3.3. FT-IR Characterization
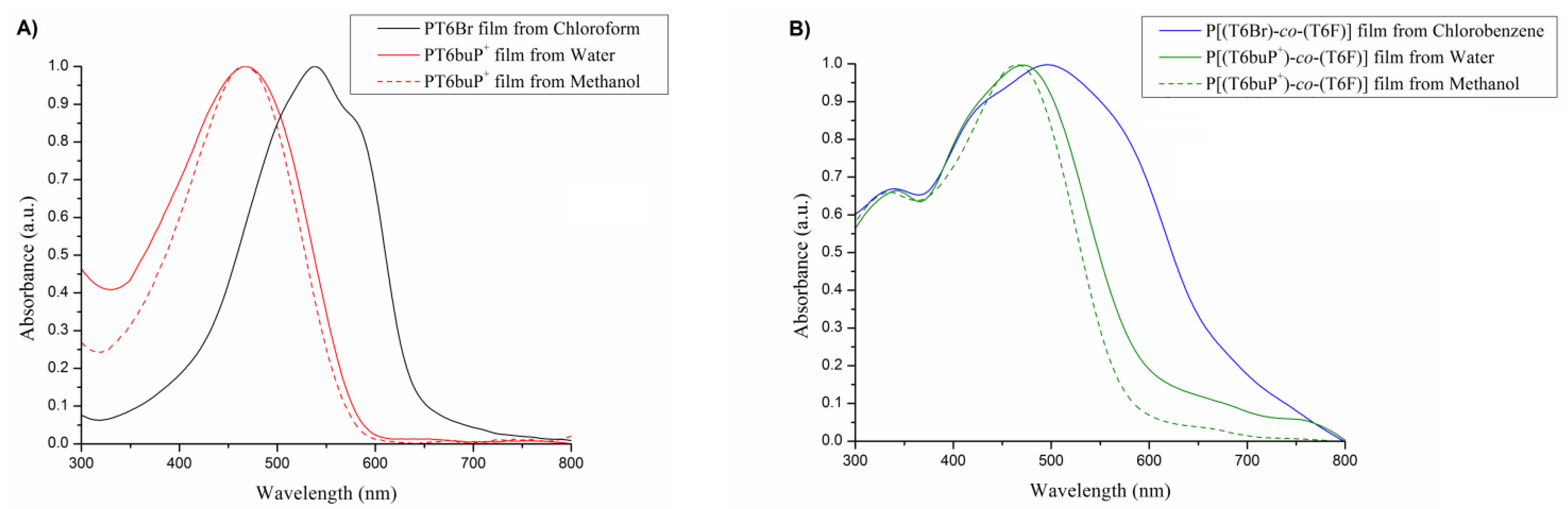
3.4. UV–Vis Analysis
3.5. Thermal Properties
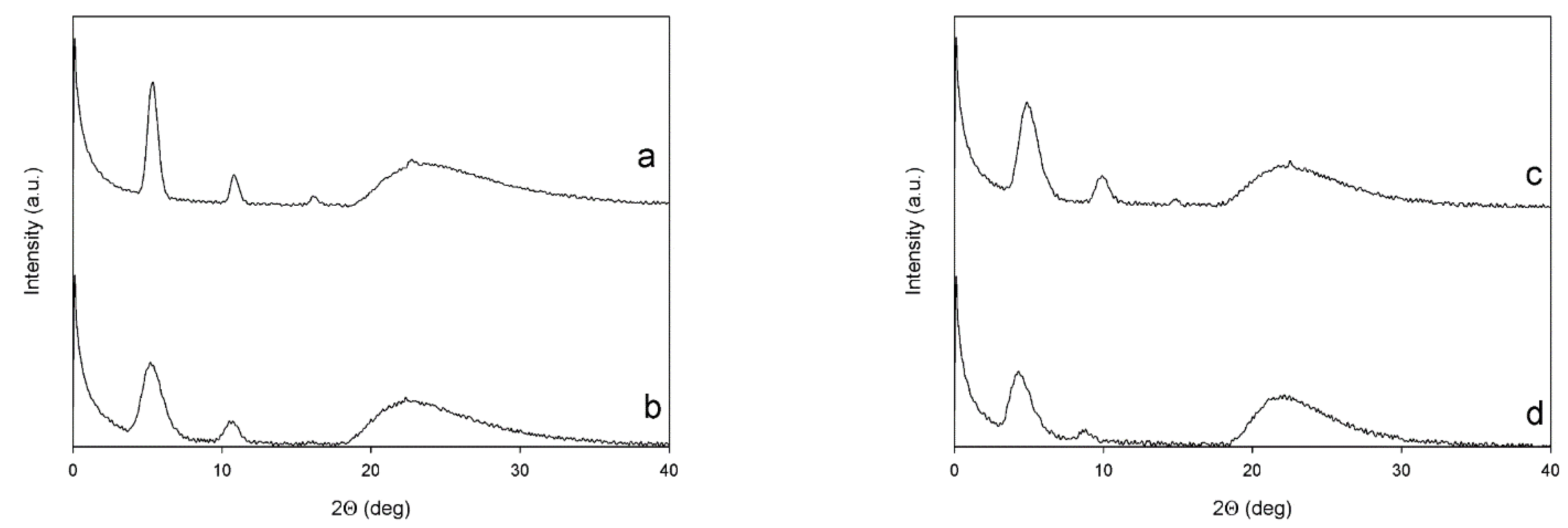
3.6. X-ray Diffraction
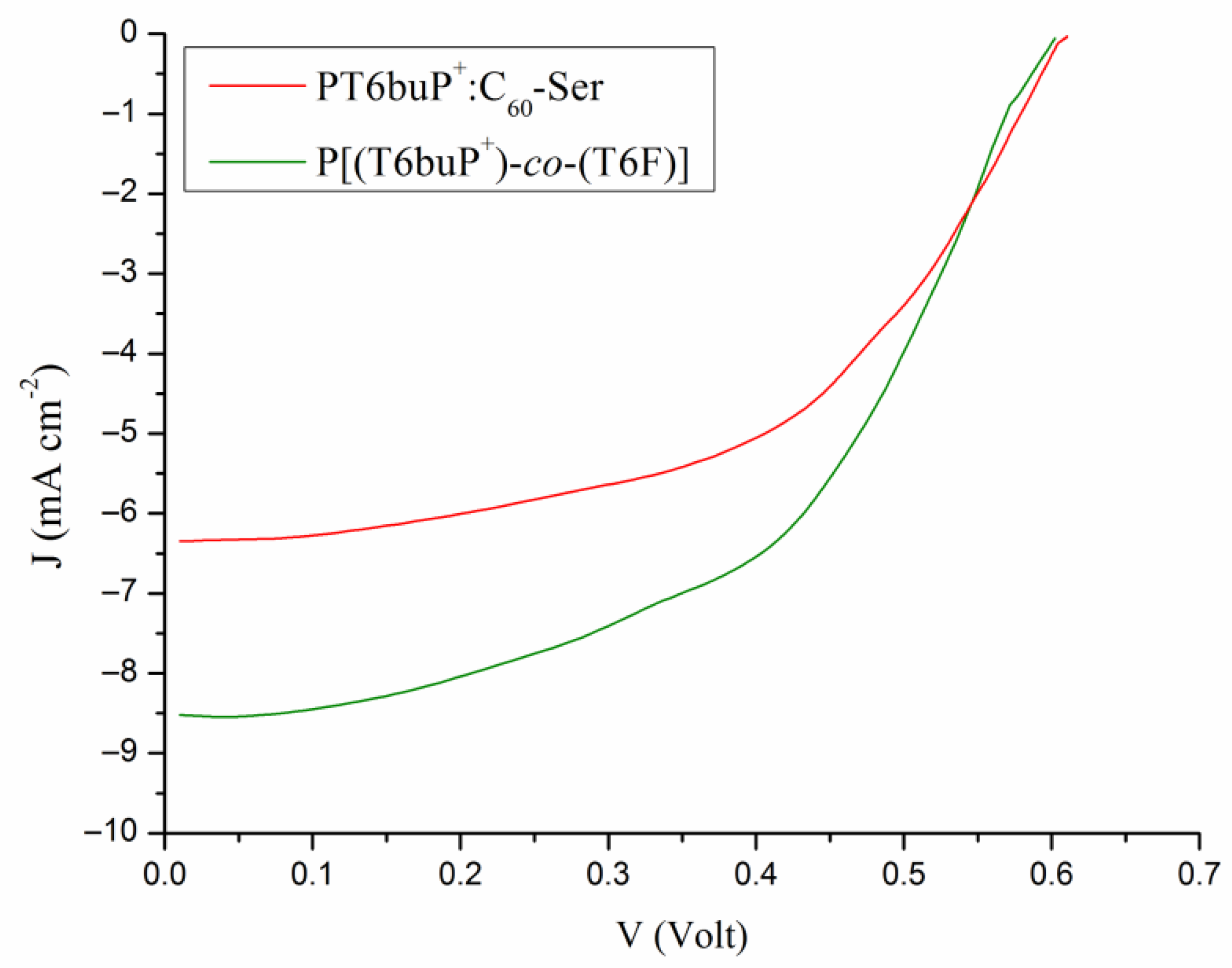
3.7. Photovoltaic Properties
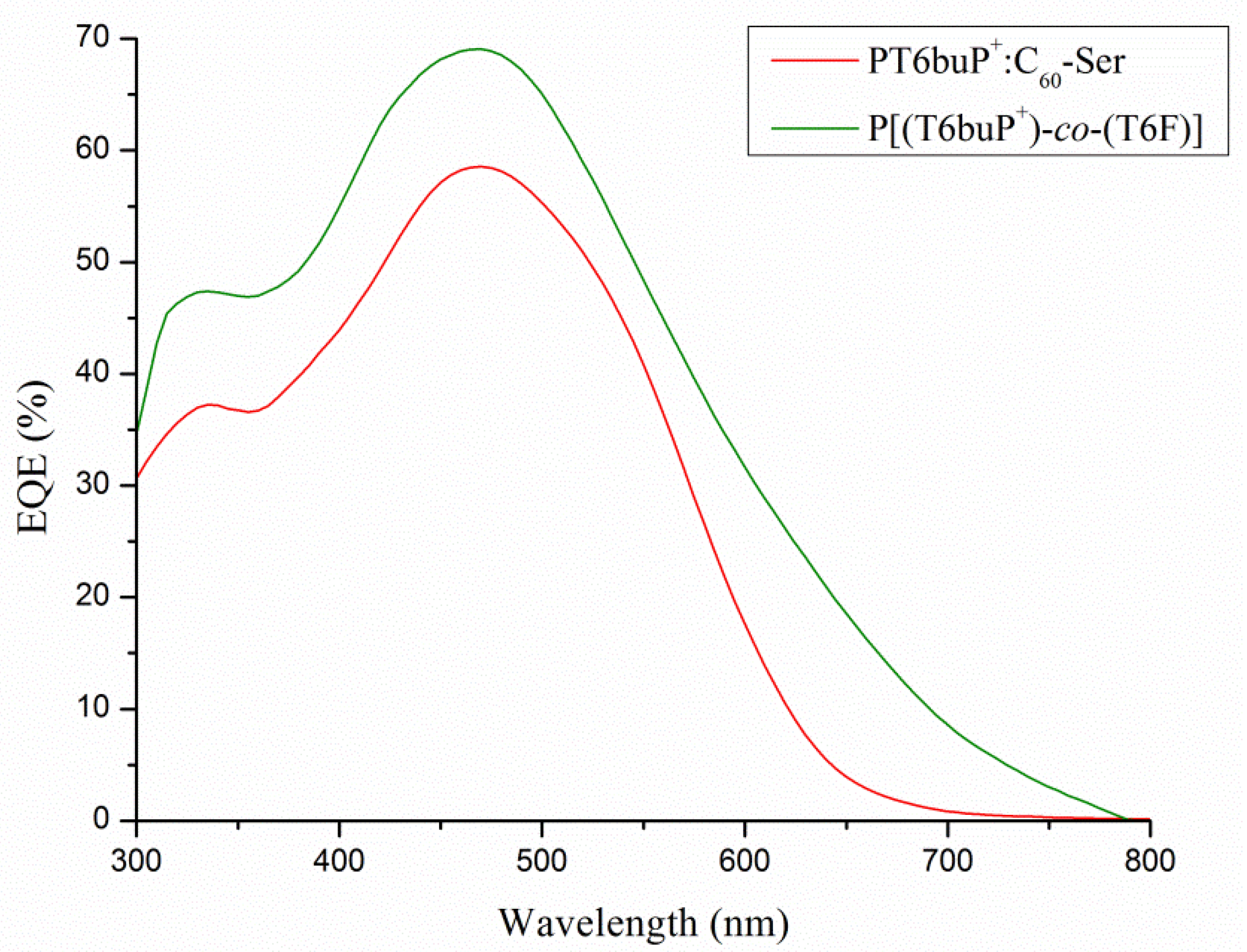
3.8. EQE Measurements
3.9. Morphological Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mazzio, K.A.; Luscombe, C.K. The future of organic photovoltaics. Chem. Soc. Rev. 2015, 44, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Yip, H.-L.; Jen, A.K.-Y. Recent advances in solution-processed interfacial materials for efficient and stable polymer solar cells. Energy Environ. Sci. 2012, 5, 5994–6011. [Google Scholar] [CrossRef]
- Lu, L.; Zheng, T.; Wu, Q.; Schneider, A.M.; Zhao, D.; Yu, L. Recent Advances in Bulk Heterojunction Polymer Solar Cells. Chem. Rev. 2015, 115, 12666–12731. [Google Scholar] [CrossRef] [PubMed]
- Lanzi, M.; Salatelli, E.; Benelli, T.; Caretti, D.; Giorgini, L.; Di-Nicola, F.P. A regioregular polythiophene-fullerene for polymeric solar cells. J. Appl. Polym. Sci. 2015, 132, 42121. [Google Scholar] [CrossRef]
- Lanzi, M.; Salatelli, E.; Marinelli, M.; Pierini, F. Effect of Photocrosslinking of D-A Thiophene Copolymers on the Performance of Single-Material Solar Cells. Macromol. Chem. Phys. 2019, 221, 1900433. [Google Scholar] [CrossRef]
- Das, S.; Chatterjee, D.P.; Ghosh, R.; Nandi, A.K. Water soluble polythiophenes: Preparation and applications. RSC Adv. 2015, 5, 20160–20177. [Google Scholar] [CrossRef]
- Duan, C.; Zhang, K.; Zhong, C.; Huang, F.; Cao, Y. Recent advances in water/alcohol-soluble π-conjugated materials: New materials and growing applications in solar cells. Chem. Soc. Rev. 2013, 42, 9071–9104. [Google Scholar] [CrossRef] [PubMed]
- Lanzi, M.; Salatelli, E.; Giorgini, L.; Mucci, A.; Pierini, F.; Di-Nicola, F.P. Water-soluble polythiophenes as efficient charge-transport layers for the improvement of photovoltaic performance in bulk heterojunction polymeric solar cells. Eur. Polym. J. 2017, 97, 378–388. [Google Scholar] [CrossRef]
- Huang, F.; Wu, H.; Cao, Y. Water/alcohol soluble conjugated polymers as highly efficient electron transporting/injection layer in optoelectronic devices. Chem. Soc. Rev. 2010, 39, 2500–2521. [Google Scholar] [CrossRef]
- Lanzi, M.; Salatelli, E.; Giorgini, L.; Marinelli, M.; Pierini, F. Effect of the incorporation of an Ag nanoparticle interlayer on the photovoltaic performance of green bulk heterojunction water-soluble polythiophene solar cells. Polymer 2018, 149, 273–285. [Google Scholar] [CrossRef]
- Ghoos, T.; Brassinne, J.; Fustin, C.-A.; Gohy, J.-F.; Defour, M.; Brande, N.V.D.; Van Mele, B.; Lutsen, L.; Vanderzande, D.J.; Maes, W. Imidazolium-substituted ionic (co)polythiophenes: Compositional influence on solution behavior and thermal properties. Polymer 2013, 54, 6293–6304. [Google Scholar] [CrossRef]
- Urbanek, P.; Di Martino, A.; Gladyš, S.; Kuritka, I.; Minařik, A.; Pavlova, E.; Bondarev, D.; Hladysh, S. Polythiophene-based conjugated polyelectrolyte: Optical properties and association behavior in solution. Synth. Met. 2015, 202, 16–24. [Google Scholar] [CrossRef]
- Hladysh, S.; Murmiliuk, A.; Vohlídal, J.; Havlíček, D.; Sedlarik, V.; Stepanek, M.; Zednik, J. Combination of phosphonium and ammonium pendant groups in cationic conjugated polyelectrolytes based on regioregular poly(3-hexylthiophene) polymer chains. Eur. Polym. J. 2018, 100, 200–208. [Google Scholar] [CrossRef]
- Hladysh, S.; Bondarev, D.; Svoboda, J.; Vohlídal, J.; Vrbata, D.; Zedník, J. Novel conjugated polyelectrolytes based on polythiophene bearing phosphonium side groups. Eur. Polym. J. 2016, 83, 367–376. [Google Scholar] [CrossRef]
- López, A.M.; Mateo-Alonso, A.; Prato, M. Materials chemistry of fullerene C60derivatives. J. Mater. Chem. 2010, 21, 1305–1318. [Google Scholar] [CrossRef]
- Markovic, Z.; Trajkovic, V. Biomedical potential of the reactive oxygen species generation and quenching by fullerenes (C60). Biomaterials 2008, 29, 3561–3573. [Google Scholar] [CrossRef]
- Kokubo, K.; Matsubayashi, K.; Tategaki, H.; Takada, H.; Oshima, T. Facile Synthesis of Highly Water-Soluble Fullerenes More Than Half-Covered by Hydroxyl Groups. ACS Nano 2008, 2, 327–333. [Google Scholar] [CrossRef]
- Li, H.; Haque, S.A.; Kitaygorodskiy, A.; Meziani, M.J.; Torres-Castillo, M.; Sun, Y.-P. Alternatively Modified Bingel Reaction for Efficient Syntheses of C60Hexakis- Adducts. Org. Lett. 2006, 8, 5641–5643. [Google Scholar] [CrossRef]
- Lanzi, M.; Costa-Bizzarri, P.; Paganin, L.; Cesari, G. Synthesis by post-polymerization functionalization of sensitive polythiophenes for selective chemo-recognition purposes. React. Funct. Polym. 2007, 67, 329–340. [Google Scholar] [CrossRef]
- Mitchell, R.H.; Lai, Y.-H.; Williams, R.V. N-Bromosuccinimide-dimethylformamide: A mild, selective nuclear monobromination reagent for reactive aromatic compounds. J. Org. Chem. 1979, 44, 4733–4735. [Google Scholar] [CrossRef]
- McCullough, R.D. The Chemistry of Conducting Polythiophenes. Adv. Mater. 1998, 10, 93–116. [Google Scholar] [CrossRef]
- Stefan, M.C.; Bhatt, M.P.; Sista, P.; Magurudeniya, H.D. Grignard metathesis (GRIM) polymerization for the synthesis of conjugated block copolymers containing regioregular poly(3-hexylthiophene). Polym. Chem. 2011, 3, 1693–1701. [Google Scholar] [CrossRef]
- Loewe, R.S.; Ewbank, P.C.; Liu, J.; Zhai, A.L.; McCullough, R.D. Regioregular, Head-to-Tail Coupled Poly(3-alkylthiophenes) Made Easy by the GRIM Method: Investigation of the Reaction and the Origin of Regioselectivity. Macromolecules 2001, 34, 4324–4333. [Google Scholar] [CrossRef]
- Hoppe, H.; Sariciftci, N.S. Morphology of polymer/fullerene bulk heterojunction solar cells. J. Mater. Chem. 2006, 16, 45–61. [Google Scholar] [CrossRef]
- Hirsch, A.; Vostrowsky, O. C60 Hexakisadducts with an Octahedral Addition Pattern—A New Structure Motif in Organic Chemistry. Eur. J. Org. Chem. 2001, 5, 829–848. [Google Scholar] [CrossRef]
- Wharton, T.; Kini, V.U.; Mortis, R.A.; Wilson, L.J. New non-ionic, highly water-soluble derivatives of C60 designed for biological compatibility. Tetrahedron Lett. 2001, 42, 5159–5162. [Google Scholar] [CrossRef]
- Bar-Shir, A.; Engel, Y.; Gozin, M. Synthesis and Water Solubility of Adamantyl-OEG-fullerene Hybrids. J. Org. Chem. 2005, 70, 2660–2666. [Google Scholar] [CrossRef] [PubMed]
- Shirosaki, T.; Harisaki, R.; Horikawa, M.; Sakurai, H.; Nagaoka, S.; Ihara, H. Synthesis of a Series of Malonic Diester–Introduced Fullerene Derivatives. Synth. Commun. 2013, 44, 275–279. [Google Scholar] [CrossRef]
- Wharton, T.; Wilson, L.J. Highly-Iodinated Fullerene as a Contrast Agent For X-ray Imaging. Bioorganic Med. Chem. 2002, 10, 3545–3554. [Google Scholar] [CrossRef]
- Sheina, E.E.; Liu, J.; Iovu, M.C.; Laird, D.W.; McCullough, R.D. Chain Growth Mechanism for Regioregular Nickel-Initiated Cross-Coupling Polymerizations. Macromolecules 2004, 37, 3526–3528. [Google Scholar] [CrossRef]
- Lohwasser, R.H.; Thelakkat, M. Toward Perfect Control of End Groups and Polydispersity in Poly(3-hexylthiophene) via Catalyst Transfer Polymerization. Macromolecules 2011, 44, 3388–3397. [Google Scholar] [CrossRef]
- Chen, T.-A.; Wu, X.; Rieke, R.D. Regiocontrolled Synthesis of Poly(3-alkylthiophenes) Mediated by Rieke Zinc: Their Characterization and Solid-State Properties. J. Am. Chem. Soc. 1995, 117, 233–244. [Google Scholar] [CrossRef]
- Koch, F.P.V.; Smith, P.; Heeney, M. “Fibonacci’s Route” to Regioregular Oligo(3-hexylthiophene)s. J. Am. Chem. Soc. 2013, 135, 13695–13698. [Google Scholar] [CrossRef] [PubMed]
- Iovu, M.C.; Sheina, E.E.; Gil, R.R.; McCullough, R.D. Experimental Evidence for the Quasi-“Living” Nature of the Grignard Metathesis Method for the Synthesis of Regioregular Poly(3-alkylthiophenes). Macromolecules 2005, 38, 8649–8656. [Google Scholar] [CrossRef]
- Zagórska, M.; Krische, B. Chemical synthesis and characterization of soluble poly(4,4′-dialkyl-2,2′-bithiophenes). Polymer 1990, 31, 1379–1383. [Google Scholar] [CrossRef]
- Jeffries-El, M.; Sauvé, G.; McCullough, R.D. Facile Synthesis of End-Functionalized Regioregular Poly(3-alkylthiophene)s via Modified Grignard Metathesis Reaction. Macromolecules 2005, 38, 10346–10352. [Google Scholar] [CrossRef]
- Mayer, A.C.; Toney, M.F.; Scully, S.R.; Rivnay, J.; Brabec, C.J.; Scharber, M.; Koppe, M.; Heeney, M.; McCulloch, I.; McGehee, M.D. Bimolecular Crystals of Fullerenes in Conjugated Polymers and the Implications of Molecular Mixing for Solar Cells. Adv. Funct. Mater. 2009, 19, 1173–1179. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Yield (%) | Ionic Group Content (%mol) a | C60Content (%mol) a | Mn (kDa) | PDI | |
|---|---|---|---|---|---|
| PT6Br | 21 | - | - | 14.9 b | 1.15 b |
| PT6buP+ | 98 | 100 | - | 27.2 c | 1.15 c |
| P[(T6Br)-co-(T6F)] | 85 | - | 7 | 17.6 c | 1.15 c |
| P[(T6buP+)-co-(T6F)] | 78 | 93 | 7 | 29.1 c | 1.15 c |
| Solvents | λmax Film (nm) | |
|---|---|---|
| PT6Br | Chloroform | 538 |
| PT6buP+ | Water | 467 |
| Methanol | 467 | |
| P[(T6Br)-co-(T6F)] | Chlorobenzene | 340, 496 |
| P[(T6buP+)-co-(T6F)] | Water | 343, 471 |
| Methanol | 334, 465 |
| Sample | Tg (°C) | Tm (°C) | Tc (°C) | Td (°C) |
|---|---|---|---|---|
| PT6Br | 55 | 164 | 95 | 282 |
| PT6buP+ | 43 | - | 42 | 355 |
| P[(T6Br)-co-(T6F)] | 77 | 148 | 86 | 260 |
| P[(T6buP+)-co-(T6F)] | 27 | - | 22 | 296 |
| Sample | Low-Angle Diffractions | High-Angle Diffractions | On Plane Th Chains Distances | Planes Stacking Distances | Crystallites Mean Size (L) |
|---|---|---|---|---|---|
| (Degrees) | (Degrees) | (Å) | (Å) | (nm) | |
| PT6Br | 5.34; 10.82; 16.19 | 22.68 | 16.5 | 3.92 | 15.6 |
| PT6buP+ | 5.24; 10.61 | 22.34 | 16.8 | 3.98 | 6.61 |
| P[(T6Br)-co-(T6F)] | 4.91; 9.89; 14.84 | 22.51 | 17.9 | 3.95 | 6.74 |
| P[(T6buP+)-co-(T6F)] | 4.34; 8.71 | 21.61 | 20.3 | 4.09 | 5.33 |
| Sample | JSC (mA cm−2) (a) | VOC (V) | FF | PCE (%) | JSC (mA cm−2) (b) |
|---|---|---|---|---|---|
| PT6buP+:C60-Ser | 6.36 ± 0.3 | 0.61 ± 0.01 | 0.59 ± 0.04 | 2.29 ± 0.13 | 6.22 |
| P[(T6buP+)-co-(T6F)] | 8.51 ± 0.4 | 0.61 ± 0.01 | 0.60 ± 0.05 | 3.11 ± 0.15 | 8.78 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanzi, M.; Quadretti, D.; Marinelli, M.; Ziai, Y.; Salatelli, E.; Pierini, F. Influence of the Active Layer Structure on the Photovoltaic Performance of Water-Soluble Polythiophene-Based Solar Cells. Polymers 2021, 13, 1640. https://doi.org/10.3390/polym13101640
Lanzi M, Quadretti D, Marinelli M, Ziai Y, Salatelli E, Pierini F. Influence of the Active Layer Structure on the Photovoltaic Performance of Water-Soluble Polythiophene-Based Solar Cells. Polymers. 2021; 13(10):1640. https://doi.org/10.3390/polym13101640
Chicago/Turabian StyleLanzi, Massimiliano, Debora Quadretti, Martina Marinelli, Yasamin Ziai, Elisabetta Salatelli, and Filippo Pierini. 2021. "Influence of the Active Layer Structure on the Photovoltaic Performance of Water-Soluble Polythiophene-Based Solar Cells" Polymers 13, no. 10: 1640. https://doi.org/10.3390/polym13101640
APA StyleLanzi, M., Quadretti, D., Marinelli, M., Ziai, Y., Salatelli, E., & Pierini, F. (2021). Influence of the Active Layer Structure on the Photovoltaic Performance of Water-Soluble Polythiophene-Based Solar Cells. Polymers, 13(10), 1640. https://doi.org/10.3390/polym13101640