iRGD Peptide as a Tumor-Penetrating Enhancer for Tumor-Targeted Drug Delivery

Abstract
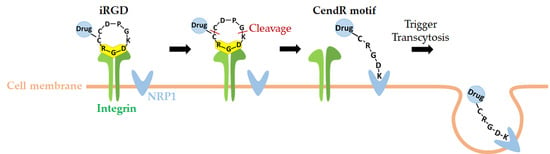
1. Introduction
2. iRGD Peptide
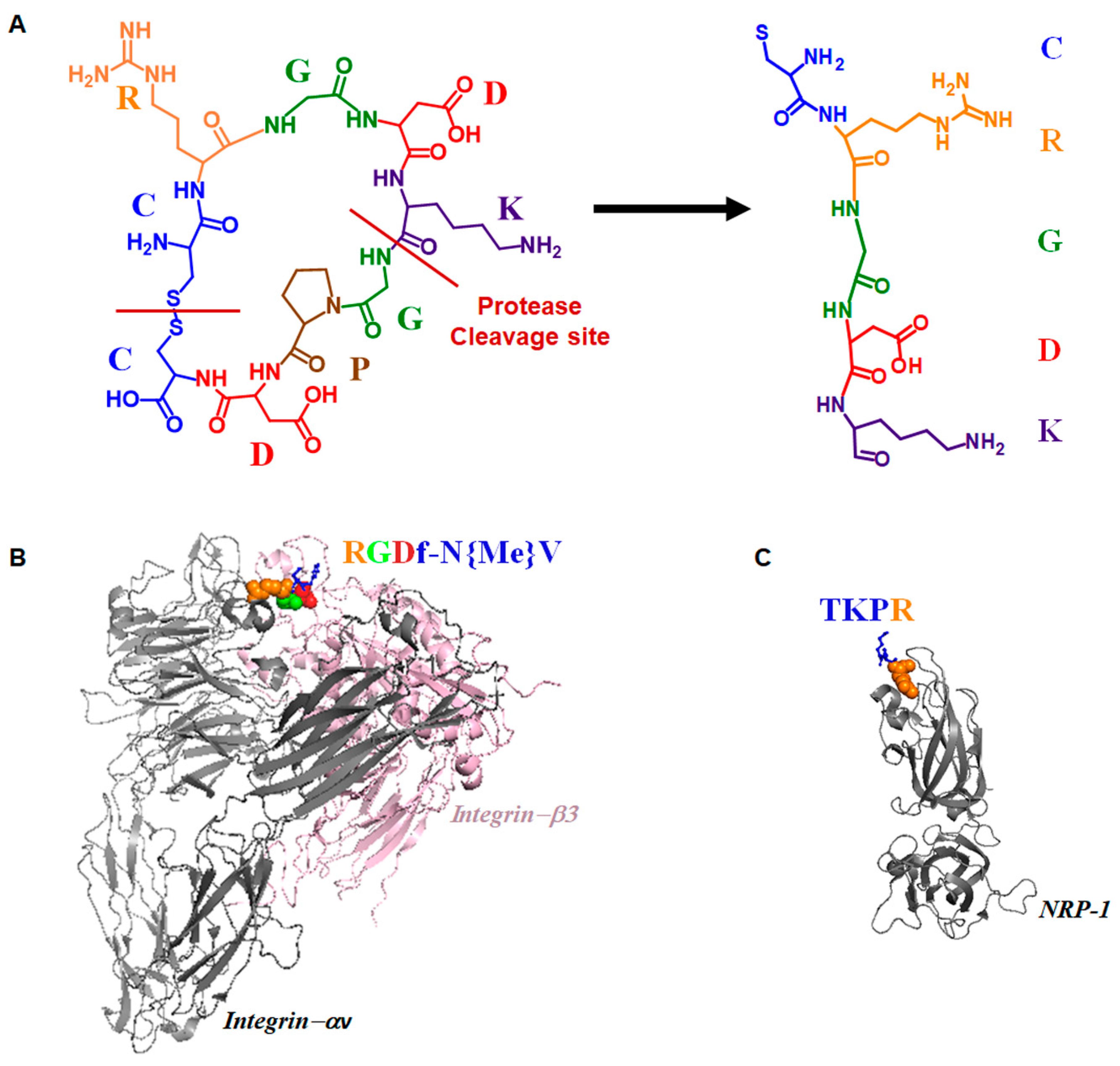
2.1. Discovery of iRGD Using Phage Display Screening
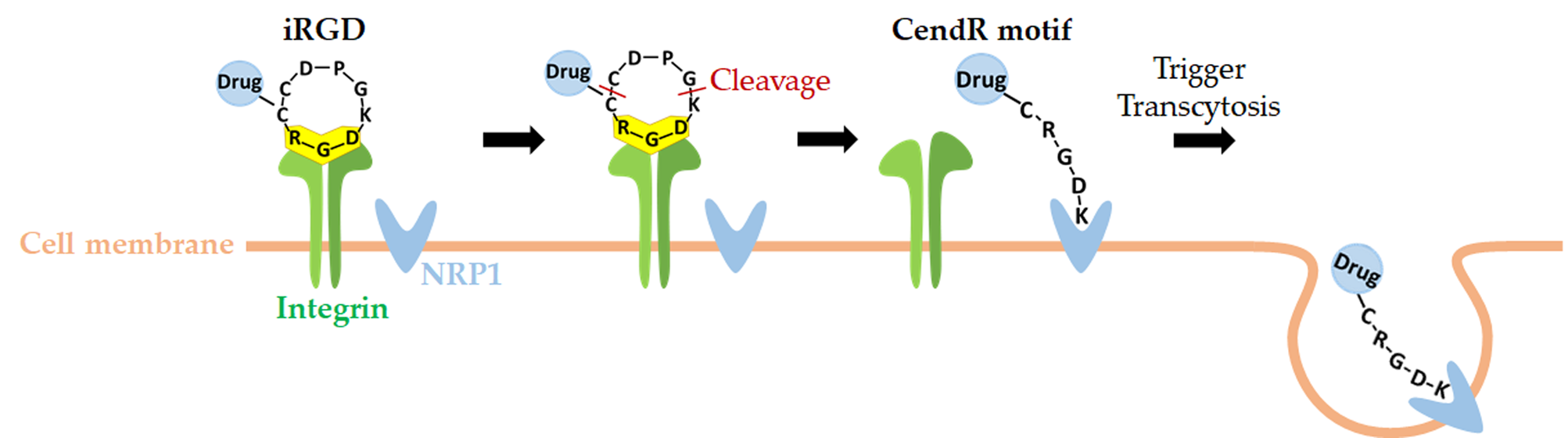
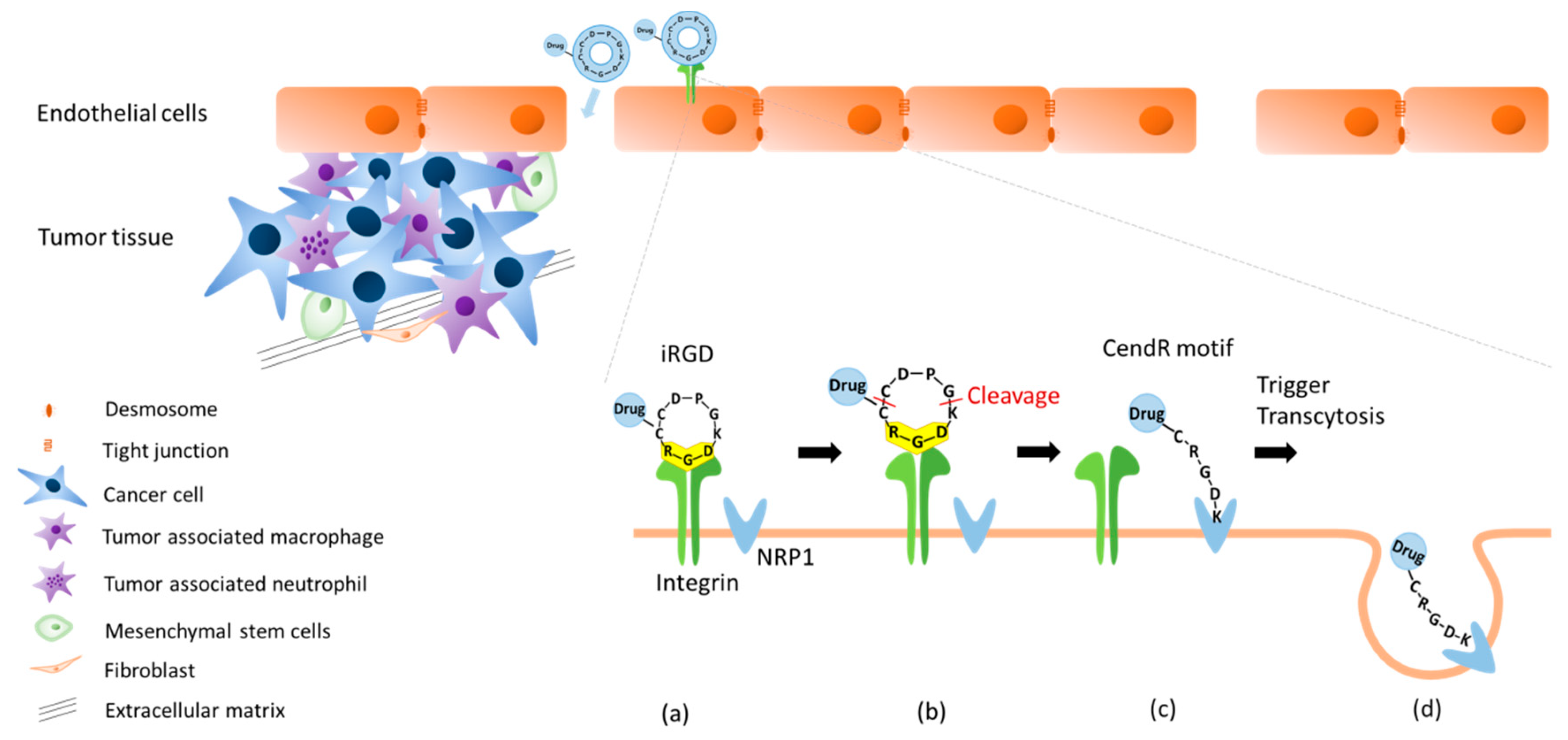
2.2. Dual Targeting Mechanism of iRGD in Tumor Treatment
2.3. Active Targeting of iRGD Peptide to Exploit the Tumor Microenvironment
3. Applications of iRGD in Cancer Therapy
3.1. Implementation Strategies in iRGD Technology
3.2. Recent Clinical Trials with Co-Administration of iRGD in Pancreatic Cancer
3.3. iRGD Application with Immunotherapy
3.4. iRGD Application in Brain Pathology
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APCs | Antigen-presenting cells |
| API | Active pharmaceutical ingredient |
| BBB | Blood–brain barrier |
| CendR | C-end Rule |
| CTLA-4 | Cytotoxic T lymphocyte-associated antigen-4 |
| DSG2 | Desmoglein 2 |
| ECM | Extracellular matrix |
| EPR | Enhanced permeability and retention |
| FAM | Fluorescein |
| GF | Gland formation |
| nGF | No gland formation |
| HA-PLA | Self-assembled amphiphilic block copolymer NPs |
| HPRP-A1 | Anti-microbial peptide derived from the N-terminus of ribosomal protein L1 of H. pylori |
| IFP | Interstitial fluid pressure |
| iRGD-ICG-LPs | iRGD-modified indocyanine green (ICG) liposomes |
| ISL-iRGD NPs | iRGD-modified lipid–polymer hybrid NPs loaded with isoliquiritigenin |
| JO-1 | Junction opener-1 |
| LGR | Leucine-rich repeat-containing G protein receptors |
| NCs | Nanocapsules |
| NIR | Near infrared |
| NPs | Nanoparticles |
| NRP-1 | Neuropilin-1 |
| PCL-PVP | Poly (ε-caprolactone)-poly (N-vinylpyrrolidone) |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed death ligand 1 |
| PDAC | Pancreatic ductal adenocarcinoma |
| PDT | Photodynamic therapy |
| PEDF | Pigment epithelium-derived factor |
| PLGA | Paclitaxel-loaded poly (lactic-co-glycolic acid) |
| PTT | Photothermal therapy |
| R-LP | iRGD-modified liposomes |
| R-LP/PEDF | iRGD-conjugated pigment epithelium-derived factor (PEDF)-DNA-loaded liposomes |
| R-LP/shelF3i | iRGD-modified liposomes encapsulating elF3i shRNA |
| SEC2 | Staphylococcus endotoxin C2 |
| SEER | Surveillance, Epidemiology, and End Results |
| SLN | Solid–lipid nanoparticles |
| TAMs | Tumor-associated macrophages |
| TRAIL | TNF-α-related apoptosis inducing ligand |
| VEGF | Vascular endothelial growth factor |
References
- Jung, K.W.; Won, Y.J.; Kong, H.J.; Lee, E.S. Cancer Statistics in Korea: Incidence, Mortality, Survival, and Prevalence in 2016. Cancer Res. Treat. 2019, 51, 417–430. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Holman, D.M.; Boehm, J.E.; Peipins, L.A.; Grossman, M.; Henley, S.J. Age and cancer risk: A potentially modifiable relationship. Am. J. Prev. Med. 2014, 46, S7–S15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-T.; Li, H.-C.; Li, Z.-W.; Guo, C.-H. A tumor-penetrating peptide modification enhances the antitumor activity of endostatin in vivo. Anticancer Drugs 2011, 22, 409–415. [Google Scholar] [CrossRef]
- Rabbani-Chadegani, A.; Paydar, P.; Amirshenava, M.; Aramvash, A. An in vitro study on the effect of vinca alkaloid, vinorelbine, on chromatin histone, HMGB proteins and induction of apoptosis in mice non-adherent bone marrow cells. Drug Chem. Toxicol. 2015, 38, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Nyongesa, C.O.; Park, S. Chemotherapeutic resistance: A nano-mechanical point of view. Biol. Chem. 2018, 399, 1433–1446. [Google Scholar] [CrossRef] [PubMed]
- Hambley, T.W.; Hait, W.N. Is anticancer drug development heading in the right direction? Cancer Res. 2009, 69, 1259–1262. [Google Scholar] [CrossRef]
- Bissell, M.J.; Radisky, D. Putting tumours in context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef]
- Mueller, M.M.; Fusenig, N.E. Friends or foes—Bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef]
- Madara, J.L. Regulation of the movement of solutes across tight junctions. Annu. Rev. Physiol. 1998, 60, 143–159. [Google Scholar] [CrossRef]
- Harada, H.; Iwatsuki, K.; Ohtsuka, M.; Han, G.W.; Kaneko, F. Abnormal desmoglein expression by squamous cell carcinoma cells. Acta Derm. Venereol. 1996, 76, 417–420. [Google Scholar] [CrossRef]
- Biedermann, K.; Vogelsang, H.; Becker, I.; Plaschke, S.; Siewert, J.R.; Hofler, H.; Keller, G. Desmoglein 2 is expressed abnormally rather than mutated in familial and sporadic gastric cancer. J. Pathol. 2005, 207, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Pluen, A.; Boucher, Y.; Ramanujan, S.; McKee, T.D.; Gohongi, T.; di Tomaso, E.; Brown, E.B.; Izumi, Y.; Campbell, R.B.; Berk, D.A.; et al. Role of tumor-host interactions in interstitial diffusion of macromolecules: Cranial vs. subcutaneous tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 4628–4633. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Vascular and interstitial barriers to delivery of therapeutic agents in tumors. Cancer Metastasis Rev. 1990, 9, 253–266. [Google Scholar] [CrossRef]
- Wenning, L.A.; Murphy, R.M. Coupled cellular trafficking and diffusional limitations in delivery of immunotoxins to multicell tumor spheroids. Biotechnol. Bioeng. 1999, 62, 562–575. [Google Scholar] [CrossRef]
- Kuh, H.J.; Jang, S.H.; Wientjes, M.G.; Weaver, J.R.; Au, J.L. Determinants of paclitaxel penetration and accumulation in human solid tumor. J. Pharmacol. Exp. Ther. 1999, 290, 871–880. [Google Scholar]
- Huang, S.; Ingber, D.E. Cell tension, matrix mechanics, and cancer development. Cancer Cell 2005, 8, 175–176. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Lopez, J.I.; Kang, I.; You, W.K.; McDonald, D.M.; Weaver, V.M. In situ force mapping of mammary gland transformation. Integr. Biol. 2011, 3, 910–921. [Google Scholar] [CrossRef]
- Netti, P.A.; Berk, D.A.; Swartz, M.A.; Grodzinsky, A.J.; Jain, R.K. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000, 60, 2497–2503. [Google Scholar]
- Brown, E.B.; Boucher, Y.; Nasser, S.; Jain, R.K. Measurement of macromolecular diffusion coefficients in human tumors. Microvasc. Res. 2004, 67, 231–236. [Google Scholar] [CrossRef]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Maeda, H.; Sawa, T.; Konno, T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J. Control. Release 2001, 74, 47–61. [Google Scholar] [CrossRef]
- Maeda, H.; Bharate, G.Y.; Daruwalla, J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur. J. Pharm. Biopharm. 2009, 71, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. Drug targeting. Eur. J. Pharm. Sci. 2000, 11 (Suppl. 2), S81–S91. [Google Scholar] [CrossRef]
- Bae, Y.H. Drug targeting and tumor heterogeneity. J. Control. Release 2009, 133, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.; Catane, R.; Uziely, B.; Kaufman, B.; Safra, T.; Cohen, R.; Martin, F.; Huang, A.; Barenholz, Y. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994, 54, 987–992. [Google Scholar] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Li, C.P.; Bodoky, G.; Dean, A.; Shan, Y.S.; Jameson, G.; Macarulla, T.; Lee, K.H.; Cunningham, D.; Blanc, J.F.; et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet 2016, 387, 545–557. [Google Scholar] [CrossRef]
- Adams, G.P.; Schier, R.; McCall, A.M.; Simmons, H.H.; Horak, E.M.; Alpaugh, R.K.; Marks, J.D.; Weiner, L.M. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001, 61, 4750–4755. [Google Scholar]
- Gosk, S.; Moos, T.; Gottstein, C.; Bendas, G. VCAM-1 directed immunoliposomes selectively target tumor vasculature in vivo. Biochim. Biophys. Acta 2008, 1778, 854–863. [Google Scholar] [CrossRef]
- Lammers, T.; Hennink, W.E.; Storm, G. Tumour-targeted nanomedicines: Principles and practice. Br. J. Cancer 2008, 99, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Feron, O.; Preat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Park, K. Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release 2011, 153, 198–205. [Google Scholar] [CrossRef]
- Choi, I.K.; Strauss, R.; Richter, M.; Yun, C.O.; Lieber, A. Strategies to increase drug penetration in solid tumors. Front. Oncol. 2013, 3, 193. [Google Scholar] [CrossRef] [PubMed]
- Van Der Westhuizen, E.T.; Summers, R.J.; Halls, M.L.; Bathgate, R.A.; Sexton, P.M. Relaxin receptors—New drug targets for multiple disease states. Curr. Drug Targets 2007, 8, 91–104. [Google Scholar] [CrossRef]
- Amento, E.P.; Bhan, A.K.; McCullagh, K.G.; Krane, S.M. Influences of gamma interferon on synovial fibroblast-like cells. Ia induction and inhibition of collagen synthesis. J. Clin. Investig. 1985, 76, 837–848. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, Y.S.; Kim, H.; Huang, J.H.; Yoon, A.R.; Yun, C.O. Relaxin expression from tumor-targeting adenoviruses and its intratumoral spread, apoptosis induction, and efficacy. J. Natl. Cancer Inst. 2006, 98, 1482–1493. [Google Scholar] [CrossRef]
- Silvertown, J.D.; Ng, J.; Sato, T.; Summerlee, A.J.; Medin, J.A. H2 relaxin overexpression increases in vivo prostate xenograft tumor growth and angiogenesis. Int. J. Cancer 2006, 118, 62–73. [Google Scholar] [CrossRef]
- Binder, C.; Hagemann, T.; Husen, B.; Schulz, M.; Einspanier, A. Relaxin enhances in-vitro invasiveness of breast cancer cell lines by up-regulation of matrix metalloproteases. Mol. Hum. Reprod 2002, 8, 789–796. [Google Scholar] [CrossRef]
- Wang, H.; Li, Z.; Yumul, R.; Lara, S.; Hemminki, A.; Fender, P.; Lieber, A. Multimerization of adenovirus serotype 3 fiber knob domains is required for efficient binding of virus to desmoglein 2 and subsequent opening of epithelial junctions. J. Virol. 2011, 85, 6390–6402. [Google Scholar] [CrossRef] [PubMed]
- Beyer, I.; van Rensburg, R.; Strauss, R.; Li, Z.; Wang, H.; Persson, J.; Yumul, R.; Feng, Q.; Song, H.; Bartek, J.; et al. Epithelial junction opener JO-1 improves monoclonal antibody therapy of cancer. Cancer Res. 2011, 71, 7080–7090. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Z.Y.; Liu, Y.; Persson, J.; Beyer, I.; Moller, T.; Koyuncu, D.; Drescher, M.R.; Strauss, R.; Zhang, X.B.; et al. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat. Med. 2011, 17, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Beyer, I.; Cao, H.; Persson, J.; Song, H.; Richter, M.; Feng, Q.; Yumul, R.; van Rensburg, R.; Li, Z.; Berenson, R.; et al. Coadministration of epithelial junction opener JO-1 improves the efficacy and safety of chemotherapeutic drugs. Clin. Cancer Res. 2012, 18, 3340–3351. [Google Scholar] [CrossRef]
- Li, Z.; Di, C.; Li, S.; Yang, X.; Nie, G. Smart Nanotherapeutic Targeting of Tumor Vasculature. Acc. Chem. Res. 2019, 52, 2703–2712. [Google Scholar] [CrossRef]
- Zuo, H.D.; Yao, W.W.; Chen, T.W.; Zhu, J.; Zhang, J.J.; Pu, Y.; Liu, G.; Zhang, X.M. The effect of superparamagnetic iron oxide with iRGD peptide on the labeling of pancreatic cancer cells in vitro: A preliminary study. BioMed Res. Int. 2014, 2014, 852352. [Google Scholar] [CrossRef]
- Yin, H.; Yang, J.; Zhang, Q.; Yang, J.; Wang, H.; Xu, J.; Zheng, J. iRGD as a tumorpenetrating peptide for cancer therapy (Review). Mol. Med. Rep. 2017, 15, 2925–2930. [Google Scholar] [CrossRef]
- Hu, C.; Chen, X.; Huang, Y.; Chen, Y. Co-administration of iRGD with peptide HPRP-A1 to improve anticancer activity and membrane penetrability. Sci. Rep. 2018, 8, 2274. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Scodeller, P.; Braun, G.B.; De Mendoza, T.H.; Yamazaki, C.M.; Kluger, M.D.; Kitayama, J.; Alvarez, E.; Howell, S.B.; Teesalu, T. A tumor-penetrating peptide enhances circulation-independent targeting of peritoneal carcinomatosis. J. Control. Release 2015, 212, 59–69. [Google Scholar] [CrossRef]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 1998, 279, 377–380. [Google Scholar] [CrossRef]
- Sipkins, D.A.; Cheresh, D.A.; Kazemi, M.R.; Nevin, L.M.; Bednarski, M.D.; Li, K.C. Detection of tumor angiogenesis in vivo by alphaVbeta3-targeted magnetic resonance imaging. Nat. Med. 1998, 4, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.A.; Giraudo, E.; Singh, M.; Zhang, L.; Inoue, M.; Porkka, K.; Hanahan, D.; Ruoslahti, E. Progressive vascular changes in a transgenic mouse model of squamous cell carcinoma. Cancer Cell 2003, 4, 383–391. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Greenwald, D.R.; Ruoslahti, E. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 2010, 328, 1031–1035. [Google Scholar] [CrossRef]
- Teesalu, T.; Sugahara, K.N.; Kotamraju, V.R.; Ruoslahti, E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. USA 2009, 106, 16157–16162. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, D.A. Integrin activation. J. Cell Sci. 2004, 117, 657–666. [Google Scholar] [CrossRef]
- Nieberler, M.; Reuning, U.; Reichart, F.; Notni, J.; Wester, H.J.; Schwaiger, M.; Weinmuller, M.; Rader, A.; Steiger, K.; Kessler, H. Exploring the Role of RGD-Recognizing Integrins in Cancer. Cancers 2017, 9, 116. [Google Scholar] [CrossRef]
- Huveneers, S.; Danen, E.H. Adhesion signaling—Crosstalk between integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef]
- Geiger, B.; Spatz, J.P.; Bershadsky, A.D. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 2009, 10, 21–33. [Google Scholar] [CrossRef]
- Schittenhelm, J.; Klein, A.; Tatagiba, M.S.; Meyermann, R.; Fend, F.; Goodman, S.L.; Sipos, B. Comparing the expression of integrins alphavbeta3, alphavbeta5, alphavbeta6, alphavbeta8, fibronectin and fibrinogen in human brain metastases and their corresponding primary tumors. Int. J. Clin. Exp. Pathol. 2013, 6, 2719–2732. [Google Scholar]
- Pytela, R.; Pierschbacher, M.D.; Ruoslahti, E. Identification and isolation of a 140 kd cell surface glycoprotein with properties expected of a fibronectin receptor. Cell 1985, 40, 191–198. [Google Scholar] [CrossRef]
- Boger, C.; Warneke, V.S.; Behrens, H.M.; Kalthoff, H.; Goodman, S.L.; Becker, T.; Rocken, C. Integrins alphavbeta3 and alphavbeta5 as prognostic, diagnostic, and therapeutic targets in gastric cancer. Gastric Cancer 2015, 18, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Schnell, O.; Krebs, B.; Wagner, E.; Romagna, A.; Beer, A.J.; Grau, S.J.; Thon, N.; Goetz, C.; Kretzschmar, H.A.; Tonn, J.C.; et al. Expression of integrin alphavbeta3 in gliomas correlates with tumor grade and is not restricted to tumor vasculature. Brain Pathol. 2008, 18, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Boger, C.; Kalthoff, H.; Goodman, S.L.; Behrens, H.M.; Rocken, C. Integrins and their ligands are expressed in non-small cell lung cancer but not correlated with parameters of disease progression. Virchows Arch. 2014, 464, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Hosotani, R.; Kawaguchi, M.; Masui, T.; Koshiba, T.; Ida, J.; Fujimoto, K.; Wada, M.; Doi, R.; Imamura, M. Expression of integrin alphaVbeta3 in pancreatic carcinoma: Relation to MMP-2 activation and lymph node metastasis. Pancreas 2002, 25, e30–e35. [Google Scholar] [CrossRef] [PubMed]
- Hess, K.; Boger, C.; Behrens, H.M.; Rocken, C. Correlation between the expression of integrins in prostate cancer and clinical outcome in 1284 patients. Ann. Diagn Pathol. 2014, 18, 343–350. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Kovanda, A.K.; Melchardt, T.; Bartsch, R.; Hainfellner, J.A.; Sipos, B.; Schittenhelm, J.; Zielinski, C.C.; Widhalm, G.; Dieckmann, K.; et al. Alphavbeta3, alphavbeta5 and alphavbeta6 integrins in brain metastases of lung cancer. Clin. Exp. Metastasis 2014, 31, 841–851. [Google Scholar] [CrossRef]
- Xiong, J.P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellular segment of integrin alpha Vbeta3 in complex with an Arg-Gly-Asp ligand. Science 2002, 296, 151–155. [Google Scholar] [CrossRef]
- Vander Kooi, C.W.; Jusino, M.A.; Perman, B.; Neau, D.B.; Bellamy, H.D.; Leahy, D.J. Structural basis for ligand and heparin binding to neuropilin B domains. Proc. Natl. Acad. Sci. USA 2007, 104, 6152–6157. [Google Scholar] [CrossRef]
- Haspel, N.; Zanuy, D.; Nussinov, R.; Teesalu, T.; Ruoslahti, E.; Aleman, C. Binding of a C-end rule peptide to the neuropilin-1 receptor: A molecular modeling approach. Biochemistry 2011, 50, 1755–1762. [Google Scholar] [CrossRef]
- Koivunen, E.; Gay, D.A.; Ruoslahti, E. Selection of peptides binding to the alpha 5 beta 1 integrin from phage display library. J. Biol. Chem. 1993, 268, 20205–20210. [Google Scholar] [PubMed]
- Koivunen, E.; Wang, B.; Ruoslahti, E. Phage libraries displaying cyclic peptides with different ring sizes: Ligand specificities of the RGD-directed integrins. Biotechnology 1995, 13, 265–270. [Google Scholar] [CrossRef]
- Salikhova, A.; Wang, L.; Lanahan, A.A.; Liu, M.; Simons, M.; Leenders, W.P.; Mukhopadhyay, D.; Horowitz, A. Vascular endothelial growth factor and semaphorin induce neuropilin-1 endocytosis via separate pathways. Circ. Res. 2008, 103, e71–e79. [Google Scholar] [CrossRef] [PubMed]
- Feron, O. Tumor-penetrating peptides: A shift from magic bullets to magic guns. Sci. Transl. Med. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Hirata, T.; Agata, K.; Mochii, M.; Eguchi, G.; Fujisawa, H. The A5 antigen, a candidate for the neuronal recognition molecule, has homologies to complement components and coagulation factors. Neuron 1991, 7, 295–307. [Google Scholar] [CrossRef]
- Neufeld, G.; Kessler, O. The semaphorins: Versatile regulators of tumour progression and tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 632–645. [Google Scholar] [CrossRef]
- Gaur, P.; Bielenberg, D.R.; Samuel, S.; Bose, D.; Zhou, Y.; Gray, M.J.; Dallas, N.A.; Fan, F.; Xia, L.; Lu, J.; et al. Role of class 3 semaphorins and their receptors in tumor growth and angiogenesis. Clin. Cancer Res. 2009, 15, 6763–6770. [Google Scholar] [CrossRef]
- Kitsukawa, T.; Shimono, A.; Kawakami, A.; Kondoh, H.; Fujisawa, H. Overexpression of a membrane protein, neuropilin, in chimeric mice causes anomalies in the cardiovascular system, nervous system and limbs. Development 1995, 121, 4309–4318. [Google Scholar]
- Kawasaki, T.; Kitsukawa, T.; Bekku, Y.; Matsuda, Y.; Sanbo, M.; Yagi, T.; Fujisawa, H. A requirement for neuropilin-1 in embryonic vessel formation. Development 1999, 126, 4895–4902. [Google Scholar]
- Soker, S.; Takashima, S.; Miao, H.Q.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef]
- Poon, R.T.; Fan, S.T.; Wong, J. Clinical implications of circulating angiogenic factors in cancer patients. J. Clin. Oncol. 2001, 19, 1207–1225. [Google Scholar] [CrossRef] [PubMed]
- Bachelder, R.E.; Crago, A.; Chung, J.; Wendt, M.A.; Shaw, L.M.; Robinson, G.; Mercurio, A.M. Vascular endothelial growth factor is an autocrine survival factor for neuropilin-expressing breast carcinoma cells. Cancer Res. 2001, 61, 5736–5740. [Google Scholar] [PubMed]
- Zhang, H.; He, C.; Han, S.; Zhu, M.; Ke, W. The prognostic value of neuropilin-1 in various cancers: A meta-analysis. Int. J. Clin. Exp. Med. 2016, 9, 13942–13949. [Google Scholar]
- Akashi, Y.; Oda, T.; Ohara, Y.; Miyamoto, R.; Kurokawa, T.; Hashimoto, S.; Enomoto, T.; Yamada, K.; Satake, M.; Ohkohchi, N. Anticancer effects of gemcitabine are enhanced by co-administered iRGD peptide in murine pancreatic cancer models that overexpressed neuropilin-1. Br. J. Cancer 2014, 110, 1481–1487. [Google Scholar] [CrossRef]
- Seo, H.S.; Hyeon, J.; Song, I.H.; Lee, H.H. Relationship between neuropilin-1 expression and prognosis, according to gastric cancer histology. J. Mol. Histol. 2020, 51, 199–208. [Google Scholar] [CrossRef]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Raghunand, N.; Gatenby, R.A.; Gillies, R.J. Microenvironmental and cellular consequences of altered blood flow in tumours. Br. J. Radiol. 2003, 76, S11–S22. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef]
- Avraamides, C.J.; Garmy-Susini, B.; Varner, J.A. Integrins in angiogenesis and lymphangiogenesis. Nat. Rev. Cancer 2008, 8, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Stollman, T.H.; Ruers, T.J.; Oyen, W.J.; Boerman, O.C. New targeted probes for radioimaging of angiogenesis. Methods 2009, 48, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, X.; Liu, Y.; Liu, C.; Jiang, B.; Jiang, Y. Tumor penetrability and anti-angiogenesis using iRGD-mediated delivery of doxorubicin-polymer conjugates. Biomaterials 2014, 35, 8735–8747. [Google Scholar] [CrossRef] [PubMed]
- Laakkonen, P.; Porkka, K.; Hoffman, J.A.; Ruoslahti, E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat. Med. 2002, 8, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Alberici, L.; Roth, L.; Sugahara, K.N.; Agemy, L.; Kotamraju, V.R.; Teesalu, T.; Bordignon, C.; Traversari, C.; Rizzardi, G.P.; Ruoslahti, E. De novo design of a tumor-penetrating peptide. Cancer Res. 2013, 73, 804–812. [Google Scholar] [CrossRef]
- Xiao, W.; Zhang, W.; Huang, H.; Xie, Y.; Zhang, Y.; Guo, X.; Jin, C.; Liao, X.; Yao, S.; Chen, G.; et al. Cancer Targeted Gene Therapy for Inhibition of Melanoma Lung Metastasis with eIF3i shRNA Loaded Liposomes. Mol. Pharm. 2020, 17, 229–238. [Google Scholar] [CrossRef]
- Bao, X.; Zeng, J.; Huang, H.; Ma, C.; Wang, L.; Wang, F.; Liao, X.; Song, X. Cancer-targeted PEDF-DNA therapy for metastatic colorectal cancer. Int. J. Pharm. 2020, 576, 118999. [Google Scholar] [CrossRef]
- Yan, F.; Wu, H.; Liu, H.; Deng, Z.; Liu, H.; Duan, W.; Liu, X.; Zheng, H. Molecular imaging-guided photothermal/photodynamic therapy against tumor by iRGD-modified indocyanine green nanoparticles. J. Control. Release 2016, 224, 217–228. [Google Scholar] [CrossRef]
- Shen, J.; Meng, Q.; Sui, H.; Yin, Q.; Zhang, Z.; Yu, H.; Li, Y. iRGD conjugated TPGS mediates codelivery of paclitaxel and survivin shRNA for the reversal of lung cancer resistance. Mol. Pharm. 2014, 11, 2579–2591. [Google Scholar] [CrossRef]
- Wang, C.-F.; Sarparanta, M.P.; Mäkilä, E.M.; Hyvönen, M.L.; Laakkonen, P.M.; Salonen, J.J.; Hirvonen, J.T.; Airaksinen, A.J.; Santos, H.A. Multifunctional porous silicon nanoparticles for cancer theranostics. Biomaterials 2015, 48, 108–118. [Google Scholar] [CrossRef]
- Li, X.; Wu, M.; Pan, L.; Shi, J. Tumor vascular-targeted co-delivery of anti-angiogenesis and chemotherapeutic agents by mesoporous silica nanoparticle-based drug delivery system for synergetic therapy of tumor. Int. J. Nanomed. 2016, 11, 93. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, J.; Ji, Y.; Lu, J.; Chan, R.; Meng, H. Targeted drug delivery using iRGD peptide for solid cancer treatment. Mol. Syst. Des. Eng. 2017, 2, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Takenaka, T.; Toh, K.; Wu, S.; Nishihara, H.; Kano, M.R.; Ino, Y.; Nomoto, T.; Matsumoto, Y.; Koyama, H.; et al. Cyclic RGD-linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood-brain tumor barrier. ACS Nano 2013, 7, 8583–8592. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhao, X.; Fu, T.; Li, K.; He, Y.; Luo, Z.; Dai, L.; Zeng, R.; Cai, K. An iRGD-conjugated prodrug micelle with blood-brain-barrier penetrability for anti-glioma therapy. Biomaterials 2020, 230, 119666. [Google Scholar] [CrossRef]
- Irby, D.; Du, C.; Li, F. Lipid-Drug Conjugate for Enhancing Drug Delivery. Mol. Pharm. 2017, 14, 1325–1338. [Google Scholar] [CrossRef]
- Han, S.-M.; Na, Y.-G.; Lee, H.-S.; Son, G.-H.; Jeon, S.-H.; Bang, K.-H.; Kim, S.-J.; Lee, H.-J.; Cho, C.-W. Improvement of cellular uptake of hydrophilic molecule, calcein, formulated by liposome. J. Pharm. Investig. 2018, 48, 595–601. [Google Scholar] [CrossRef]
- Song, X.; Wan, Z.; Chen, T.; Fu, Y.; Jiang, K.; Yi, X.; Ke, H.; Dong, J.; Yang, L.; Li, L.; et al. Development of a multi-target peptide for potentiating chemotherapy by modulating tumor microenvironment. Biomaterials 2016, 108, 44–56. [Google Scholar] [CrossRef]
- Wang, L.; Xie, X.; Liu, D.; Fang, X.-B.; Li, P.; Wan, J.-B.; He, C.-W.; Chen, M.-W. iRGD-mediated reduction-responsive DSPE–PEG/LA–PLGA–TPGS mixed micelles used in the targeted delivery and triggered release of docetaxel in cancer. RSC Adv. 2016, 6, 28331–28342. [Google Scholar] [CrossRef]
- Liu, Y.; Ji, M.; Wong, M.K.; Joo, K.I.; Wang, P. Enhanced therapeutic efficacy of iRGD-conjugated crosslinked multilayer liposomes for drug delivery. BioMed Res. Int. 2013, 2013, 378380. [Google Scholar] [CrossRef]
- Yu, K.F.; Zhang, W.Q.; Luo, L.M.; Song, P.; Li, D.; Du, R.; Ren, W.; Huang, D.; Lu, W.L.; Zhang, X.; et al. The antitumor activity of a doxorubicin loaded, iRGD-modified sterically-stabilized liposome on B16-F10 melanoma cells: In vitro and in vivo evaluation. Int. J. Nanomed. 2013, 8, 2473–2485. [Google Scholar] [CrossRef]
- Dai, W.; Fan, Y.; Zhang, H.; Wang, X.; Zhang, Q.; Wang, X. A comprehensive study of iRGD-modified liposomes with improved chemotherapeutic efficacy on B16 melanoma. Drug Deliv. 2015, 22, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Song, X.; Gong, T.; Fu, Y.; Yang, L.; Zhang, Z.; Gong, T. nRGD modified lycobetaine and octreotide combination delivery system to overcome multiple barriers and enhance anti-glioma efficacy. Colloids Surf. B Biointerfaces 2017, 156, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Zhang, J.; Xu, Q.; Liu, X.; Li, Y.; Wu, Y.; Chen, C. Targeting peptide iRGD-conjugated amphiphilic chitosan-co-PLA/DPPE drug delivery system for enhanced tumor therapy. J. Mater. Chem. B 2014, 2, 3232–3242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Lip, H.; He, C.; Cai, P.; Wang, Z.; Henderson, J.T.; Rauth, A.M.; Wu, X.Y. Multitargeted Nanoparticles Deliver Synergistic Drugs across the Blood-Brain Barrier to Brain Metastases of Triple Negative Breast Cancer Cells and Tumor-Associated Macrophages. Adv. Health Mater. 2019, 8, e1900543. [Google Scholar] [CrossRef]
- Chen, J.; Yang, Q.; Liu, M.; Lin, M.; Wang, T.; Zhang, Z.; Zhong, X.; Guo, N.; Lu, Y.; Xu, J.; et al. Remarkable Boron Delivery Of iRGD-Modified Polymeric Nanoparticles for Boron Neutron Capture Therapy. Int. J. Nanomed. 2019, 14, 8161–8177. [Google Scholar] [CrossRef]
- Xie, Y.; Qi, X.; Xu, K.; Meng, X.; Chen, X.; Wang, F.; Zhong, H. Transarterial Infusion of iRGD-Modified ZrO(2) Nanoparticles with Lipiodol Improves the Tissue Distribution of Doxorubicin and Its Antitumor Efficacy. J. Vasc. Interv. Radiol. 2019, 30, 2026–2035. [Google Scholar] [CrossRef]
- Simón-Gracia, L.; Hunt, H.; Scodeller, P.; Gaitzsch, J.; Kotamraju, V.R.; Sugahara, K.N.; Tammik, O.; Ruoslahti, E.; Battaglia, G.; Teesalu, T. iRGD peptide conjugation potentiates intraperitoneal tumor delivery of paclitaxel with polymersomes. Biomaterials 2016, 104, 247–257. [Google Scholar] [CrossRef]
- Zhu, Z.; Xie, C.; Liu, Q.; Zhen, X.; Zheng, X.; Wu, W.; Li, R.; Ding, Y.; Jiang, X.; Liu, B. The effect of hydrophilic chain length and iRGD on drug delivery from poly(ε-caprolactone)-poly(N-vinylpyrrolidone) nanoparticles. Biomaterials 2011, 32, 9525–9535. [Google Scholar] [CrossRef]
- Kang, T.; Li, Y.; Wang, Y.; Zhu, J.; Yang, L.; Huang, Y.; Xiong, M.; Liu, J.; Wang, S.; Huang, M.; et al. Modular Engineering of Targeted Dual-Drug Nanoassemblies for Cancer Chemoimmunotherapy. ACS Appl. Mater. Interfaces 2019, 11, 36371–36382. [Google Scholar] [CrossRef]
- Liu, C.; Yao, S.; Li, X.; Wang, F.; Jiang, Y. iRGD-mediated core-shell nanoparticles loading carmustine and O(6)-benzylguanine for glioma therapy. J. Drug Target. 2017, 25, 235–246. [Google Scholar] [CrossRef]
- Bessone, M.I.D.; Simón-Gracia, L.; Scodeller, P.; de los Angeles, R.M.; Huvelle, M.A.L.; Soler-Illia, G.J.; Simian, M. iRGD-guided tamoxifen polymersomes inhibit estrogen receptor transcriptional activity and decrease the number of breast cancer cells with self-renewing capacity. J. Nanobiotechnol. 2019, 17, 1–14. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, J.; Fu, C.; Xie, X.; Peng, F.; You, J.; Tang, H.; Wang, Z.; Li, P.; Chen, J. iRGD-modified lipid-polymer hybrid nanoparticles loaded with isoliquiritigenin to enhance anti-breast cancer effect and tumor-targeting ability. Int. J. Nanomed. 2017, 12, 4147–4162. [Google Scholar] [CrossRef]
- Lee, J.; Oh, E.-T.; Lee, J.; Kang, T.; Kim, H.G.; Kang, H.; Park, H.J.; Kim, C. Cyclic iRGD peptide as a dual-functional on–off gatekeeper of mesoporous nanocontainers for targeting NRP-1 and selective drug release triggered by conformational conversion. New J. Chem. 2019, 43, 1517–1522. [Google Scholar] [CrossRef]
- Su, S.; Wang, H.; Liu, X.; Wu, Y.; Nie, G. iRGD-coupled responsive fluorescent nanogel for targeted drug delivery. Biomaterials 2013, 34, 3523–3533. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, N.; Luo, C.; Zhu, J.; Bao, C. Photosensitizer-loaded cell membrane biomimetic nanoparticles for enhanced tumor synergetic targeted therapy. RSC Adv. 2020, 10, 9378–9386. [Google Scholar] [CrossRef]
- Erel-Akbaba, G.; Carvalho, L.A.; Tian, T.; Zinter, M.; Akbaba, H.; Obeid, P.J.; Chiocca, E.A.; Weissleder, R.; Kantarci, A.G.; Tannous, B.A. Radiation-Induced Targeted Nanoparticle-Based Gene Delivery for Brain Tumor Therapy. ACS Nano 2019, 13, 4028–4040. [Google Scholar] [CrossRef]
- Jin, Z.; Lv, Y.; Cao, H.; Yao, J.; Zhou, J.; He, W.; Yin, L. Core-shell nanocarriers with high paclitaxel loading for passive and active targeting. Sci. Rep. 2016, 6, 27559. [Google Scholar] [CrossRef]
- Gu, G.; Gao, X.; Hu, Q.; Kang, T.; Liu, Z.; Jiang, M.; Miao, D.; Song, Q.; Yao, L.; Tu, Y.; et al. The influence of the penetrating peptide iRGD on the effect of paclitaxel-loaded MT1-AF7p-conjugated nanoparticles on glioma cells. Biomaterials 2013, 34, 5138–5148. [Google Scholar] [CrossRef]
- Zhong, Y.; Su, T.; Shi, Q.; Feng, Y.; Tao, Z.; Huang, Q.; Li, L.; Hu, L.; Li, S.; Tan, H.; et al. Co-Administration of iRGD Enhances Tumor-Targeted Delivery and Anti-Tumor Effects of Paclitaxel-Loaded PLGA Nanoparticles for Colorectal Cancer Treatment. Int. J. Nanomed. 2019, 14, 8543–8560. [Google Scholar] [CrossRef]
- Deng, C.; Xu, X.; Tashi, D.; Wu, Y.; Su, B.; Zhang, Q. Co-administration of biocompatible self-assembled polylactic acid-hyaluronic acid block copolymer nanoparticles with tumor-penetrating peptide-iRGD for metastatic breast cancer therapy. J. Mater. Chem. B 2018, 6, 3163–3180. [Google Scholar] [CrossRef]
- Liu, X.; Lin, P.; Perrett, I.; Lin, J.; Liao, Y.P.; Chang, C.H.; Jiang, J.; Wu, N.; Donahue, T.; Wainberg, Z.; et al. Tumor-penetrating peptide enhances transcytosis of silicasome-based chemotherapy for pancreatic cancer. J. Clin. Investig. 2017, 127, 2007–2018. [Google Scholar] [CrossRef]
- Deng, C.; Zhang, Q.; Fu, Y.; Sun, X.; Gong, T.; Zhang, Z. Coadministration of Oligomeric Hyaluronic Acid-Modified Liposomes with Tumor-Penetrating Peptide-iRGD Enhances the Antitumor Efficacy of Doxorubicin against Melanoma. ACS Appl. Mater. Interfaces 2017, 9, 1280–1292. [Google Scholar] [CrossRef]
- Yu, H.; Tang, Z.; Song, W.; Zhang, D.; Zhang, Y.; Chen, X. Co-administration of iRGD enhancing the anticancer efficacy of cisplatin-loaded polypeptide nanoparticles. J. Control. Release 2015, 213, e145–e146. [Google Scholar] [CrossRef]
- Cun, X.; Chen, J.; Ruan, S.; Zhang, L.; Wan, J.; He, Q.; Gao, H. A Novel Strategy through Combining iRGD Peptide with Tumor-Microenvironment-Responsive and Multistage Nanoparticles for Deep Tumor Penetration. ACS Appl. Mater. Interfaces 2015, 7, 27458–27466. [Google Scholar] [CrossRef]
- Umeshappa, C.S.; Shao, K. Comment on “Coadministration of iRGD with Multistage Responsive Nanoparticles Enhanced Tumor Targeting and Penetration Abilities for Breast Cancer Therapy”. ACS Appl. Mater. Interfaces 2018, 10, 38659–38662. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Y.; Li, K.; Wang, H.; Li, H.; Zheng, J. A Novel Strategy to Improve the Therapeutic Efficacy of Gemcitabine for Non-Small Cell Lung Cancer by the Tumor-Penetrating Peptide iRGD. PLoS ONE 2015, 10, e0129865. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, J.; Ding, M.; Li, L.; Lu, Z.; Zhang, Q.; Zheng, J. Tumor-penetration and antitumor efficacy of cetuximab are enhanced by co-administered iRGD in a murine model of human NSCLC. Oncol. Lett. 2016, 12, 3241–3249. [Google Scholar] [CrossRef]
- Fadeev, R.; Chekanov, A.; Solovieva, M.; Bezborodova, O.; Nemtsova, E.; Dolgikh, N.; Fadeeva, I.; Senotov, A.; Kobyakova, M.; Evstratova, Y.; et al. Improved Anticancer Effect of Recombinant Protein izTRAIL Combined with Sorafenib and Peptide iRGD. Int. J. Mol. Sci. 2019, 20, 525. [Google Scholar] [CrossRef]
- Zhang, D.; Chu, Y.; Qian, H.; Qian, L.; Shao, J.; Xu, Q.; Yu, L.; Li, R.; Zhang, Q.; Wu, F.; et al. Antitumor Activity of Thermosensitive Hydrogels Packaging Gambogic Acid Nanoparticles and Tumor-Penetrating Peptide iRGD Against Gastric Cancer. Int. J. Nanomed. 2020, 15, 735–747. [Google Scholar] [CrossRef]
- Saad, A.M.; Turk, T.; Al-Husseini, M.J.; Abdel-Rahman, O. Trends in pancreatic adenocarcinoma incidence and mortality in the United States in the last four decades; a SEER-based study. BMC Cancer 2018, 18, 688. [Google Scholar] [CrossRef]
- Khalaf, N.; El-Serag, H.B.; Abrams, H.R.; Thrift, A.P. Burden of Pancreatic Cancer—From Epidemiology to Practice. Clin. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Oberstein, P.E.; Olive, K.P. Pancreatic cancer: Why is it so hard to treat? Ther. Adv. Gastroenterol. 2013, 6, 321–337. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Frese, K.K.; Neesse, A.; Cook, N.; Bapiro, T.E.; Lolkema, M.P.; Jodrell, D.I.; Tuveson, D.A. Nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012, 2, 260–269. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ramanathan, R.K.; Borad, M.J.; Laheru, D.A.; Smith, L.S.; Wood, T.E.; Korn, R.L.; Desai, N.; Trieu, V.; Iglesias, J.L.; et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: A phase I/II trial. J. Clin. Oncol. 2011, 29, 4548–4554. [Google Scholar] [CrossRef]
- Fukahi, K.; Fukasawa, M.; Neufeld, G.; Itakura, J.; Korc, M. Aberrant expression of neuropilin-1 and -2 in human pancreatic cancer cells. Clin. Cancer Res. 2004, 10, 581–590. [Google Scholar] [CrossRef]
- Wey, J.S.; Gray, M.J.; Fan, F.; Belcheva, A.; McCarty, M.F.; Stoeltzing, O.; Somcio, R.; Liu, W.; Evans, D.B.; Klagsbrun, M.; et al. Overexpression of neuropilin-1 promotes constitutive MAPK signalling and chemoresistance in pancreatic cancer cells. Br. J. Cancer 2005, 93, 233–241. [Google Scholar] [CrossRef]
- Fukasawa, M.; Matsushita, A.; Korc, M. Neuropilin-1 interacts with integrin beta1 and modulates pancreatic cancer cell growth, survival and invasion. Cancer Biol. Ther. 2007, 6, 1173–1180. [Google Scholar] [CrossRef]
- Matkar, P.N.; Singh, K.K.; Rudenko, D.; Kim, Y.J.; Kuliszewski, M.A.; Prud’homme, G.J.; Hedley, D.W.; Leong-Poi, H. Novel regulatory role of neuropilin-1 in endothelial-to-mesenchymal transition and fibrosis in pancreatic ductal adenocarcinoma. Oncotarget 2016, 7, 69489–69506. [Google Scholar] [CrossRef]
- DrugCendR. CEND-1 in Combination with Nabpaclitaxel and Gemcitabine in Metastatic Pancreatic Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03517176 (accessed on 12 July 2020).
- Slaney, C.Y.; Kershaw, M.H.; Darcy, P.K. Trafficking of T cells into tumors. Cancer Res. 2014, 74, 7168–7174. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, H.; Wang, P.; Zhang, K.; Geng, X.; Liu, Q.; Wang, X. Tumor targeting DVDMS-nanoliposomes for an enhanced sonodynamic therapy of gliomas. Biomater. Sci. 2019, 7, 985–994. [Google Scholar] [CrossRef]
- Martin, M.; Paul, D.; Orwin, M.; Schlievert, P. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 2000, 13, 16–34. [Google Scholar]
- Song, Y.; Xu, M.; Li, Y.; Li, Y.; Gu, W.; Halimu, G.; Fu, X.; Zhang, H.; Zhang, C. An iRGD peptide fused superantigen mutant induced tumor-targeting and T lymphocyte infiltrating in cancer immunotherapy. Int. J. Pharm. 2020, 586, 119498. [Google Scholar] [CrossRef]
- Ricci, M.S.; Zong, W.-X. Chemotherapeutic approaches for targeting cell death pathways. Oncologist 2006, 11, 342. [Google Scholar] [CrossRef]
- Li, Y.; Ahmed, F.; Ali, S.; Philip, P.A.; Kucuk, O.; Sarkar, F.H. Inactivation of nuclear factor κB by soy isoflavone genistein contributes to increased apoptosis induced by chemotherapeutic agents in human cancer cells. Cancer Res. 2005, 65, 6934–6942. [Google Scholar] [CrossRef]
- Dudek-Perić, A.M.; Ferreira, G.B.; Muchowicz, A.; Wouters, J.; Prada, N.; Martin, S.; Kiviluoto, S.; Winiarska, M.; Boon, L.; Mathieu, C. Antitumor immunity triggered by melphalan is potentiated by melanoma cell surface–associated calreticulin. Cancer Res. 2015, 75, 1603–1614. [Google Scholar] [CrossRef]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Ding, Z.C.; Munn, D.H.; Zhou, G. Chemotherapy-induced myeloid suppressor cells and antitumor immunity: The Janus face of chemotherapy in immunomodulation. Oncoimmunology 2014, 3, e954471. [Google Scholar] [CrossRef]
- Pruneri, G.; Gray, K.P.; Vingiani, A.; Viale, G.; Curigliano, G.; Criscitiello, C.; Láng, I.; Ruhstaller, T.; Gianni, L.; Goldhirsch, A.; et al. Tumor-infiltrating lymphocytes (TILs) are a powerful prognostic marker in patients with triple-negative breast cancer enrolled in the IBCSG phase III randomized clinical trial 22-00. Breast Cancer Res. Treat. 2016, 158, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Lake, R.A.; Robinson, B.W. Immunotherapy and chemotherapy—A practical partnership. Nat. Rev. Cancer 2005, 5, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.L.; Hsu, Y.T.; Wu, C.C.; Lai, Y.Z.; Wang, C.; Yang, Y.C.; Wu, T.C.; Hung, C.F. Dose-dense chemotherapy improves mechanisms of antitumor immune response. Cancer Res. 2013, 73, 119–127. [Google Scholar] [CrossRef]
- Deng, C.; Jia, M.; Wei, G.; Tan, T.; Fu, Y.; Gao, H.; Sun, X.; Zhang, Q.; Gong, T.; Zhang, Z. Inducing Optimal Antitumor Immune Response through Coadministering iRGD with Pirarubicin Loaded Nanostructured Lipid Carriers for Breast Cancer Therapy. Mol. Pharm. 2017, 14, 296–309. [Google Scholar] [CrossRef]
- Kolishetti, N.; Dhar, S.; Valencia, P.M.; Lin, L.Q.; Karnik, R.; Lippard, S.J.; Langer, R.; Farokhzad, O.C. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 17939–17944. [Google Scholar] [CrossRef]
- Ferrari, M. Cancer nanotechnology: Opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar] [CrossRef]
- Han, W.; Shi, L.; Ren, L.; Zhou, L.; Li, T.; Qiao, Y.; Wang, H. A nanomedicine approach enables co-delivery of cyclosporin A and gefitinib to potentiate the therapeutic efficacy in drug-resistant lung cancer. Signal. Transduct Target. Ther. 2018, 3, 16. [Google Scholar] [CrossRef]
- Le, Q.-V.; Choi, J.; Oh, Y.-K. Nano delivery systems and cancer immunotherapy. J. Pharm. Investig. 2018, 48, 527–539. [Google Scholar] [CrossRef]
- Tran, P.; Lee, S.-E.; Kim, D.-H.; Pyo, Y.-C.; Park, J.-S. Recent advances of nanotechnology for the delivery of anticancer drugs for breast cancer treatment. J. Pharm. Investig. 2020, 50, 261–270. [Google Scholar] [CrossRef]
- Park, Y.J.; Kuen, D.S.; Chung, Y. Future prospects of immune checkpoint blockade in cancer: From response prediction to overcoming resistance. Exp. Mol. Med. 2018, 50, 109. [Google Scholar] [CrossRef]
- Kunimasa, K.; Goto, T. Immunosurveillance and Immunoediting of Lung Cancer: Current Perspectives and Challenges. Int. J. Mol. Sci. 2020, 21, 597. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, B.; O’Brien, S.; Lee, D.; Hou, Y.; Weinberg, V.; Rini, B.; Allison, J.P.; Small, E.J.; Fong, L. CTLA4 blockade expands FoxP3+ regulatory and activated effector CD4+ T cells in a dose-dependent fashion. Blood 2008, 112, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.H. Costimulation of T lymphocytes: The role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell 1992, 71, 1065–1068. [Google Scholar] [CrossRef]
- Linsley, P.S.; Brady, W.; Urnes, M.; Grosmaire, L.S.; Damle, N.K.; Ledbetter, J.A. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 1991, 174, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Downey, J.; Smith, A.; Zinselmeyer, B.H.; Rush, C.; Brewer, J.M.; Wei, B.; Hogg, N.; Garside, P.; Rudd, C.E. Reversal of the TCR stop signal by CTLA-4. Science 2006, 313, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Kwiecien, I.; Stelmaszczyk-Emmel, A.; Polubiec-Kownacka, M.; Dziedzic, D.; Domagala-Kulawik, J. Elevated regulatory T cells, surface and intracellular CTLA-4 expression and interleukin-17 in the lung cancer microenvironment in humans. Cancer Immunol. Immunother. 2017, 66, 161–170. [Google Scholar] [CrossRef]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, C.C.; Jin, L.; Zhang, X.D. Regulation of PD-L1: A novel role of pro-survival signalling in cancer. Ann. Oncol. 2016, 27, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Yang, J.; Hu, L. Immunomodulators targeting the PD-1/PD-L1 protein-protein interaction: From antibodies to small molecules. Med. Res. Rev. 2019, 39, 265–301. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: Mechanisms of action, efficacy, and limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Yang, J.; Yang, J.; Wei, Y.; Yin, H.; Fang, L.; Chai, D.; Li, H.; Li, H.; Zhang, Q.; Zheng, J. Modification of IL-24 by tumor penetrating peptide iRGD enhanced its antitumor efficacy against non-small cell lung cancer. Int. Immunopharmacol. 2019, 70, 125–134. [Google Scholar] [CrossRef]
- Jarvelainen, H.; Botta, G.P.; De Mendoza, T.H.; Ruoslahti, E. Co-administration of the iRGD tumor-penetrating peptide improves the tumor immunostimulatory effects of low-dose IL-2. In Proceedings of the AACR Annual Meeting, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar]
- Zhang, F.; Stephan, S.B.; Ene, C.I.; Smith, T.T.; Holland, E.C.; Stephan, M.T. Nanoparticles That Reshape the Tumor Milieu Create a Therapeutic Window for Effective T-cell Therapy in Solid Malignancies. Cancer Res. 2018, 78, 3718–3730. [Google Scholar] [CrossRef]
- Ding, N.; Zou, Z.; Sha, H.; Su, S.; Qian, H.; Meng, F.; Chen, F.; Du, S.; Zhou, S.; Chen, H.; et al. iRGD synergizes with PD-1 knockout immunotherapy by enhancing lymphocyte infiltration in gastric cancer. Nat. Commun. 2019, 10, 1336. [Google Scholar] [CrossRef]
- Zhao, M.; van Straten, D.; Broekman, M.L.D.; Preat, V.; Schiffelers, R.M. Nanocarrier-based drug combination therapy for glioblastoma. Theranostics 2020, 10, 1355–1372. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.L.; Staddon, J.M. The cell biology of the blood-brain barrier. Annu. Rev. Neurosci. 1999, 22, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Luissint, A.C.; Federici, C.; Guillonneau, F.; Chretien, F.; Camoin, L.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.O. Guanine nucleotide-binding protein Galphai2: A new partner of claudin-5 that regulates tight junction integrity in human brain endothelial cells. J. Cereb. Blood Flow Metab. 2012, 32, 860–873. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Nishio, S.; Ohta, M.; Abe, M.; Kitamura, K. Microvascular abnormalities in ethylnitrosourea (ENU)-induced rat brain tumors: Structural basis for altered blood-brain barrier function. Acta Neuropathol. 1983, 59, 1–10. [Google Scholar] [CrossRef]
- Wolburg, H.; Noell, S.; Fallier-Becker, P.; Mack, A.F.; Wolburg-Buchholz, K. The disturbed blood-brain barrier in human glioblastoma. Mol. Asp. Med. 2012, 33, 579–589. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, H.; Yang, K.; Xu, W.; Tang, W.; Li, X. The structure and functions of P-glycoprotein. Curr. Med. Chem. 2010, 17, 786–800. [Google Scholar] [CrossRef]
- Siegal, T.; Rubinstein, R.; Bokstein, F.; Schwartz, A.; Lossos, A.; Shalom, E.; Chisin, R.; Gomori, J.M. In vivo assessment of the window of barrier opening after osmotic blood-brain barrier disruption in humans. J Neurosurg. 2000, 92, 599–605. [Google Scholar] [CrossRef]
- Van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Raymond, J.J.; Robertson, D.M.; Dinsdale, H.B. Pharmacological modification of bradykinin induced breakdown of the blood-brain barrier. Can. J. Neurol. Sci. 1986, 13, 214–220. [Google Scholar] [CrossRef]
- Lu, C.T.; Zhao, Y.Z.; Wong, H.L.; Cai, J.; Peng, L.; Tian, X.Q. Current approaches to enhance CNS delivery of drugs across the brain barriers. Int. J. Nanomed. 2014, 9, 2241–2257. [Google Scholar] [CrossRef] [PubMed]
- Vogelbaum, M.A.; Aghi, M.K. Convection-enhanced delivery for the treatment of glioblastoma. Neuro-Oncology 2015, 17 (Suppl. 2). [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.M.; Aidoudi-Ahmed, S.; Sharma, S.; Kotamraju, V.R.; Foster, P.J.; Sugahara, K.N.; Ruoslahti, E.; Rutt, B.K. Nanoparticles coated with the tumor-penetrating peptide iRGD reduce experimental breast cancer metastasis in the brain. J. Mol. Med. 2015, 93, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Moncelet, D.; Bouchaud, V.; Mellet, P.; Ribot, E.; Miraux, S.; Franconi, J.M.; Voisin, P. Cellular density effect on RGD ligand internalization in glioblastoma for MRI application. PLoS ONE 2013, 8, e82777. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, H.; Jin, Q.; Ji, J. Surface tailoring of nanoparticles via mixed-charge monolayers and their biomedical applications. Small 2014, 10, 4230–4242. [Google Scholar] [CrossRef]
- Ruoslahti, E. Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv. Mater. 2012, 24, 3747–3756. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Vehicles | APIa | Cancer Type | Ref |
|---|---|---|---|
| Liposomes | PEDFb DNA | Colorectal cancer (CT26) | [97] |
| none | Breast cancer (4T1) | [98] | |
| Doxorubicin | Breast cancer (4T1), melanoma (B16-F10) | [107,113,114,115] | |
| Lycobetaine Octreotide | Glioma (C6) | [116] | |
| Polymeric NPsc | Doxorubicin | Breast cancer (4T1), liver cancer (VX2) | [112,117,118,119] |
| Paclitaxel | Gastric cancer (MKN-45P), colon cancer (CT26), hepatoma (H22), breast cancer (4T1) | [110,120,121] | |
| Carmustine O6-enzylguanine | Glioma (F98, C6, U87) | [122] | |
| Tamoxifen | Breast cancer (MCF-7, T47D) | [123] | |
| Isoliquiritigenin | Breast cancer (4T1) | [111] | |
| Silica NPsc | Doxorubicin | Colorectal cancer (HT-29) | [124] |
| Micelles | Paclitaxel | Prostate cancer (PC-3, PPC1), pancreatic cancer (MIA PaCa-2), breast cancer (BT474) | [52] |
| Platinum complex | Glioblastoma (U87) | [103] | |
| Camptothecin | Glioblastoma (U87) | [104] | |
| Docetaxel | HeLa | [108] | |
| Hydrogels | Doxorubicin | Melanoma (B16) | [125] |
| Gambogic acid | Gastric cancer (MKN-45) | [126] | |
| Solid lipid NPsc | siRNA | Glioblastoma (GL261) | [109] |
| Protein NCsd | Paclitaxel | Hepatoma (H22) | [127] |
| Drug | Vehicles | APIa | Cancer Type | Ref |
|---|---|---|---|---|
| drug delivery systems | Polymeric NPsb | Paclitaxel | Glioma (C6), breast cancer (BT474), colorectal cancer (LS174T) | [128,129] |
| Doxorubicin | Breast cancer (4T1) | [130] | ||
| Silica NPsb | Irinotecan | Pancreatic KPC-derived cancer | [131] | |
| Liposomes | Doxorubicin | Prostate cancer (22Rv1), melanoma (B16F10) | [54,132] | |
| Polypeptide NPsb | Cisplatin | Melanoma (B16F1) | [133] | |
| Gold NPsb (Au NPs) | Doxorubicin | Breast cancer (4T1) | [134] | |
| Dendrimers | Doxorubicin | Prostate cancer (22Rv1), Melanoma (B16F10) | [135] | |
| Drugs only | None | HPRP-A1c | Non-small cell lung cancer (A549) | [48] |
| Gemcitabine | Non-small cell lung cancer (A549) | [136] | ||
| Cetuximab | Non-small cell lung cancer (A549) | [137] | ||
| izTRAILd & Sorafenib | Fibrosarcoma (HT-1080) | [138] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, S.; Lee, S.; Park, S. iRGD Peptide as a Tumor-Penetrating Enhancer for Tumor-Targeted Drug Delivery. Polymers 2020, 12, 1906. https://doi.org/10.3390/polym12091906
Kang S, Lee S, Park S. iRGD Peptide as a Tumor-Penetrating Enhancer for Tumor-Targeted Drug Delivery. Polymers. 2020; 12(9):1906. https://doi.org/10.3390/polym12091906
Chicago/Turabian StyleKang, Sujin, Sooyeun Lee, and Soyeun Park. 2020. "iRGD Peptide as a Tumor-Penetrating Enhancer for Tumor-Targeted Drug Delivery" Polymers 12, no. 9: 1906. https://doi.org/10.3390/polym12091906
APA StyleKang, S., Lee, S., & Park, S. (2020). iRGD Peptide as a Tumor-Penetrating Enhancer for Tumor-Targeted Drug Delivery. Polymers, 12(9), 1906. https://doi.org/10.3390/polym12091906