Atomistic Investigation on the Wetting Behavior and Interfacial Joining of Polymer-Metal Interface



Abstract
1. Introduction
2. Materials and Methods
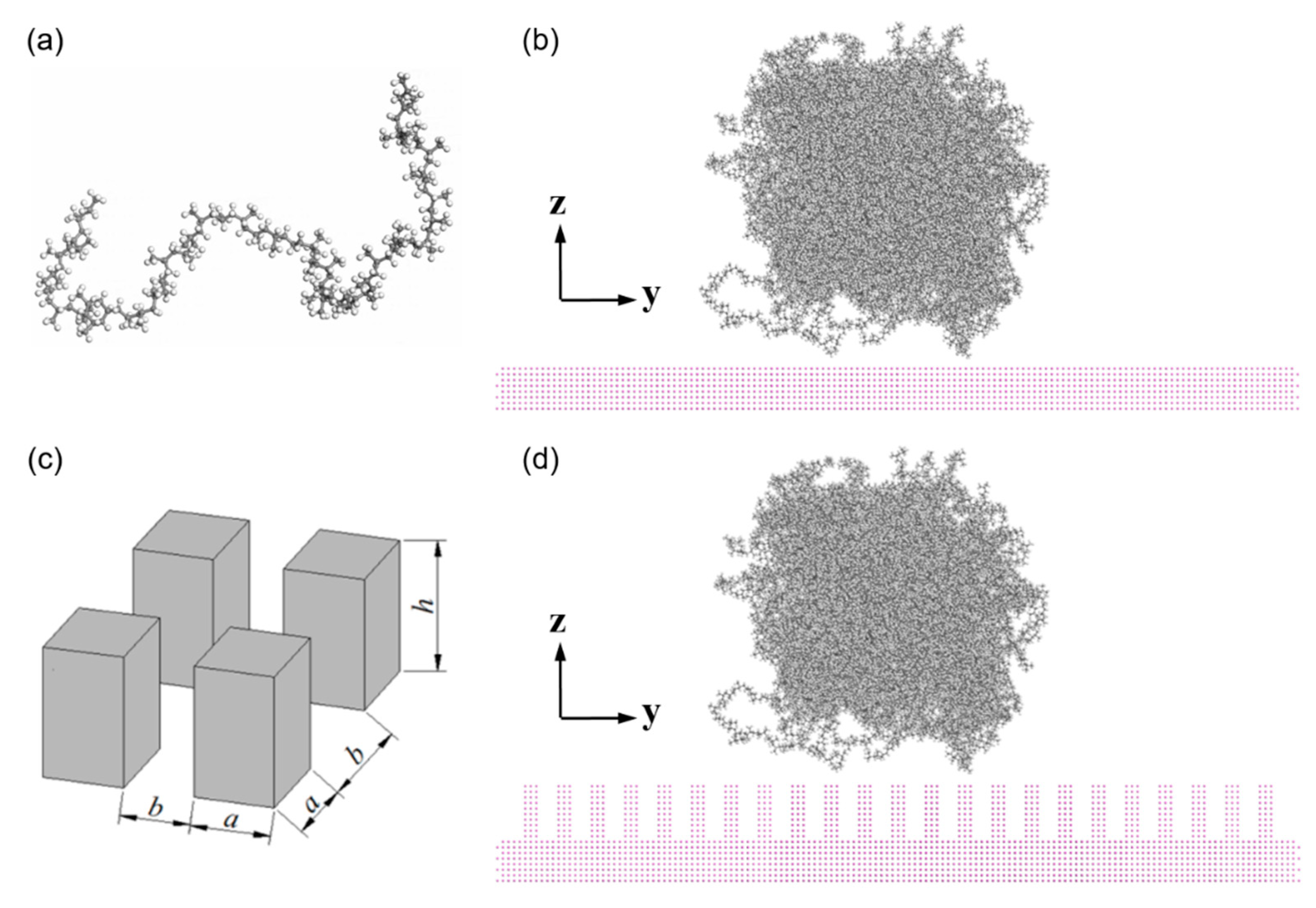
2.1. Materials and Model Constructing
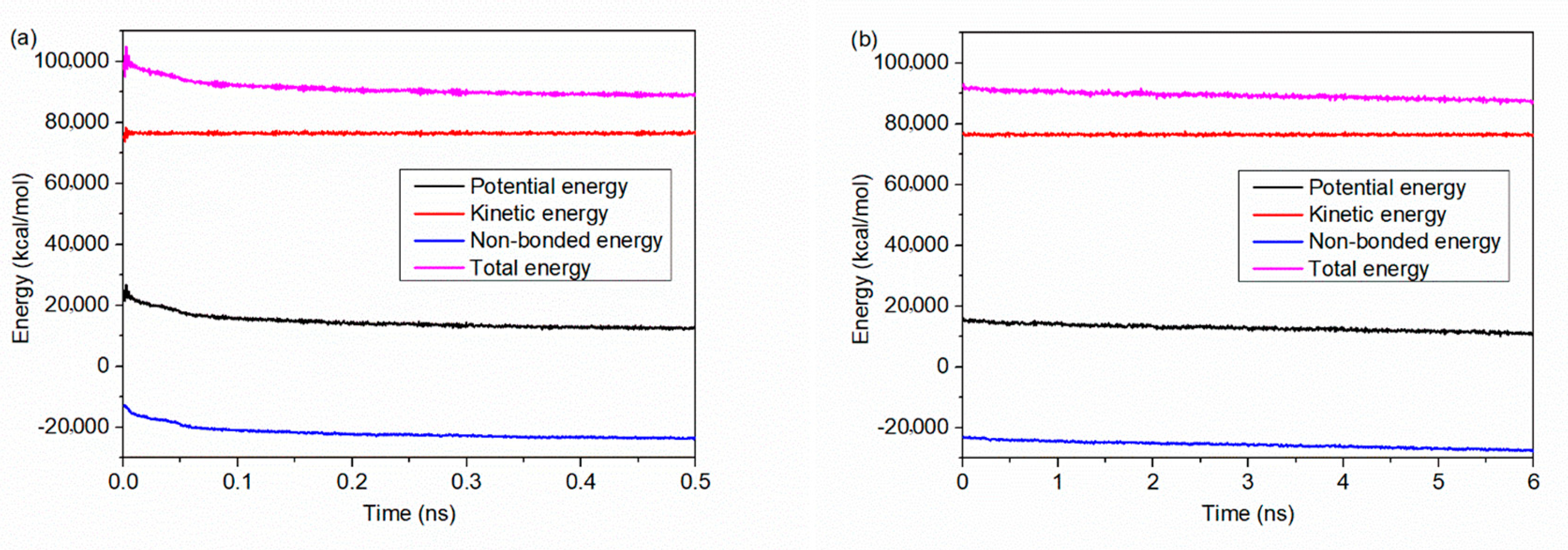
2.2. Force Field and Simulation Procedure
3. Results and Discussion
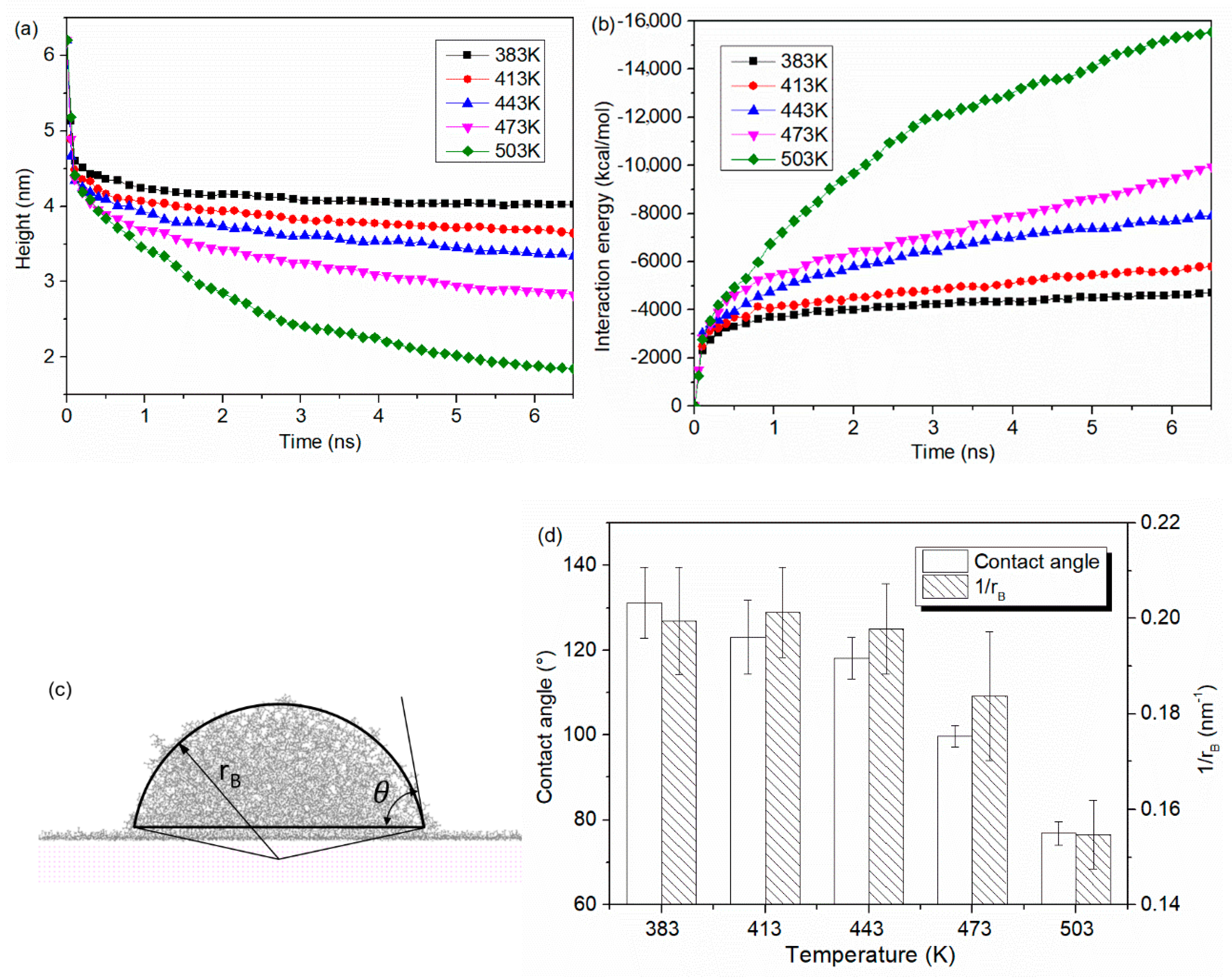
3.1. Wetting Behavior of PP Droplet on Smooth Surface
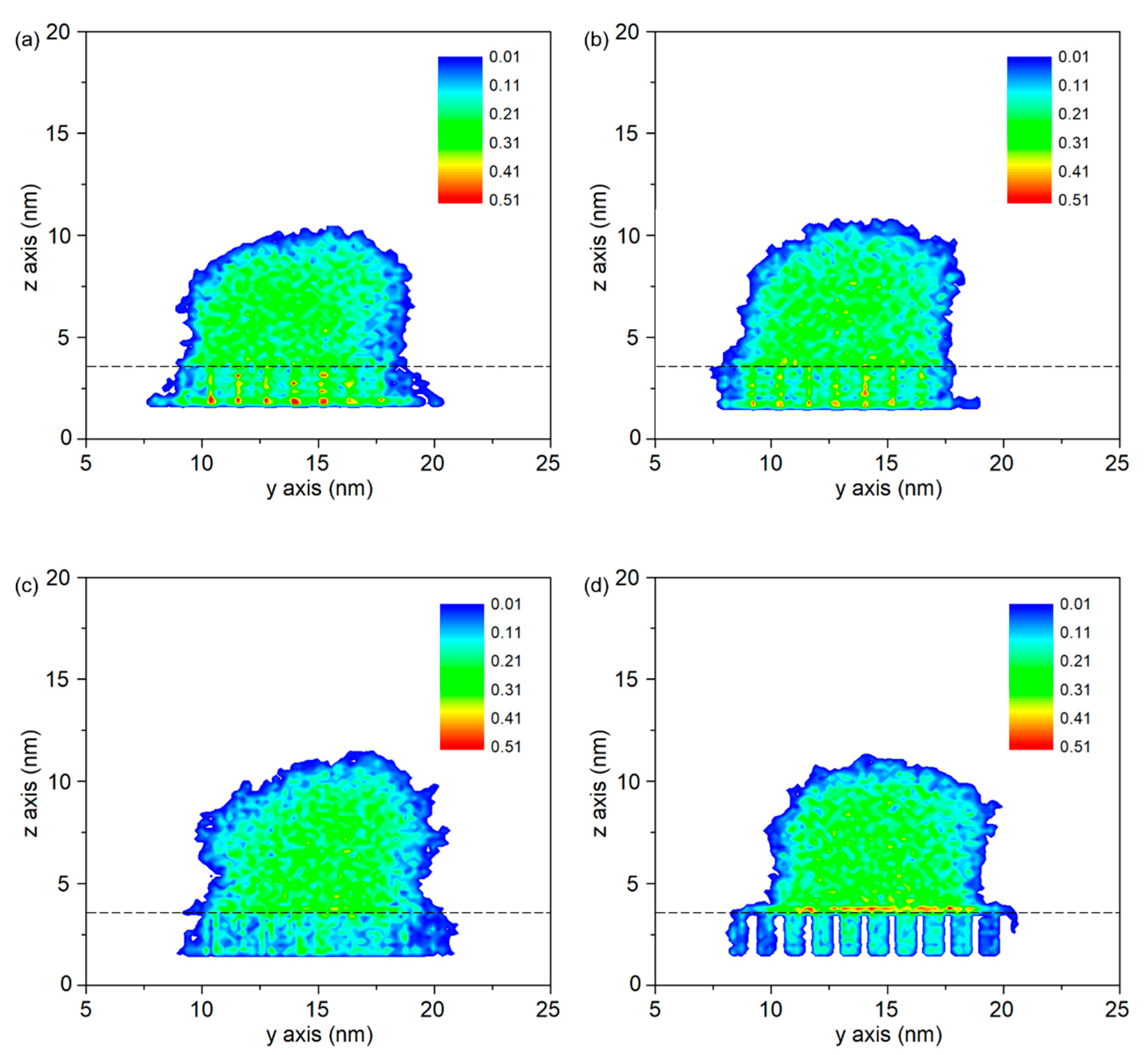
3.2. Wetting Behavior of PP Droplet on Rough Surface
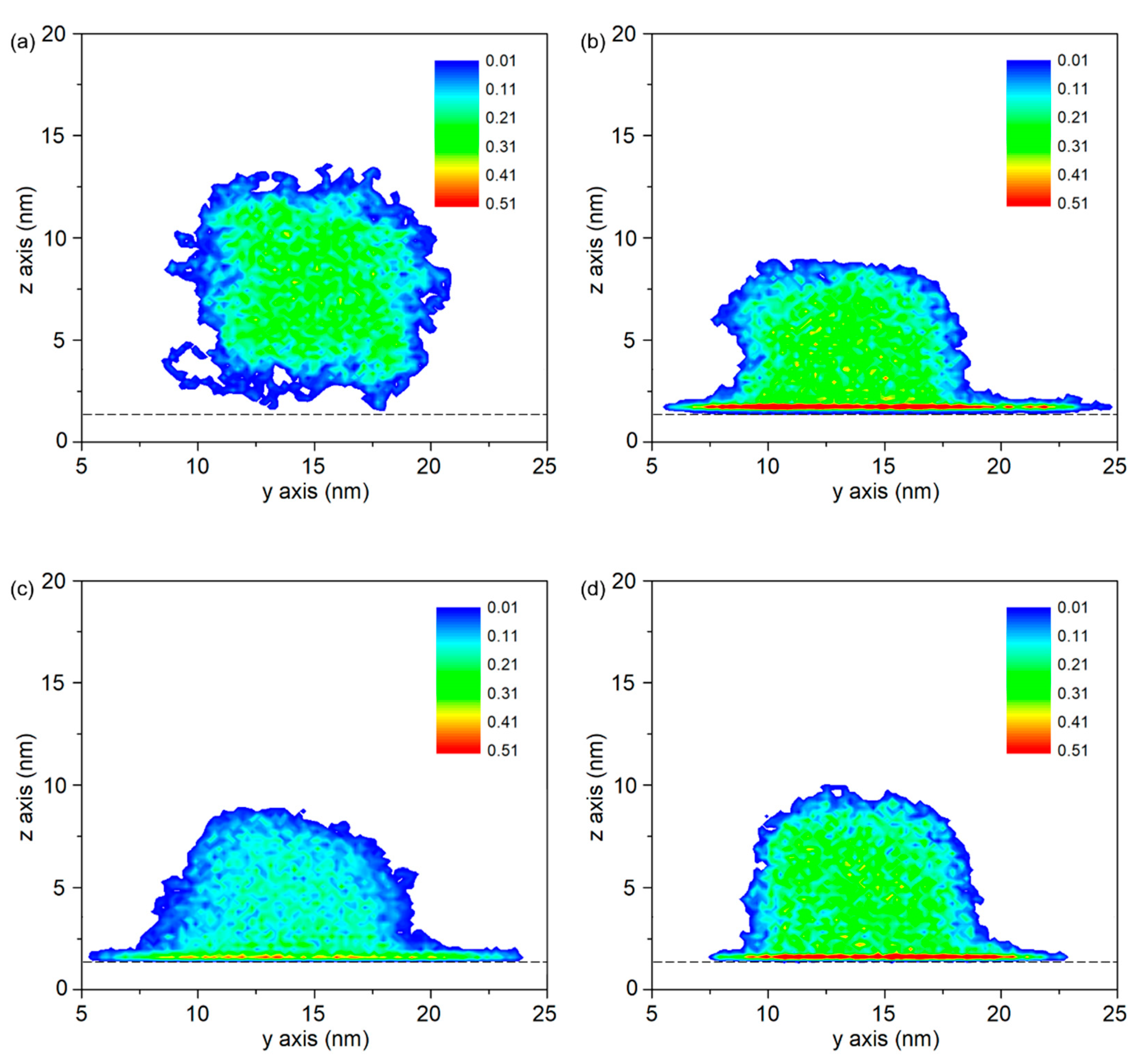
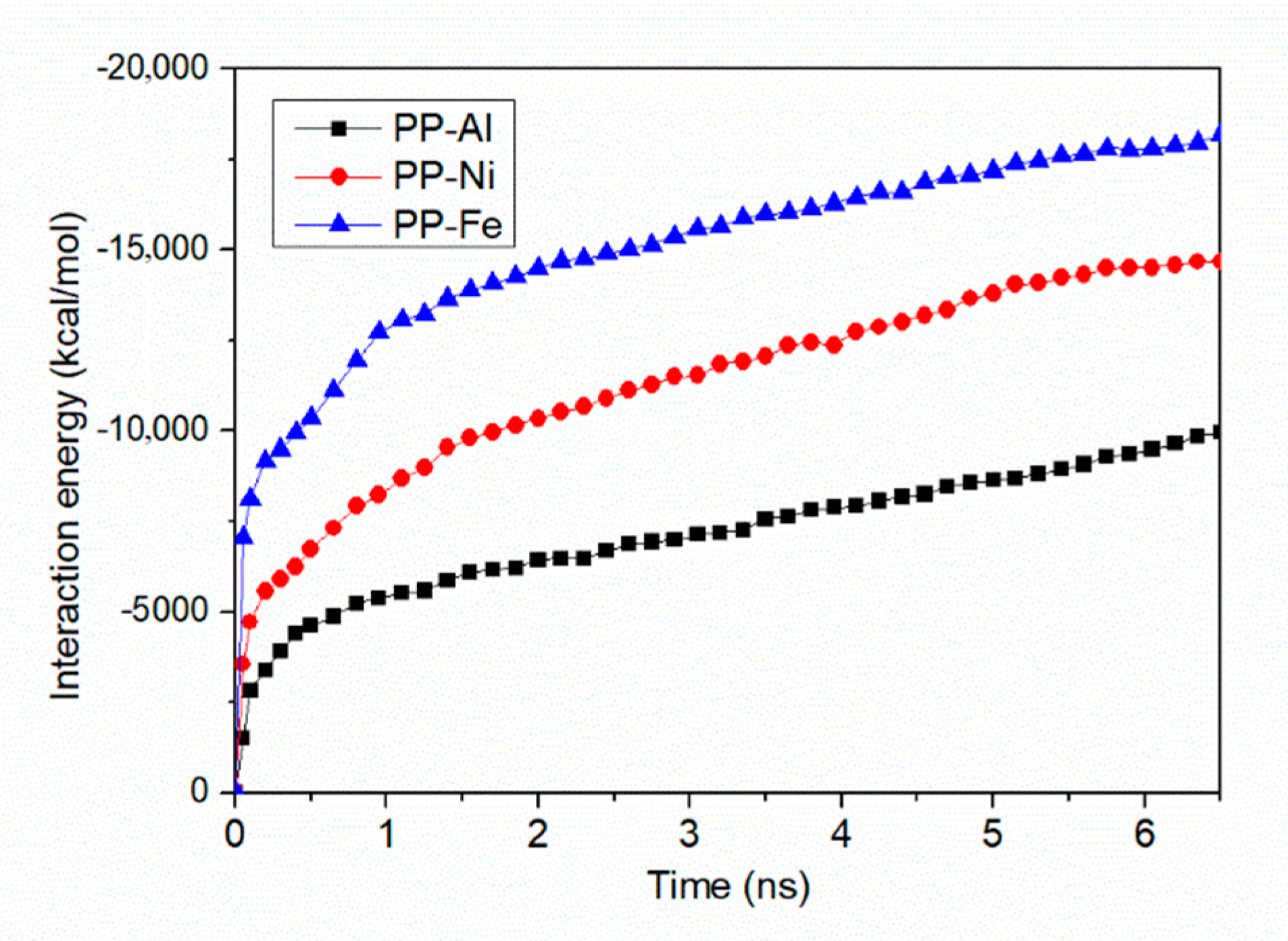
3.3. Dependence of Metal Substrate
3.4. Influence of Metal Substrate on the Joining Strength of Injection-Molded Hybrid Structure
4. Conclusions
- (1)
- Molecules in PP model are easier to spread along the surface with a higher temperature. At the beginning, the PP droplet forms a sphere-like structure while the atoms in the bottom of PP model start to spread out along the surface. With the time goes on, the whole model gradually collapses and more atoms are attached to the Al surface. The decrease in the height of mass center reflects an increase in the spreading ability of PP droplet.
- (2)
- Molecule chains are able to penetrate into the gap in the rough substrate. High density is observed at the bottom of the column or on the upper surface of the column arrays. The contact state is transitioning from Wenzel state to Cassie–Baxter state with the decrease of void fraction. The interaction energy increases with the increased interaction area.
- (3)
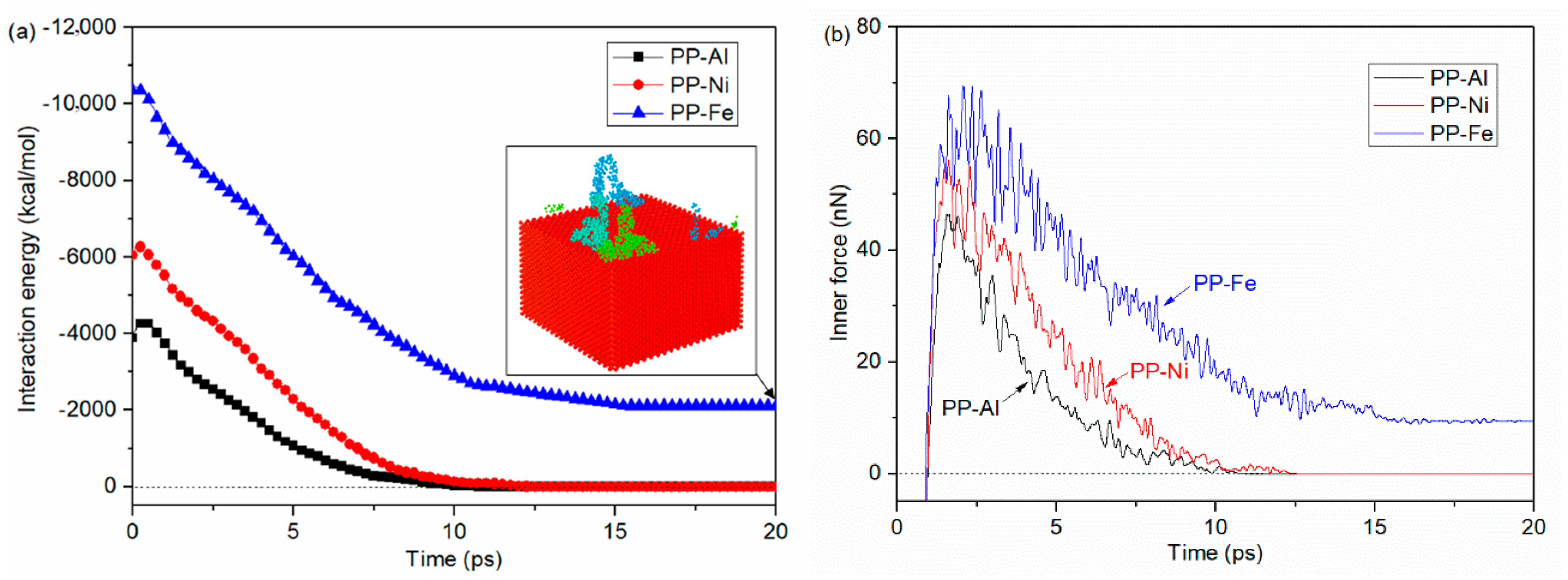
- Maximum densities in PP-Al model, PP-Ni model and PP-Fe model are found at the interface, indicating the molecules are adsorbed on the surface. From perspective of interfacial interaction, the PP-Fe hybrid system have a higher potential for the tight joining in direct molding process.
- (4)
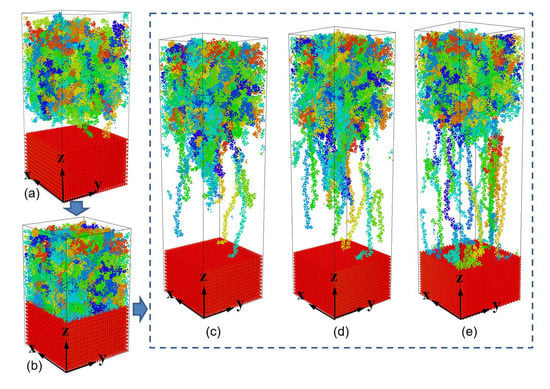
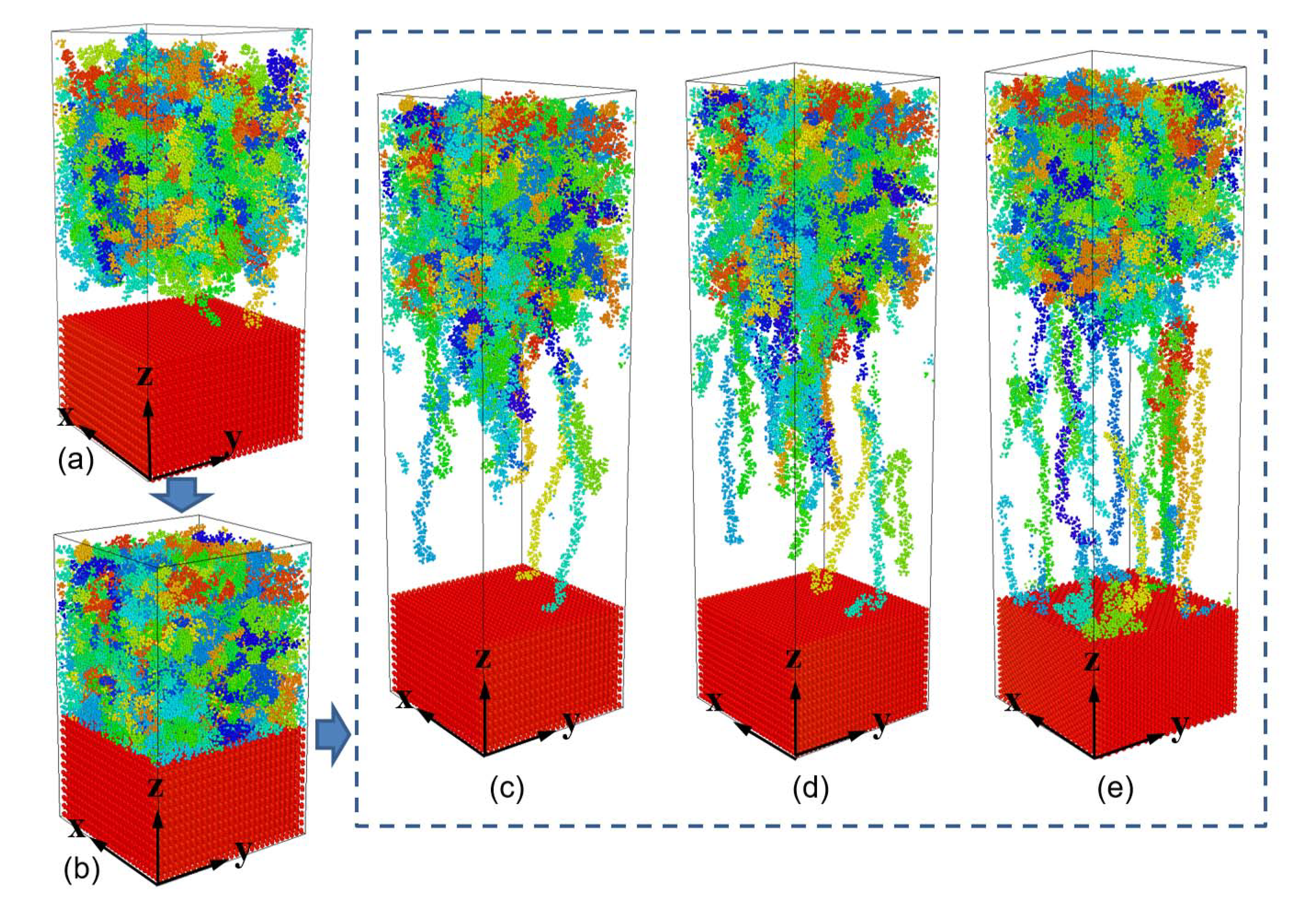
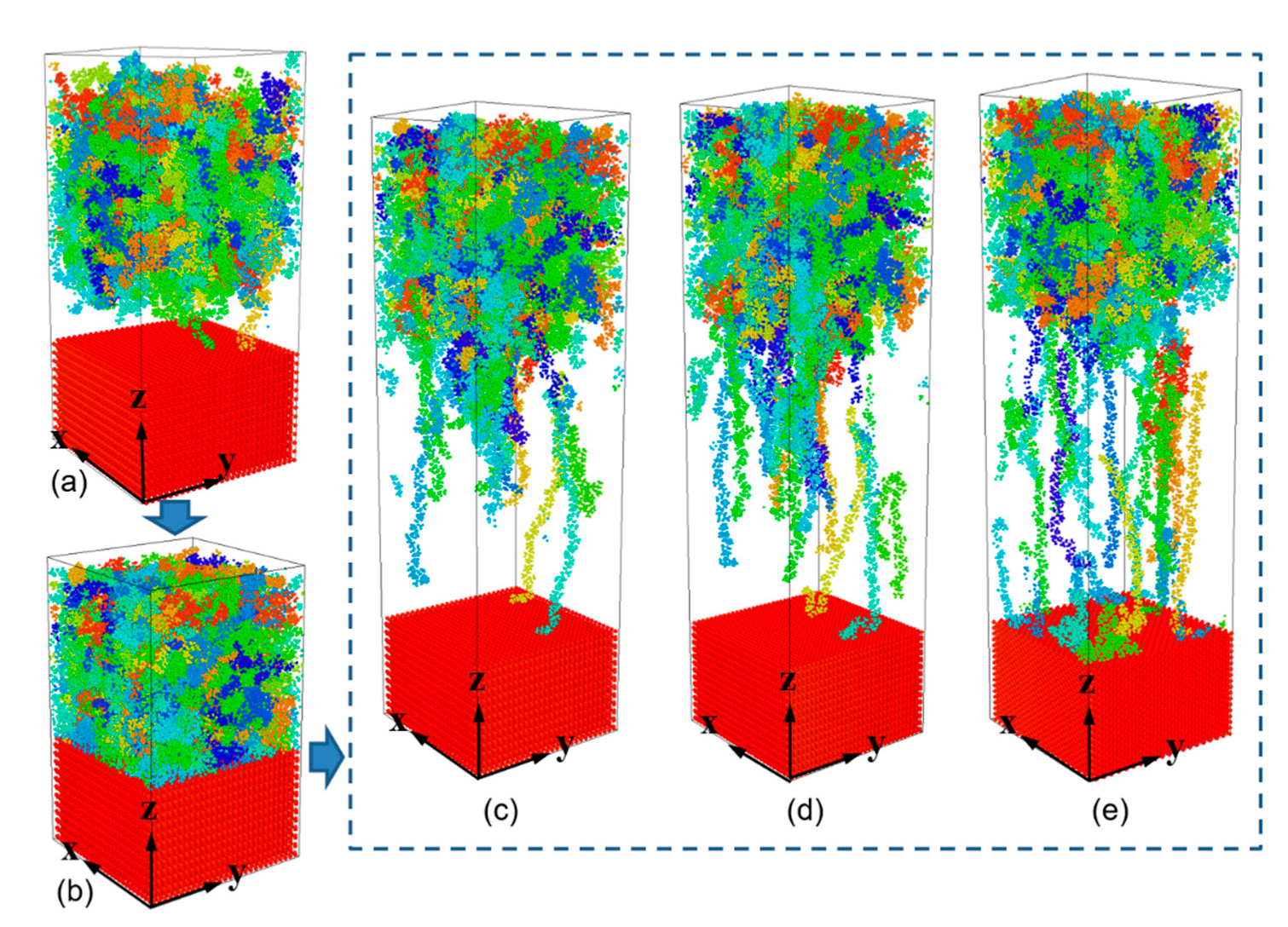
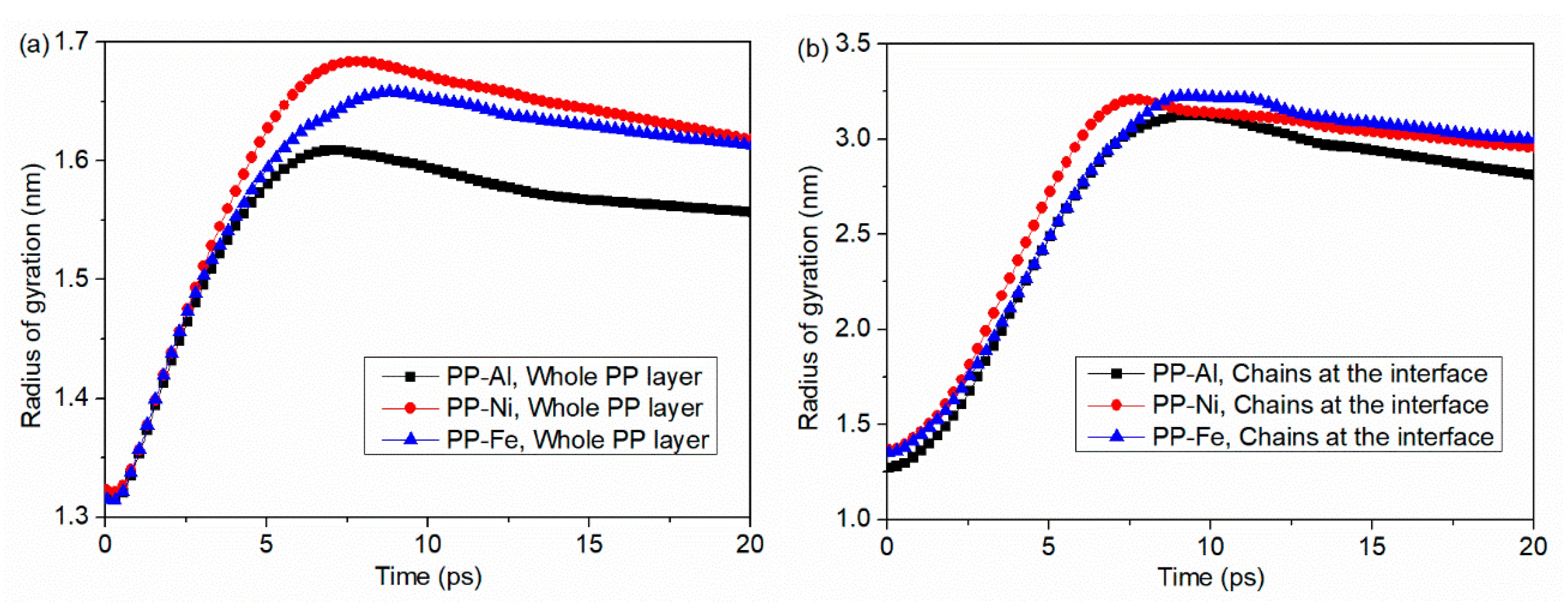
- During the separation process, molecule chains near the interface are greatly stretched, with the highly increased radius of gyration. While molecule chains far away from the interface show little conformation changes. For both PP-Al and PP-Ni hybrid structures, the PP layers are totally separated from the substrates finally. For PP-Fe hybrid structure, the majority of the chains are pulled out from the substrate, while some still remain close to the surface. The inner force of PP-Fe hybrid structure during the separation is obviously higher than other cases.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grujicic, M.; Sellappan, V.; Omar, M.A.; Seyr, N.; Obieglo, A.; Erdmann, M.; Holzleitner, J. An overview of the polymer-to-metal direct-adhesion hybrid technologies for load-bearing automotive components. J. Mater. Process. Technol. 2008, 197, 363–373. [Google Scholar] [CrossRef]
- Grujicic, M. Injection over molding of polymer-metal hybrid structures. Am. J. Sci. Technol. 2014, 1, 168–181. [Google Scholar] [CrossRef]
- Kimura, F.; Kadoya, S.; Kajihara, Y. Effects of molding conditions on injection molded direct joining using a metal with nano-structured surface. Precis. Eng. 2016, 45, 203–208. [Google Scholar] [CrossRef]
- Kleffel, T.; Drummer, D. Investigating the suitability of roughness parameters to assess the bond strength of polymer-metal hybrid structures with mechanical adhesion. Compos. Part B Eng. 2017, 117, 20–25. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, J.; Zhou, T. Large-area mechanical interlocking via nanopores: Ultra-high-strength direct bonding of polymer and metal materials. Appl. Surf. Sci. 2019, 492, 558–570. [Google Scholar] [CrossRef]
- Li, X.; Xu, D.; Gong, N.; Xu, Z.; Wang, L.; Dong, W. Improving the strength of injection molded aluminum/polyphenylene sulfide lap joints dependence on surface microstructure and composition. Mater. Des. 2019, 179, 107875. [Google Scholar] [CrossRef]
- Loaldi, D.; Regi, F.; Baruffi, F.; Calaon, M.; Quagliotti, D.; Zhang, Y.; Tosello, G. Experimental validation of injection molding simulations of 3D microparts and microstructured components using virtual design of experiments and multi-scale modeling. Micromachines 2020, 11, 614. [Google Scholar] [CrossRef]
- Zhao, S.; Kimura, F.; Kadoya, S.; Kajihara, Y. Experimental analysis on mechanical interlocking of metal–polymer direct joining. Precis. Eng. 2020, 61, 120–125. [Google Scholar] [CrossRef]
- Liparoti, S.; Pantani, R.; Sorrentino, A.; Speranza, V.; Titomanlio, G. Hydrophobicity tuning by the fast evolution of mold temperature during injection molding. Polymers 2018, 10, 322. [Google Scholar] [CrossRef]
- Jiao, J.; Jia, S.; Xu, Z.; Ye, Y.; Sheng, L.; Zhang, W. Laser direct joining of CFRTP and aluminium alloy with a hybrid surface pre-treating method. Compos. Part B Eng. 2019, 173, 106911. [Google Scholar] [CrossRef]
- Xu, D.; Yang, W.; Li, X.; Hu, Z.; Li, M.; Wang, L. Surface nanostructure and wettability inducing high bonding strength of polyphenylene sulfide-aluminum composite structure. Appl. Surf. Sci. 2020, 515, 145996. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, L.; Li, L.; Huang, B.; Wang, T.; Wang, Y. Direct injection molding and mechanical properties of high strength steel/composite hybrids. Compos. Struct. 2019, 210, 70–81. [Google Scholar] [CrossRef]
- Li, Y.; Meng, S.; Gong, Q.; Huang, Y.; Gan, J.; Zhao, M.; Liu, B.; Liu, L.; Zou, G.; Zhuang, D. Experimental and theoretical investigation of laser pretreatment on strengthening the heterojunction between carbon fiber-reinforced plastic and aluminum alloy. ACS Appl. Mater. Interfaces 2019, 11, 22005–22014. [Google Scholar] [CrossRef] [PubMed]
- Amancio-Filho, S.T.; dos Santos, J.F. Joining of polymers and polymer-metal hybrid structures: Recent developments and trends. Polym. Eng. Sci. 2009, 49, 1461–1476. [Google Scholar] [CrossRef]
- Li, X.; Gong, N.; Yang, C.; Zeng, S.; Fu, S.; Zhang, K. Aluminum/polypropylene composites produced through injection molding. J. Mater. Process. Technol. 2018, 255, 635–643. [Google Scholar] [CrossRef]
- Zaenudin, M.; Mohammed, M.N.; Al-Zubaidi, S. Molecular dynamics simulation of welding and joining processes: An overview. Int. J. Eng. Technol. 2018, 7, 3816–3825. [Google Scholar] [CrossRef]
- Mori, H.; Matubayasi, N. Resin filling into nano-sized pore on metal surface analyzed by all-atom molecular dynamics simulation over a variety of resin and pore sizes. Polymer 2018, 150, 360–370. [Google Scholar] [CrossRef]
- Zhou, M.; Xiong, X.; Drummer, D.; Jiang, B. Molecular dynamics simulation on the effect of bonding pressure on thermal bonding of polymer microfluidic chip. Polymers 2019, 11, 557. [Google Scholar] [CrossRef]
- Yang, J.; Weng, C.; Lai, J.; Ding, T.; Wang, H. Molecular dynamics simulation on the influences of nanostructure shape, interfacial adhesion energy, and mold insert material on the demolding process of micro-injection molding. Polymers 2019, 11, 1573. [Google Scholar] [CrossRef]
- Moyassari, A.; Gkourmpis, T.; Hedenqvist, M.S.; Gedde, U.W. Molecular dynamics simulations of short-chain branched bimodal polyethylene: Topological characteristics and mechanical behavior. Macromolecules 2019, 52, 807–818. [Google Scholar] [CrossRef]
- Moyassari, A.; Gkourmpis, T.; Hedenqvist, M.S.; Gedde, U.W. Molecular dynamics simulation of linear polyethylene blends: Effect of molar mass bimodality on topological characteristics and mechanical behavior. Polymer 2019, 161, 139–150. [Google Scholar] [CrossRef]
- Rout, A.; Pandey, P.; Oliveira, E.F.; da Silva Autreto, P.A.; Gumaste, A.; Singh, A.; Galvão, D.S.; Arora, A.; Tiwary, C.S. Atomically locked interfaces of metal (Aluminum) and polymer (Polypropylene) using mechanical friction. Polymer 2019, 169, 148–153. [Google Scholar] [CrossRef]
- Yin, Y.; Li, W.; Shen, H.; Zhou, J.; Nan, H.; Deng, M.; Shen, X.; Tu, Z. Molecular dynamics simulations of iron/graphite interfacial behaviors: Influence of oxygen. ISIJ Int. 2018, 58, 1022–1027. [Google Scholar] [CrossRef]
- Surace, R.; Sorgato, M.; Bellantone, V.; Modica, F.; Lucchetta, G.; Fassi, I. Effect of cavity surface roughness and wettability on the filling flow in micro injection molding. J. Manuf. Process. 2019, 43, 105–111. [Google Scholar] [CrossRef]
- Khalkhali, M.; Kazemi, N.; Zhang, H.; Liu, Q.X. Wetting at the nanoscale: A molecular dynamics study. J. Chem. Phys. 2017, 146, 114704. [Google Scholar] [CrossRef]
- Heine, D.R.; Grest, G.S.; Webb, E.B., 3rd. Spreading dynamics of polymer nanodroplets. Phys. Rev. E 2003, 68, 061603. [Google Scholar] [CrossRef]
- Yaneva, J.; Milchev, A.; Binder, K. Polymer droplets on substrates with striped surface domains: Molecular dynamics simulations of equilibrium structure and liquid bridge rupture. J. Phys. Condens. Matter 2005, 17, S4199–S4211. [Google Scholar] [CrossRef]
- Cao, Z.; Dobrynin, A.V. Polymeric droplets on soft surfaces: From Neumann’s triangle to Young’s law. Macromolecules 2015, 48, 443–451. [Google Scholar] [CrossRef]
- Zhou, M.; Xiong, X.; Drummer, D.; Jiang, B. Interfacial interaction and joining property of direct injection-molded polymer-metal hybrid structures: A molecular dynamics simulation study. Appl. Surf. Sci. 2019, 478, 680–689. [Google Scholar] [CrossRef]
- Hosseini, S.; Savaloni, H.; Gholipour Shahraki, M. Influence of surface morphology and nano-structure on hydrophobicity: A molecular dynamics approach. Appl. Surf. Sci. 2019, 485, 536–546. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Boruvka, L.; Neumann, A.W. Generalization of the classical theory of capillarity. J. Chem. Phys. 1977, 66, 5464–5476. [Google Scholar] [CrossRef]
- Zitzenbacher, G.; Dirnberger, H.; Längauer, M.; Holzer, C. Calculation of the contact angle of polymer melts on tool surfaces from viscosity parameters. Polymers 2017, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Rytka, C.; Opara, N.; Andersen, N.K.; Kristiansen, P.M.; Neyer, A. On The role of wetting, structure width, and flow characteristics in polymer replication on micro- and nanoscale. Macromol Mater. Eng. 2016, 301, 597–609. [Google Scholar] [CrossRef]
- Liu, D.; Zhou, F.; Zhou, H. The polymer-metal interactive behavior in polyphenylene sulfide/aluminium hetero interface in nano injection molding. Compos. Interfaces 2019, 27, 1–12. [Google Scholar] [CrossRef]
- Awasthi, A.P.; Lagoudas, D.C.; Hammerand, D.C. Modeling of graphene–polymer interfacial mechanical behavior using molecular dynamics. Model. Simul. Mater. Sci. Eng. 2009, 17, 015002. [Google Scholar] [CrossRef]
- Takai, R.; Yasuda, M.; Tochino, T.; Kawata, H.; Hirai, Y. Computational study of the demolding process in nanoimprint lithography. J. Vac. Sci. Technol. B 2014, 32, 06FG02. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Degree of Polymerization | Number of Chains | Total Amount of Atoms | Initial Density (g/cm3) | Model Size (nm) |
|---|---|---|---|---|---|
| PP | 50 | 120 | 54,240 | 0.85 | 8.0 × 8.0 × 8.0 |
| Substrate Structure | Side Length (nm) | Height (nm) | Distance (nm) | Total Amount of Atoms | Model Size (nm) |
|---|---|---|---|---|---|
| Case P0 | / | / | / | 39,764 | 20.0 × 20.0 × 1.4 |
| Case P1 | 0.40 | 2.02 | 1.72 | 48,532 | 20.0 × 20.0 × 3.4 |
| Case P2 | 0.64 | 53,572 | |||
| Case P3 | 0.81 | 60,052 | |||
| Case P4 | 1.21 | 76,012 |
| Substrate Structure | Side Length (nm) | Void Fraction | Roughness Ratio | Interaction Energy (kcal/mol) |
|---|---|---|---|---|
| Case P1 | 0.40 | 94.6% | 2.09 | –11,234.2 |
| Case P2 | 0.64 | 86.2% | 2.75 | –12,668.1 |
| Case P3 | 0.81 | 77.8% | 3.21 | –12,702.0 |
| Case P4 | 1.21 | 50.5% | 4.30 | –15,009.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, M.; Fu, L.; Jiang, F.; Jiang, B.; Drummer, D. Atomistic Investigation on the Wetting Behavior and Interfacial Joining of Polymer-Metal Interface. Polymers 2020, 12, 1696. https://doi.org/10.3390/polym12081696
Zhou M, Fu L, Jiang F, Jiang B, Drummer D. Atomistic Investigation on the Wetting Behavior and Interfacial Joining of Polymer-Metal Interface. Polymers. 2020; 12(8):1696. https://doi.org/10.3390/polym12081696
Chicago/Turabian StyleZhou, Mingyong, Liang Fu, Fengze Jiang, Bingyan Jiang, and Dietmar Drummer. 2020. "Atomistic Investigation on the Wetting Behavior and Interfacial Joining of Polymer-Metal Interface" Polymers 12, no. 8: 1696. https://doi.org/10.3390/polym12081696
APA StyleZhou, M., Fu, L., Jiang, F., Jiang, B., & Drummer, D. (2020). Atomistic Investigation on the Wetting Behavior and Interfacial Joining of Polymer-Metal Interface. Polymers, 12(8), 1696. https://doi.org/10.3390/polym12081696