Degradation Behavior of Polypropylene during Reprocessing and Its Biocomposites: Thermal and Oxidative Degradation Kinetics




Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Methods
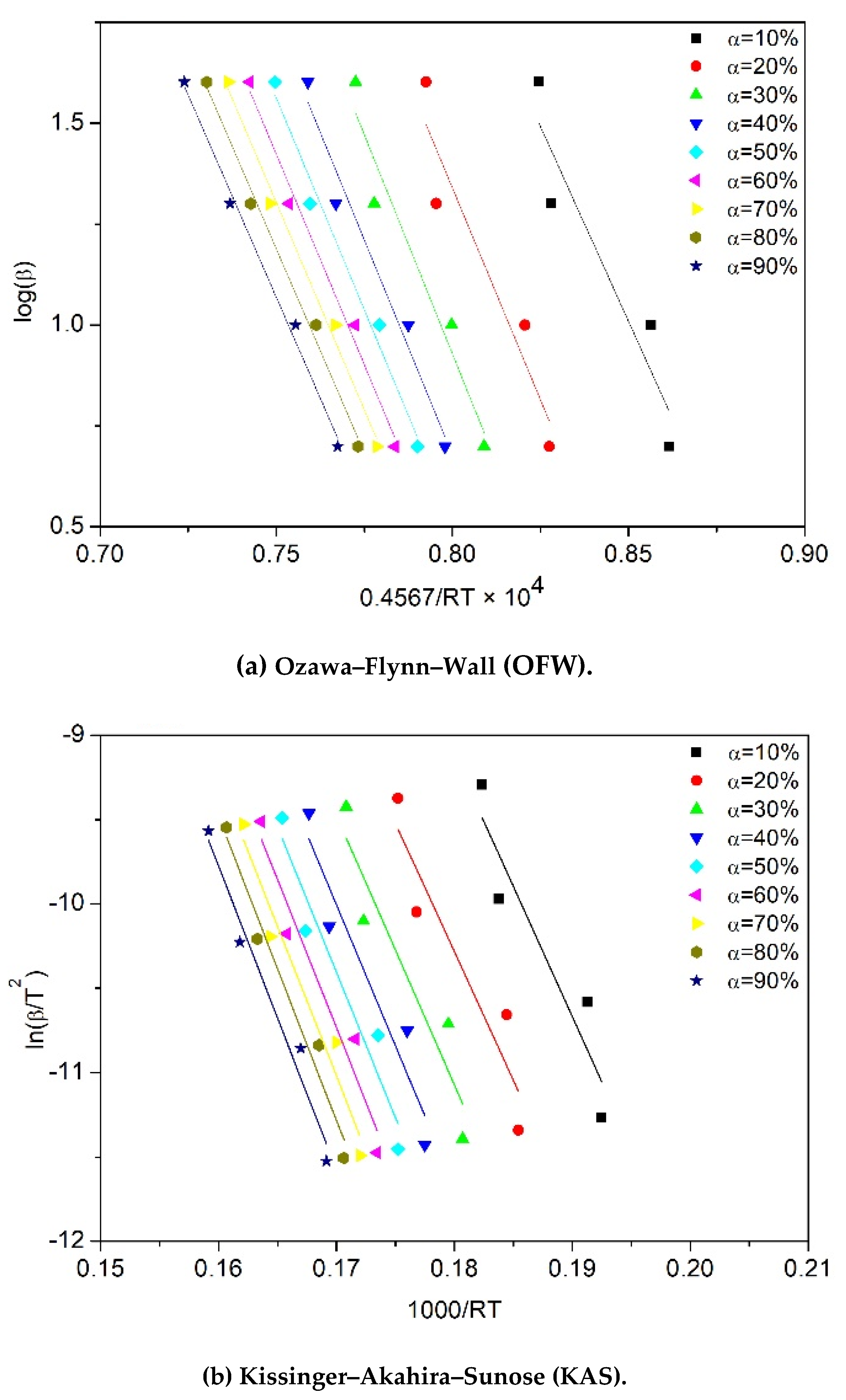
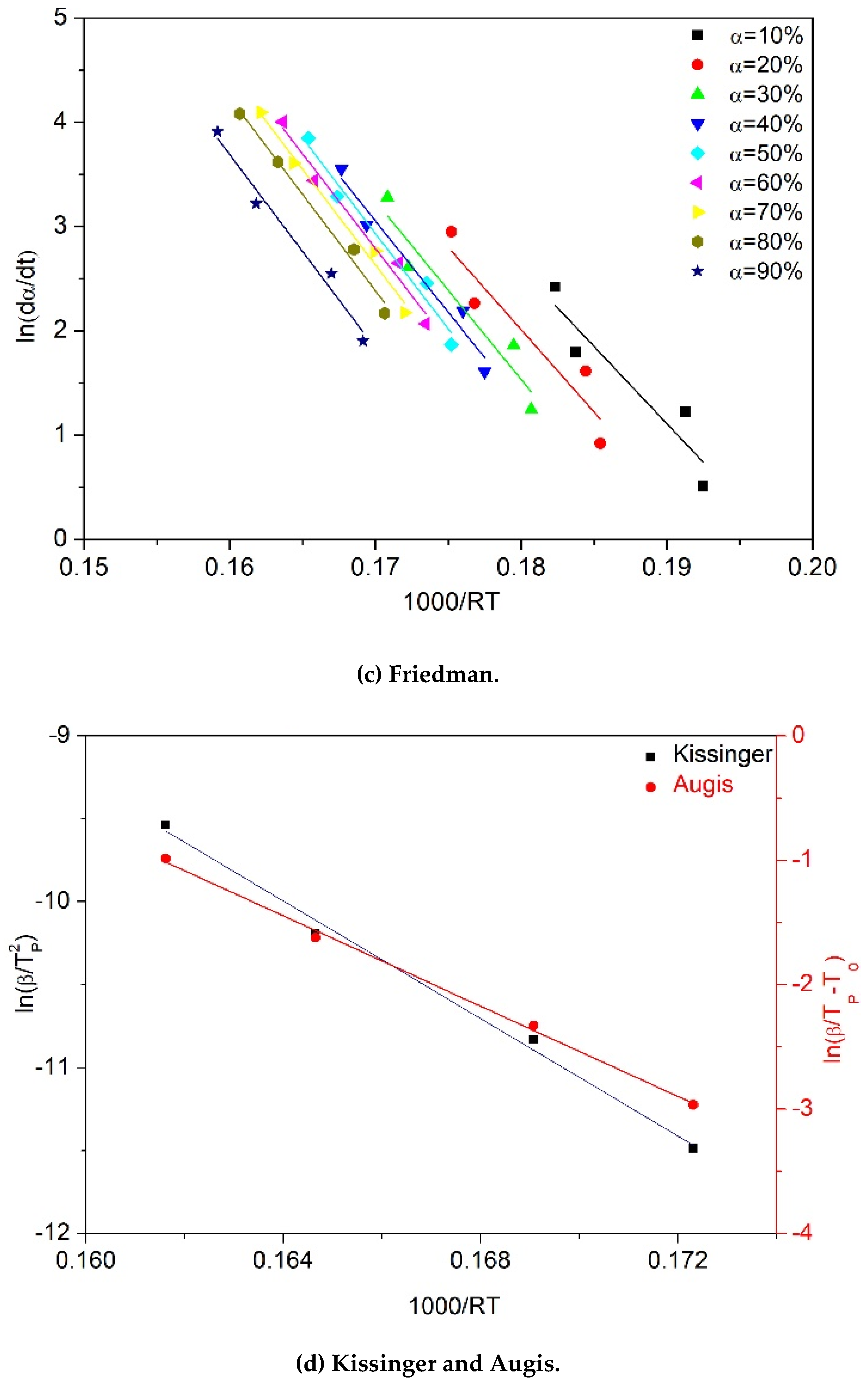
2.3. Theoretical Background
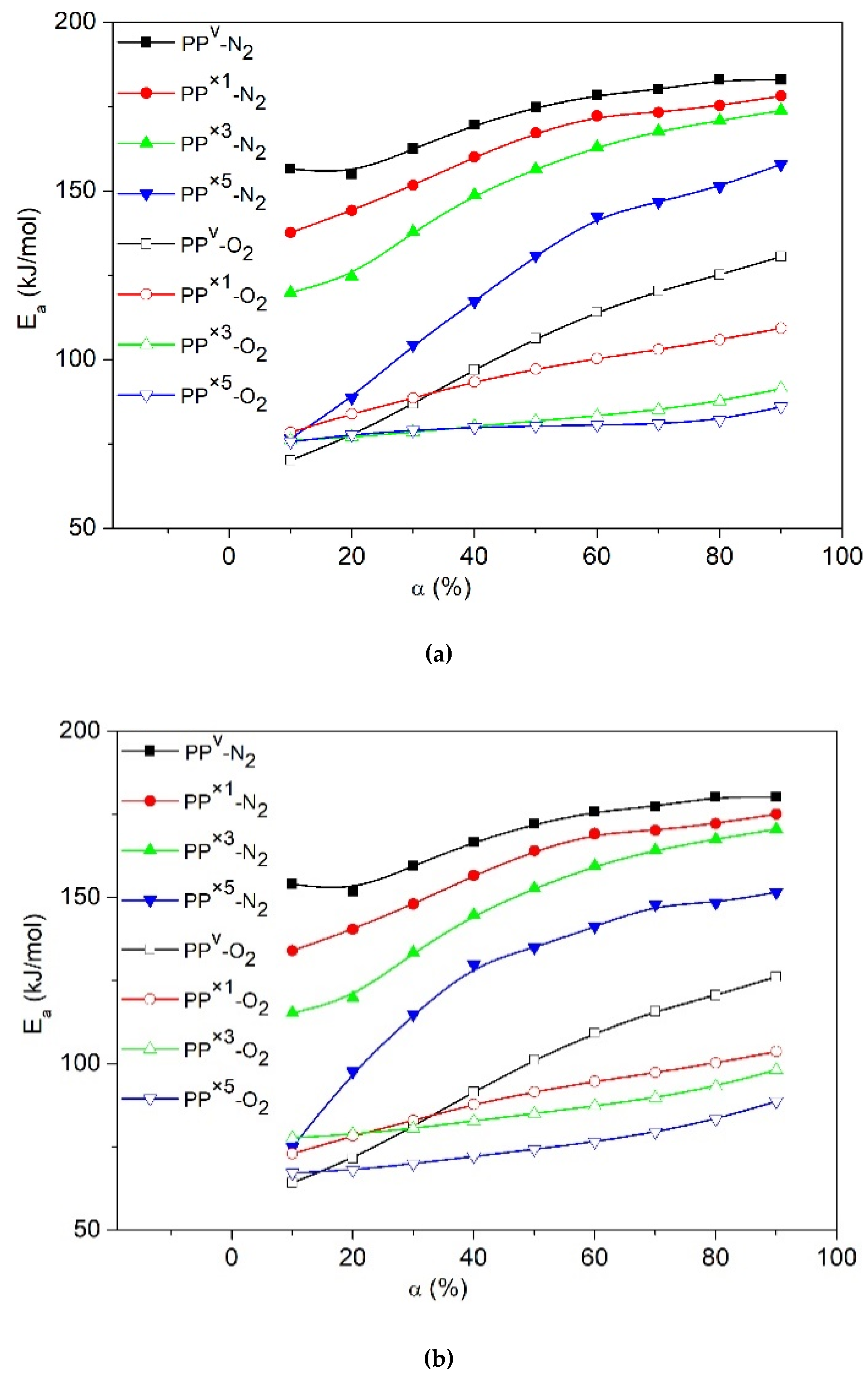
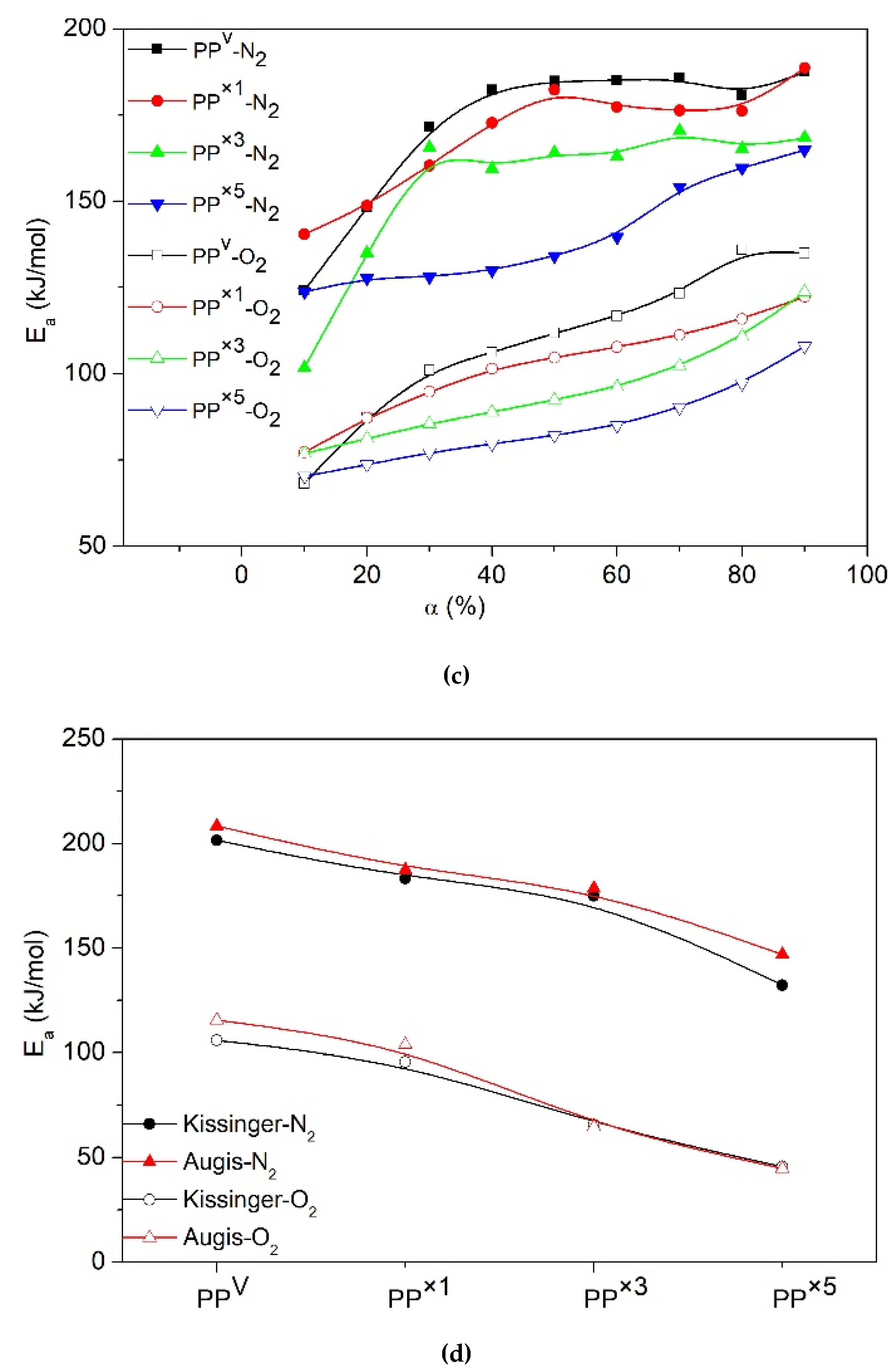
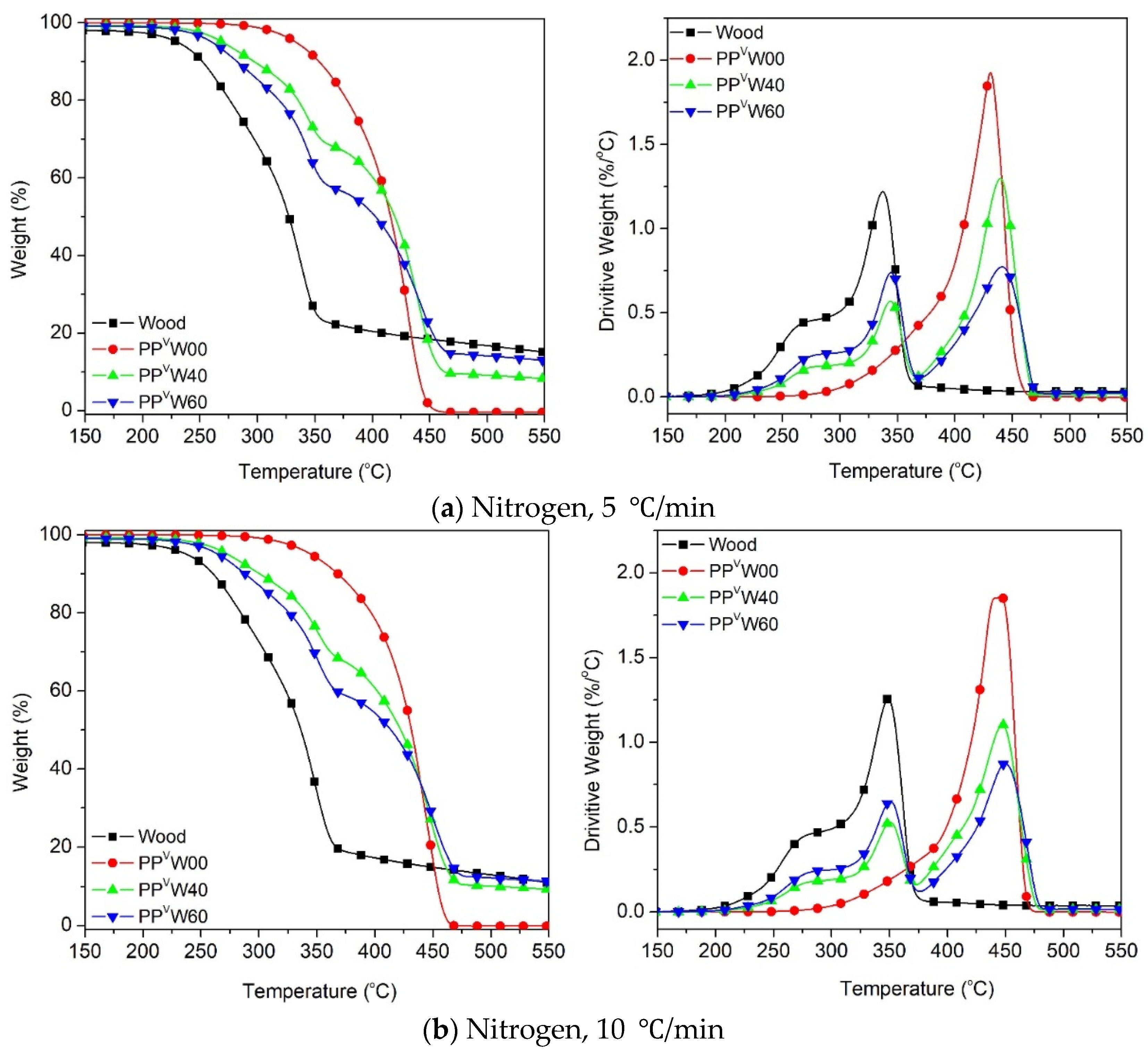
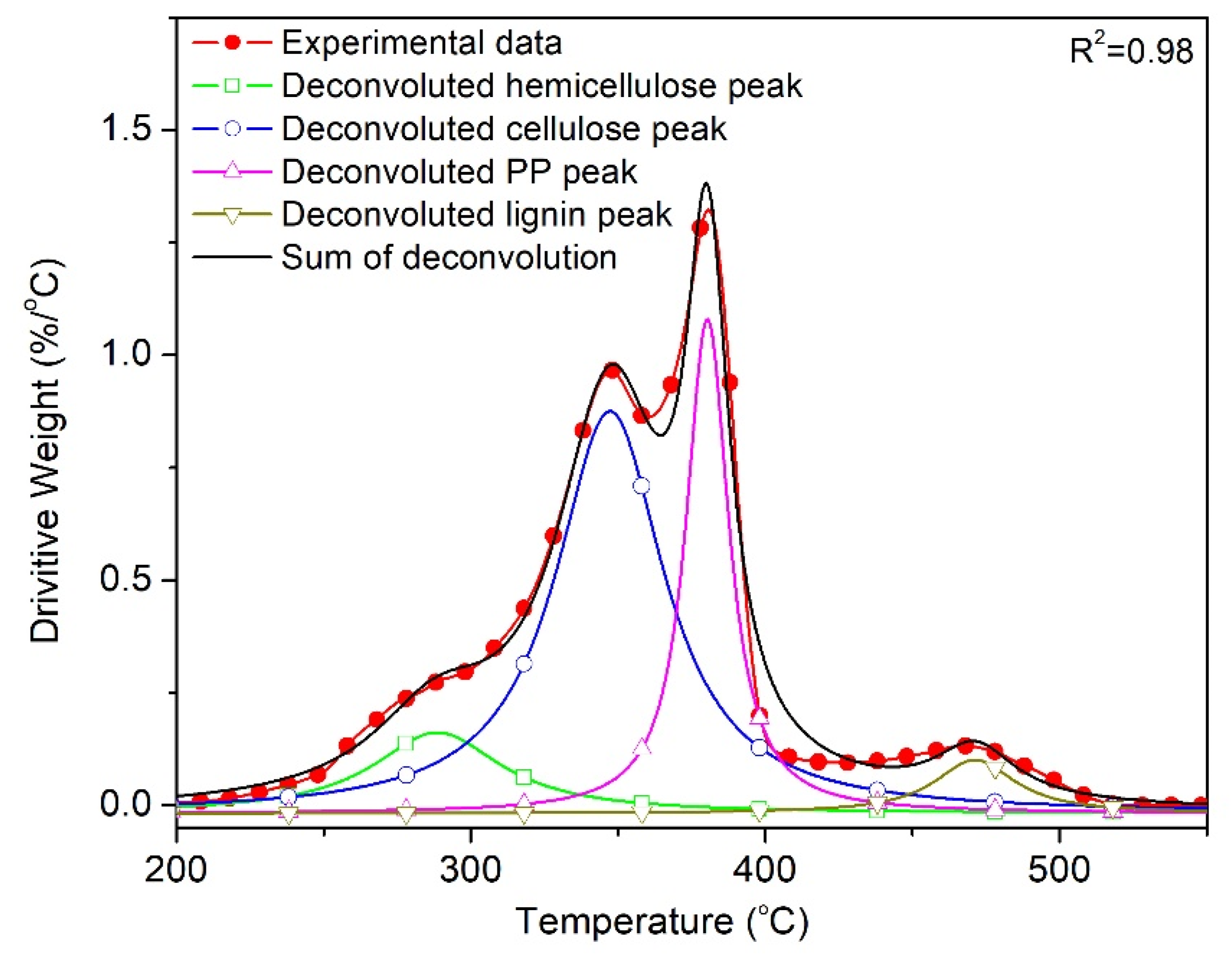
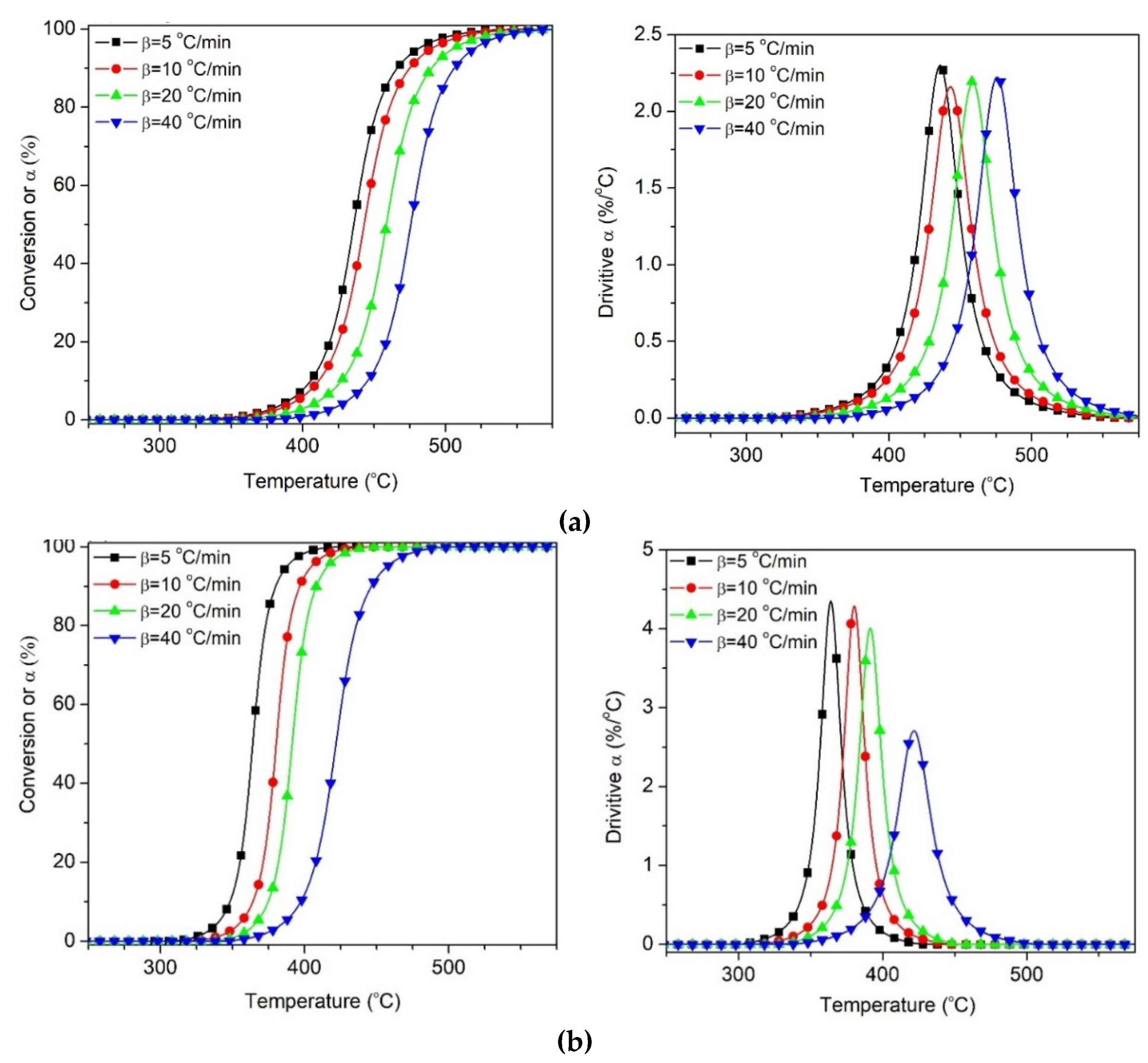
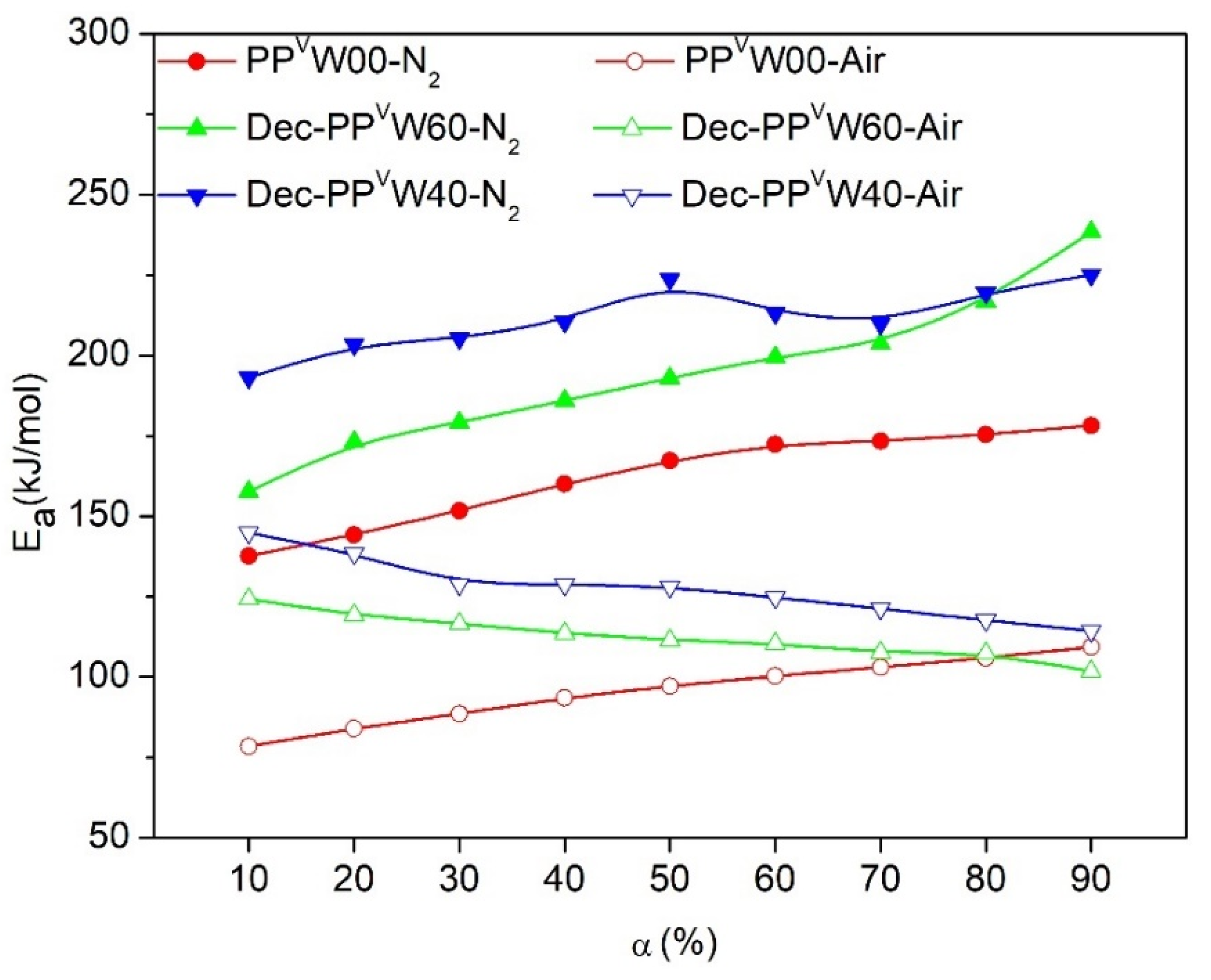
3. Results and Discussion
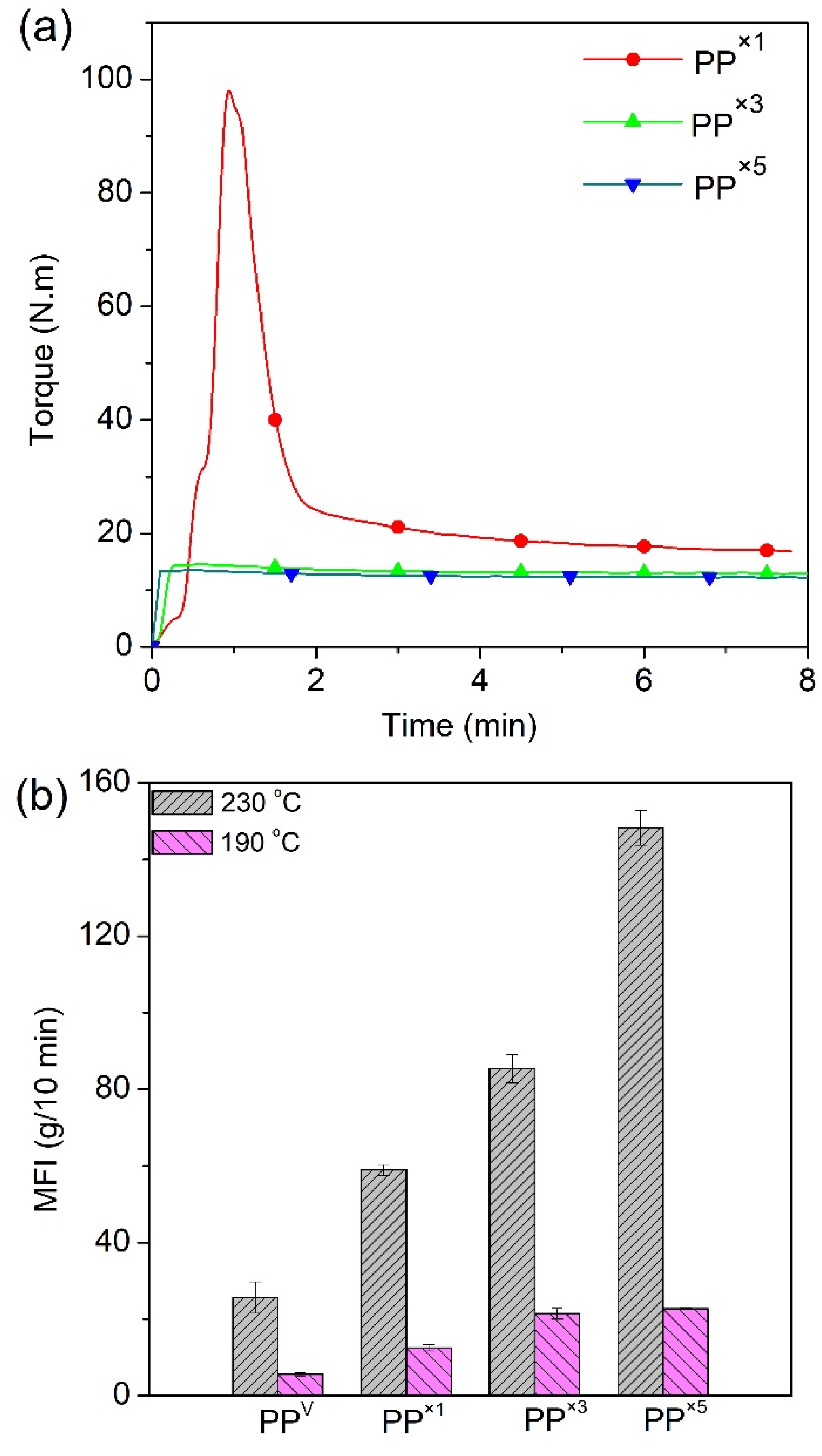
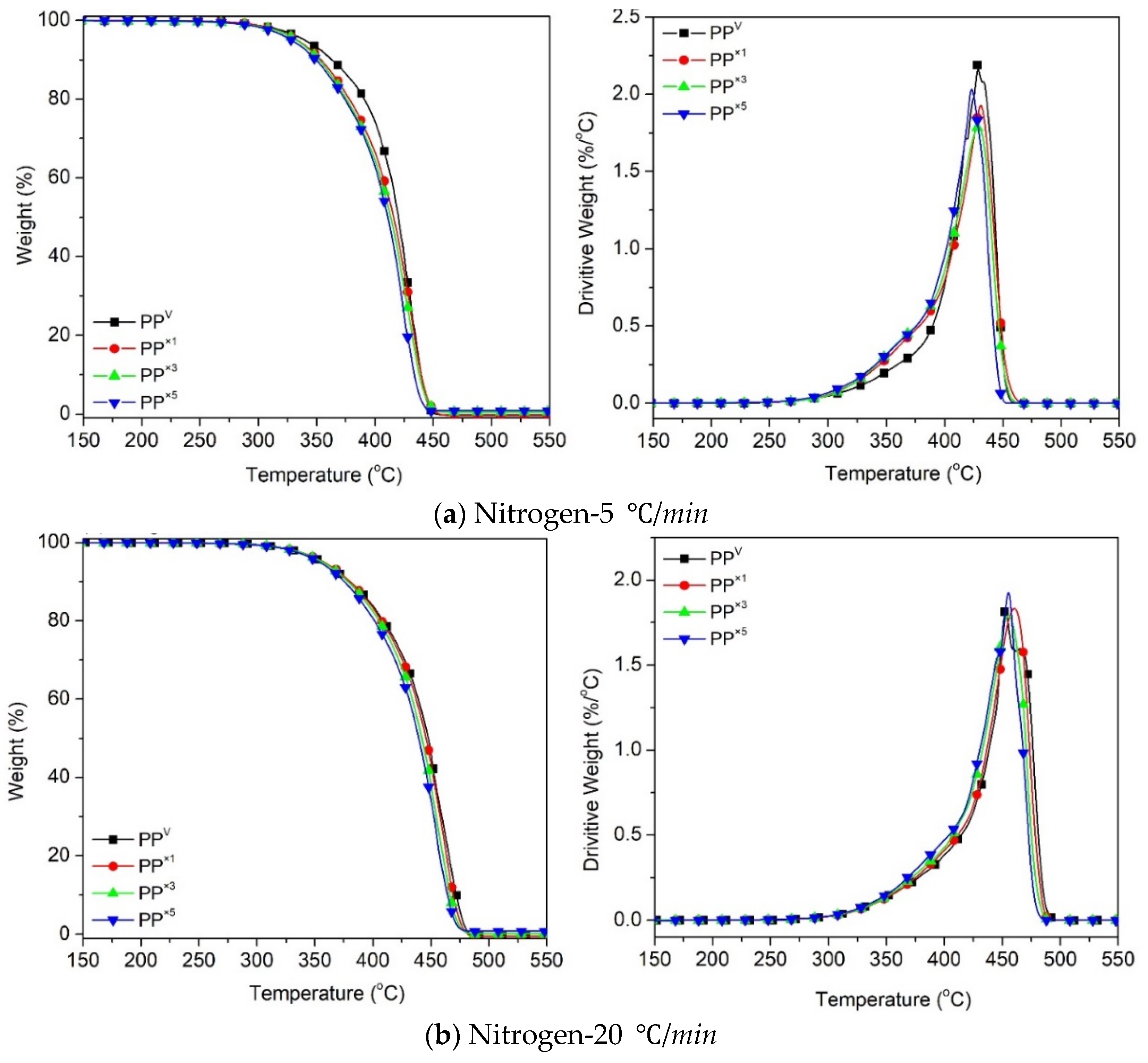
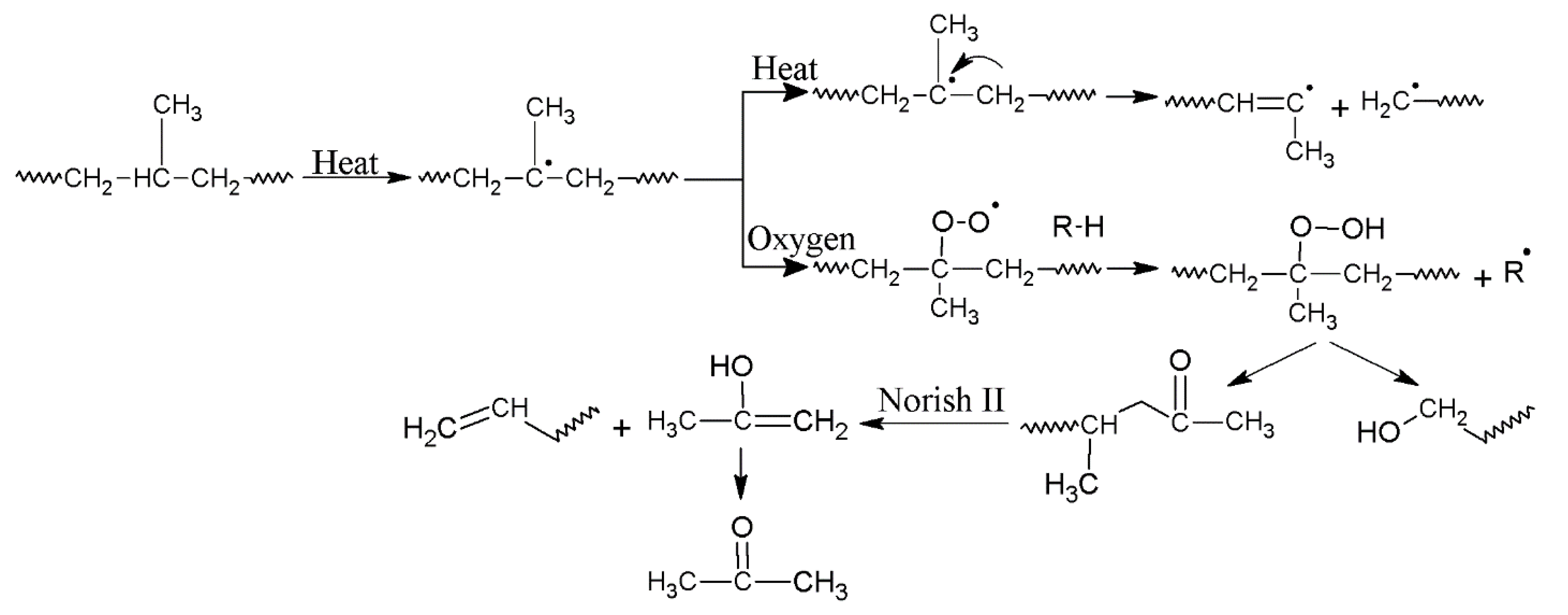
3.1. Effect of Reprocessing Cycles
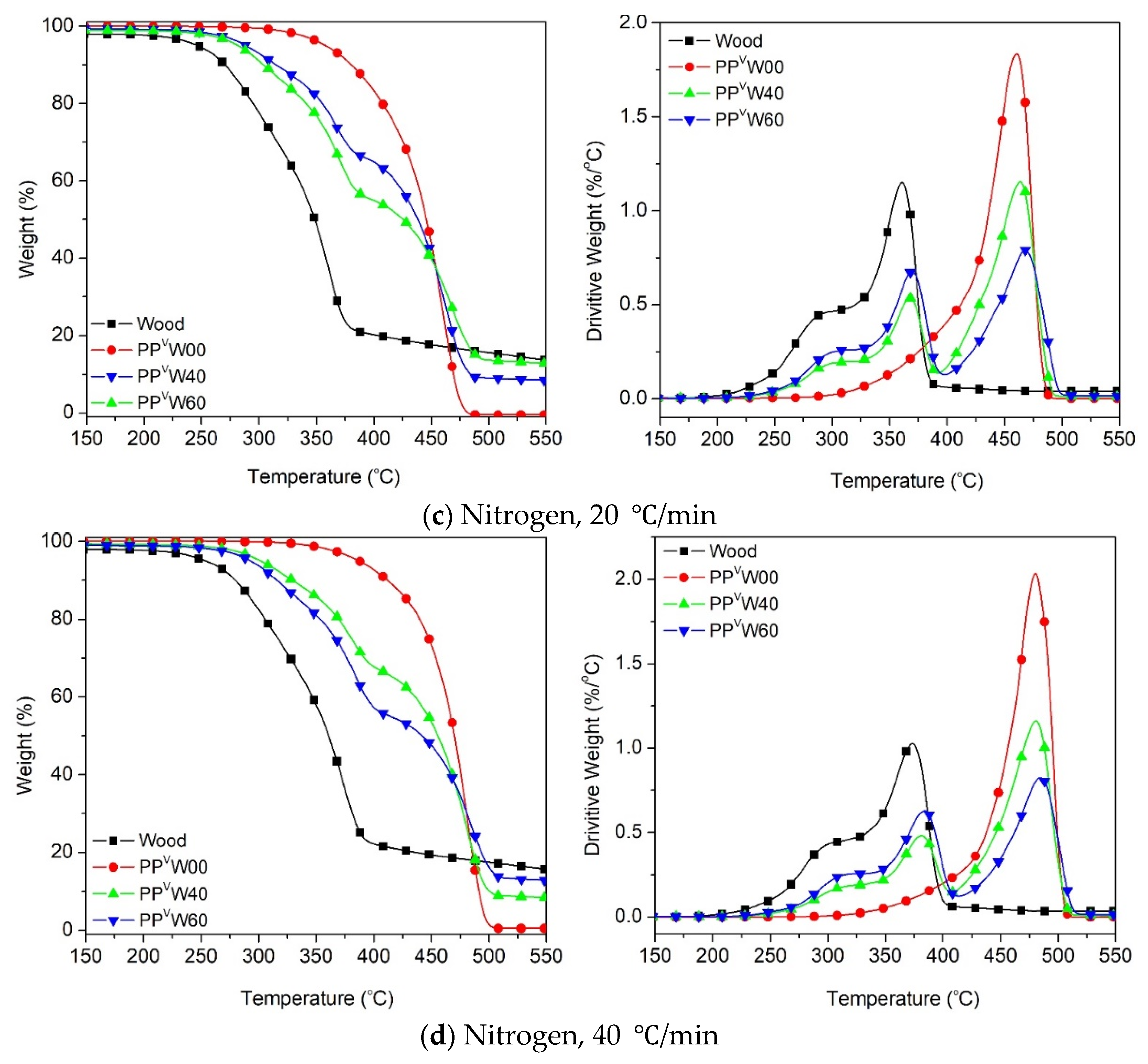
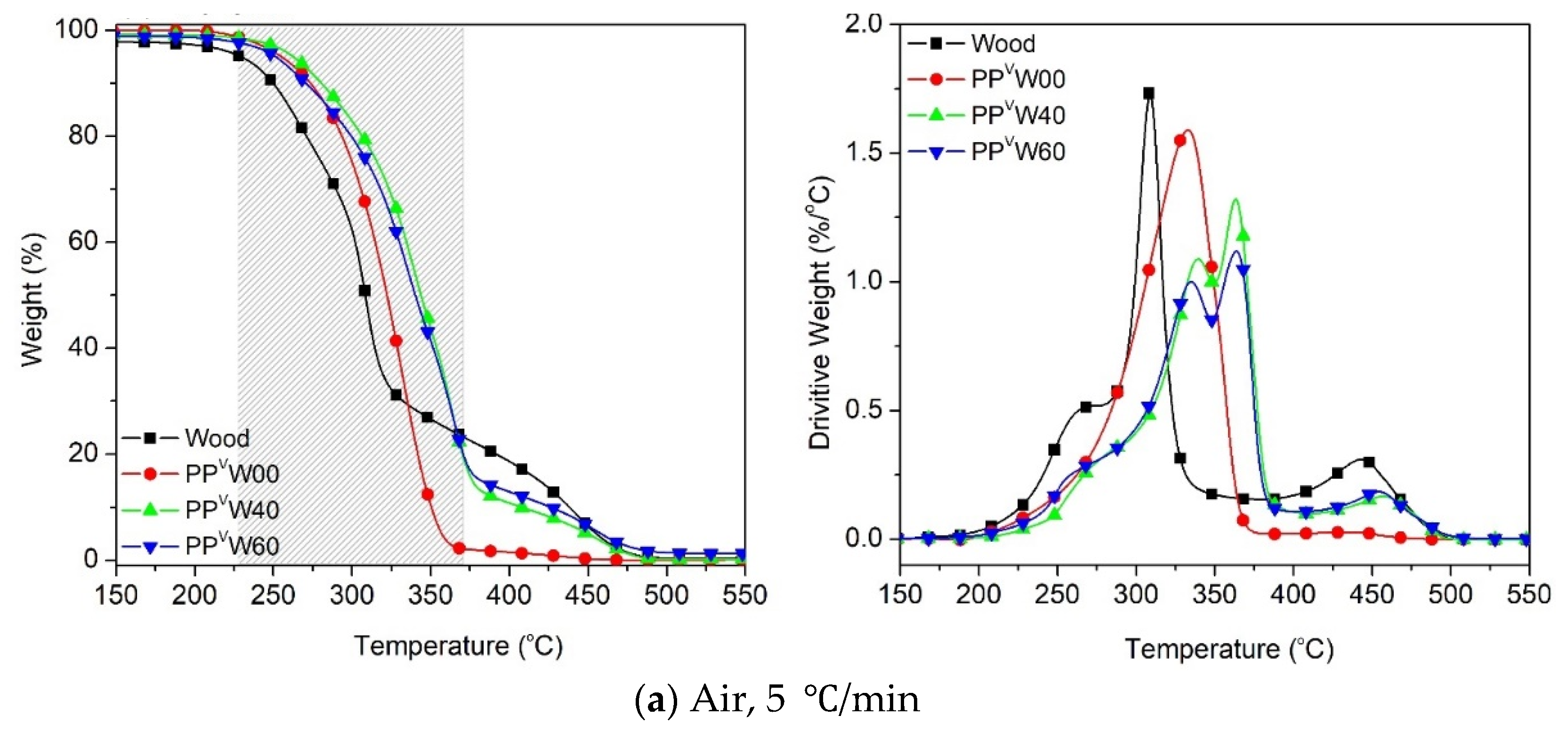
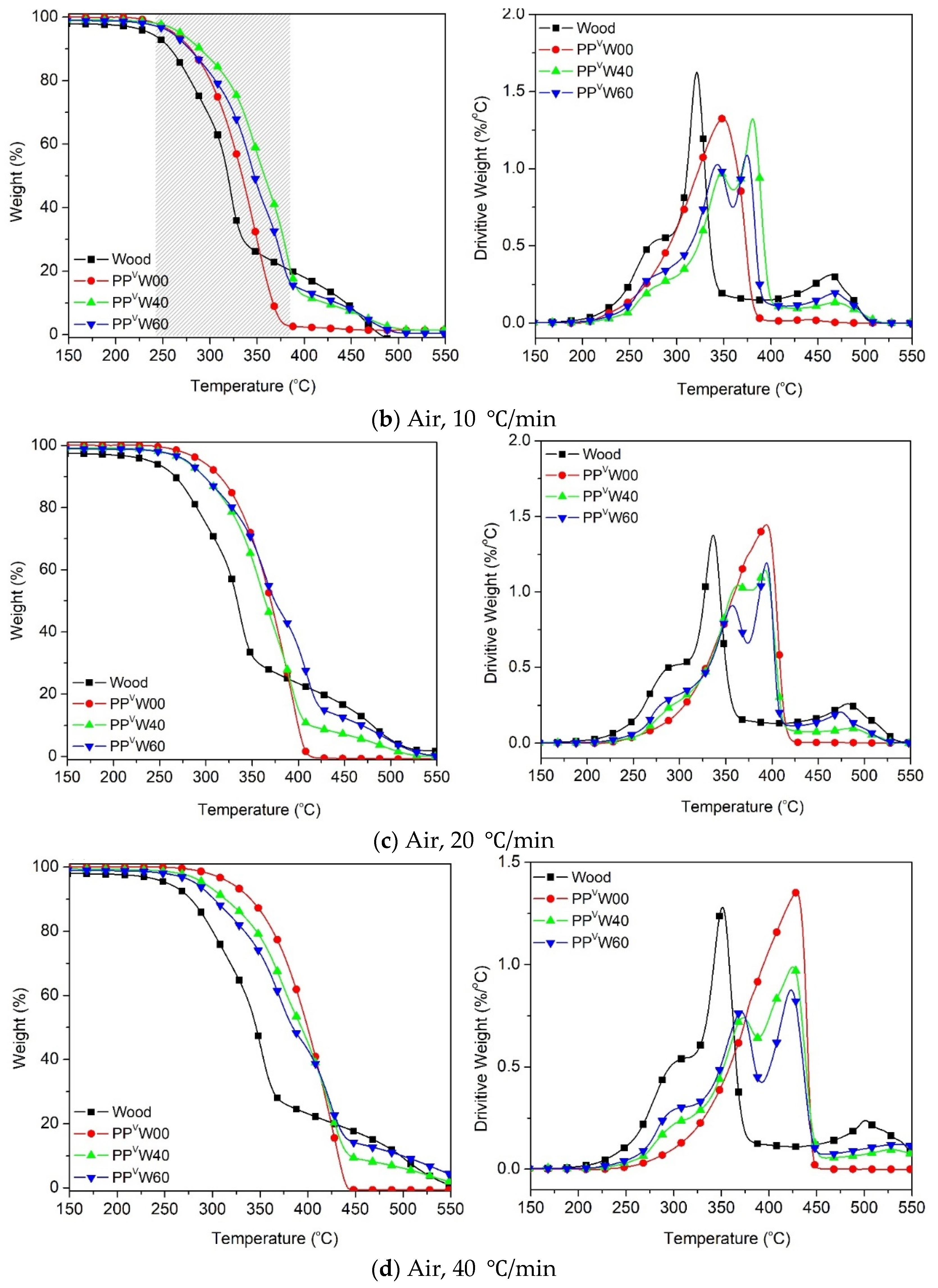
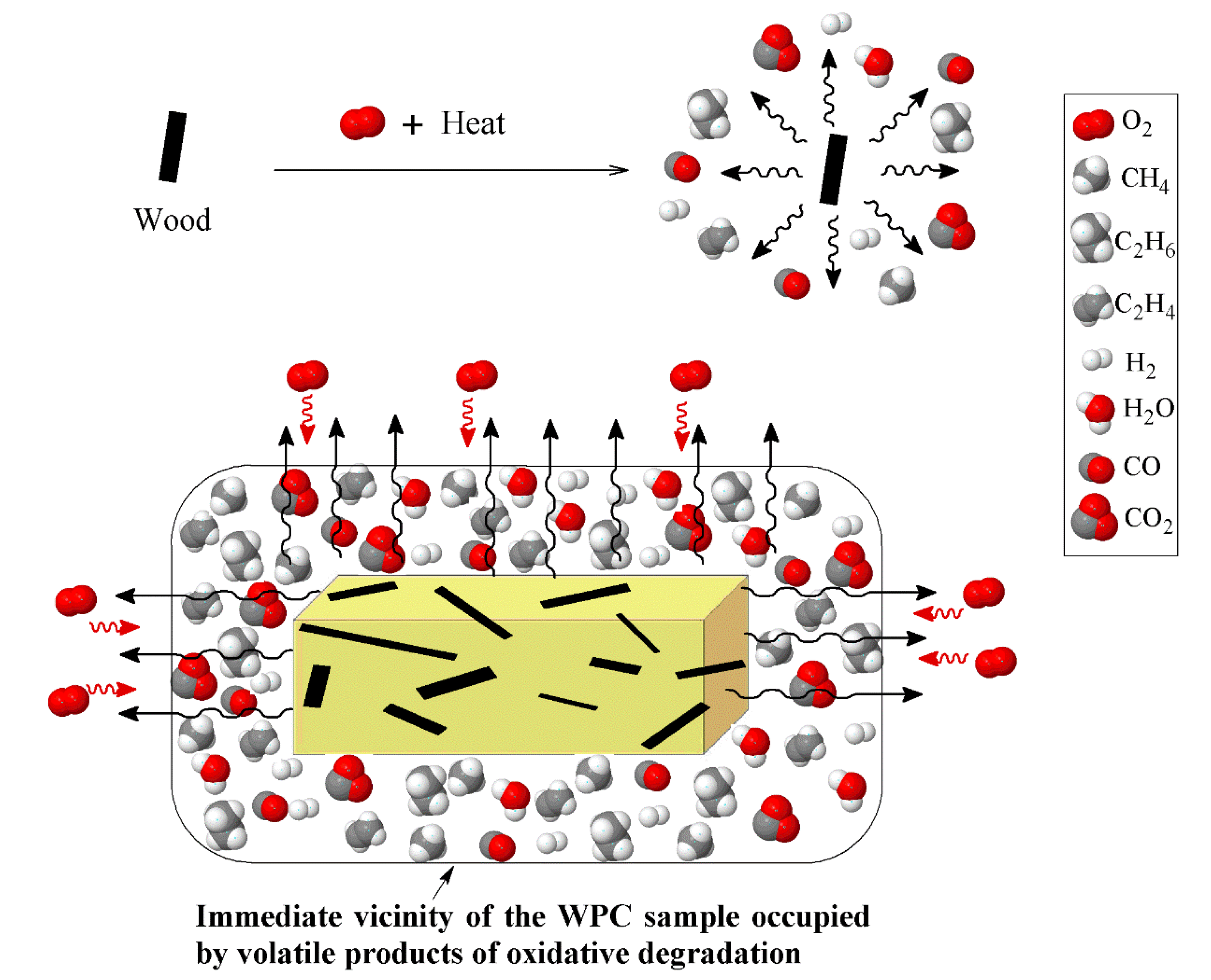
3.2. Effect of Wood
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Al-Salem, S.M.; Lettieri, P.; Baeyens, J. Recycling and recovery routes of plastic solid waste (PSW): A review. Waste Manag. 2009, 29, 2625–2643. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Tuladhar, R.; Shi, F.; Shanks, R.A.; Combe, M. Collister, Mechanical reprocessing of polyolefin waste: A review. Polym. Eng. Sci. 2015, 55, 2899–2909. [Google Scholar] [CrossRef]
- Wang, K.; Addiego, F.; Bahlouli, N.; Ahzi, S.; Rémond, Y.; Toniazzo, V.; Muller, R. Analysis of thermomechanical reprocessing effects on polypropylene/ethylene octene copolymer blends. Polym. Degrad. Stab. 2012, 97, 1475–1484. [Google Scholar] [CrossRef]
- Martín-Alfonso, J.E.; Franco, J.M. Influence of polymer reprocessing cycles on the microstructure and rheological behavior of polypropylene/mineral oil oleogels. Polym. Test. 2015, 45, 12–19. [Google Scholar] [CrossRef]
- Kurniawan, D.; Kim, B.S.; Lee, H.Y.; Lim, J.Y. Effects of repetitive processing, wood content, and coupling agent on the mechanical, thermal, and water absorption properties of wood/polypropylene green composites. J. Adhes. Sci. Technol. 2013, 27, 1301–1312. [Google Scholar] [CrossRef]
- Delva, L.; Ragaert, K.; Degrieck, J.; Cardon, L. The effect of multiple extrusions on the properties of montmorillonite filled polypropylene. Polymers 2014, 6, 2912–2927. [Google Scholar] [CrossRef]
- Aurrekoetxea, J.; Sarrionandia, M.A.; Urrutibeascoa, I.; Maspoch, M.L. Effects of recycling on the microstructure and the mechanical properties of isotactic polypropylene. J. Mater. Sci. 2001, 36, 2607–2613. [Google Scholar] [CrossRef]
- Sarrionandia, M.; Lopez-Arraiza, A.; Aurrekoetxea, J.; Arostegui, A. Structure and mechanical properties of a talc-filled polypropylene/ethylene-propylene-diene composite after reprocessing in the melt state. J. Appl. Polym. Sci. 2009, 114, 1195–1201. [Google Scholar] [CrossRef]
- Camacho, W.; Karlsson, S. Assessment of thermal and thermo-oxidative stability of multi-extruded recycled PP, HDPE and a blend thereof. Polym. Degrad. Stab. 2002, 78, 385–391. [Google Scholar] [CrossRef]
- Jiménez, A.; Torre, L.; Kenny, J.M. Processing and properties of recycled polypropylene modified with elastomers. Plast. Rubber Compos. 2003, 32, 357–367. [Google Scholar] [CrossRef]
- Thompson, D.W.; Hansen, E.N.; Knowles, C.; Muszynski, L. Opportunities for wood plastic composite products in the US highway construction sector. BioResources 2010, 5, 1336–1552. [Google Scholar]
- Ashori, A. Wood–plastic composites as promising green-composites for automotive industries. Bioresour. Technol. 2008, 99, 4661–4667. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Tzoganakis, C. An experimental study of single-screw extrusion of HDPE–wood composites. Adv. Polym. Technol. 2010, 29, 197–218. [Google Scholar] [CrossRef]
- Najafi, S.K. Use of recycled plastics in wood plastic composites—A review. Waste Manag. 2013, 33, 1898–1905. [Google Scholar] [CrossRef] [PubMed]
- Mekonnen, T.; Mussone, P.; Alemaskin, K.; Sopkow, L.; Wolodko, J.; Choi, P.; Bressler, D. Biocomposites from hydrolyzed waste proteinaceous biomass: Mechanical, thermal and moisture absorption performances. J. Mater. Chem. A 2013, 1, 13186–13196. [Google Scholar] [CrossRef]
- Ogunsona, E.O.; Panchal, P.; Mekonnen, T.H. Surface grafting of acrylonitrile butadiene rubber onto cellulose nanocrystals for nanocomposite applications. Compos. Sci. Technol. 2019, 184, 107884. [Google Scholar] [CrossRef]
- Fei, P.; Xiong, H.; Cai, J.; Liu, C.; Yu, Y. Enhanced the weatherability of bamboo fiber-based outdoor building decoration materials by rutile nano-TiO2. Constr. Build. Mater. 2016, 114, 307–316. [Google Scholar] [CrossRef]
- Tajvidi, M.; Takemura, A. Thermal degradation of natural fiber-reinforced polypropylene composites. J. Thermoplast. Compos. Mater. 2010, 23, 281–298. [Google Scholar] [CrossRef]
- Dixit, S.; Yadav, V.L. Comparative study of polystyrene/chemically modified wheat straw composite for green packaging application. Polym. Bull. 2020, 77, 1307–1326. [Google Scholar] [CrossRef]
- Zong, G.; Hao, X.; Hao, J.; Tang, W.; Fang, Y.; Ou, R.; Wang, Q. High-strength, lightweight, co-extruded wood flour-polyvinyl chloride/lumber composites: Effects of wood content in shell layer on mechanical properties, creep resistance, and dimensional stability. J. Clean. Prod. 2020, 244, 118860. [Google Scholar] [CrossRef]
- Vovk, M.; Sernek, M. Aluminium Trihydrate-filled Poly (methyl methacrylate) (PMMA/ATH) waste powder utilization in wood-plastic composite boards bonded by MUF Resin. BioResources 2020, 15, 3252–3269. [Google Scholar]
- Gardner, D.J.; Han, Y.; Wang, L. Wood–Plastic composite technology. Curr. For. Rep. 2015, 1, 139–150. [Google Scholar] [CrossRef]
- Poletto, M.; Dettenborn, J.; Pistor, V.; Zeni, M.; Zattera, A.J. Materials produced from plant biomass: Part I: Evaluation of thermal stability and pyrolysis of wood. Mater. Res. 2010, 13, 375–379. [Google Scholar] [CrossRef]
- Turku, I.; Keskisaari, A.; Kärki, T.; Puurtinen, A.; Marttila, P. Characterization of wood plastic composites manufactured from recycled plastic blends. Compos. Struct. 2017, 161, 469–476. [Google Scholar]
- Awal, A.; Ghosh, S.; Sain, M. Thermal properties and spectral characterization of wood pulp reinforced bio-composite fibers. J. Therm. Anal. Calorim. 2010, 99, 695–701. [Google Scholar] [CrossRef]
- Doh, G.-H.; Lee, S.-Y.; Kang, I.-A.; Kong, Y.-T. Thermal behavior of liquefied wood polymer composites (LWPC). Compos. Struct. 2005, 68, 103–108. [Google Scholar] [CrossRef]
- Kaboorani, A. Thermal properties of composites made of heat-treated wood and polypropylene. J. Compos. Mater. 2009, 43, 2599–2607. [Google Scholar] [CrossRef]
- Enayati, A.A.; Hosseinaei, O.; Wang, S.; Mirshokraie, S.A.; Tajvidi, M. Thermal properties of wood-plastic composites prepared from hemicellulose-extracted wood flour. Iran. J. Polym. Sci. Technol. (Persian) 2009, 22, 171–181. [Google Scholar]
- Jeske, H.; Schirp, A.; Cornelius, F. Development of a thermogravimetric analysis (TGA) method for quantitative analysis of wood flour and polypropylene in wood plastic composites (WPC). Thermochim. Acta 2012, 543, 165–171. [Google Scholar] [CrossRef]
- Jafari, S.H.; Asadinezhad, A.; Vahid, A.K.; Khonakdar, H.A.; Wagenknecht, U.; Heinrich, G. Polypropylene/Poly (trimethylene terephthalate) blend Nanocomposite: A thermal properties study. Polym. Plast. Technol. Eng. 2012, 51, 682–688. [Google Scholar] [CrossRef]
- Esmizadeh, E.; Naderi, G.; Yousefi, A.A.; Milone, C. Investigation of curing kinetics of epoxy resin/novel nanoclay–carbon nanotube hybrids by non-isothermal differential scanning calorimetry. J. Therm. Anal. Calorim. 2016, 126, 771–784. [Google Scholar] [CrossRef]
- Chiu, H.-T.; Huang, J.-K.; Kuo, M.-T.; Huang, J.-H. Characterisation of PC/ABS blend during 20 reprocessing cycles and subsequent functionality recovery by virgin additives. J. Polym. Res. 2018, 25, 124. [Google Scholar] [CrossRef]
- De Oliveira, T.A.; Barbosa, R.; Mesquita, A.B.S.; Ferreira, J.H.L.; de Carvalho, L.H.; Alves, T.S.F. degradation of reprocessed PP/PBAT/thermoplastic starch blends. J. Mater. Res. Technol. 2020, 9, 2338–2349. [Google Scholar] [CrossRef]
- Araújo, E.A.F.; Visconte, L.L.Y.; da Silva, A.L.N.; Sirelli, L.; Pacheco, É.B.A.V. Effect of clay amount and reprocessing cycles on thermal, morphological, and mechanical properties of polypropylene/organovermiculite nanocomposites. Polym. Eng. Sci. 2019, 59, 2110–2120. [Google Scholar] [CrossRef]
- Da Costa, H.M.; Ramos, V.D.; Rocha, M.C.G. Rheological properties of polypropylene during multiple extrusion. Polym. Test. 2005, 24, 86–93. [Google Scholar] [CrossRef]
- Peterson, J.D.; Vyazovkin, S.; Wight, C.A. Kinetics of the thermal and thermo-oxidative degradation of polystyrene, polyethylene and Poly (propylene). Macromol. Chem. Phys. 2001, 202, 775–784. [Google Scholar] [CrossRef]
- Selim, K.; Özkar, S.; Yilmaz, L. Thermal characterization of glycidyl azide polymer (GAP) and GAP-based binders for composite propellants. J. Appl. Polym. Sci. 2000, 77, 538–546. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Burnham, A.K.; Criado, J.M.; Pérez-Maqueda, L.A.; Popescu, C.; Sbirrazzuoli, N. ICTAC Kinetics Committee recommendations for performing kinetic computations on thermal analysis data. Thermochim. Acta. 2011, 520, 1–19. [Google Scholar] [CrossRef]
- Flynn, J.H.; Stephen, Z.D.C. Polymer Degradation. In Handbook of Thermal Analysis and Calorimetry: Applications to Polymers and Plastics; Cheng, S.Z.D., Ed.; Elsevier Science BU: Amsterdam, The Netherland, 2002; pp. 587–653. [Google Scholar]
- Abbas-Abadi, M.S.; Haghighi, M.N.; Yeganeh, H. Effect of the melt flow index and melt flow rate on the thermal degradation kinetics of commercial polyolefins. J. Appl. Polym. Sci. 2012, 126, 1739–1745. [Google Scholar] [CrossRef]
- Rao, T.R.; Sharma, A. Pyrolysis rates of biomass materials. Energy 1998, 23, 973–978. [Google Scholar] [CrossRef]
- Yang, H.; Yan, R.; Chen, H.; Lee, D.H.; Zheng, C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar] [CrossRef]
- McKendry, P. Energy production from biomass (part 1): Overview of biomass. Bioresour. Technol. 2002, 83, 37–46. [Google Scholar] [CrossRef]
- Jubinville, D.; Esmizadeh, E.; Saikrishnan, S.; Tzoganakis, C.; Mekonnen, T. A comprehensive review on global production and recycling methods of polyolefin (PO) based products and their post-recycling applications. Sustain. Mater. Technol. 2020, 25, e00188–e00222. [Google Scholar] [CrossRef]
- Ghafghazi, S.; Sowlati, T.; Sokhansanj, S.; Bi, X.; Melin, S. Particulate matter emissions from combustion of wood in district heating applications. Renew Sustain. Energy Rev. 2011, 15, 3019–3028. [Google Scholar] [CrossRef]
- Jahani, D.; Ameli, A.; Saniei, M.; Ding, W.; Park, C.B.; Naguib, H.E. Characterization of the structure, acoustic property, thermal conductivity, and mechanical property of highly expanded open-cell polycarbonate foams. Macromol. Mater. Eng. 2015, 300, 48–56. [Google Scholar] [CrossRef]
- Pouteau, C.; Dole, P.; Cathala, B.; Averous, L.; Boquillon, N. Antioxidant properties of lignin in polypropylene. Polym. Degrad. Stab. 2003, 81, 9–18. [Google Scholar] [CrossRef]
- Canetti, M.; Bertini, F.; de Chirico, A.; Audisio, G. Thermal degradation behaviour of isotactic polypropylene blended with lignin. Polym. Degrad. Stab. 2006, 91, 494–498. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | PP Resin (wt.%) | Wood (wt.%) |
|---|---|---|
| PPv* | 100 | 0 |
| PP×1 (PPvW00) | 100 | 0 |
| PP×3 | 100 | 0 |
| PP×5 | 100 | 0 |
| PPvW40 | 60 | 40 |
| PPvW60 | 40 | 60 |
| Wood | 0 | 100 |
| Kinetic Model | Equation | |
|---|---|---|
| Kissinger–Akahira–Sunose (KAS) | Slope of vs. at constant conversion (α) | |
| Kissinger | Slope of vs. | |
| Ozawa–Flynn–Wall (OFW) | Slope of vs. at constant α | |
| Friedman | Slope of vs. at constant α | |
| Augis | Slope of vs. |
| Sample | Heating Rate (°C/min) | N2 Atmosphere | Air Atmosphere | ||
|---|---|---|---|---|---|
| T5% (°C) | Tpeak (°C) | T5% (°C) | Tpeak (°C) | ||
| PPv | 5 | 339.1 | 431.2 | 263.6 | 365.2 |
| 10 | 351.9 | 440.6 | 274.1 | 398.1 | |
| 20 | 359.1 | 463.5 | 299.4 | 402.0 | |
| 40 | 416.4 | 487.9 | 324.6 | 442.8 | |
| PP×1 | 5 | 332.3 | 430.5 | 254.6 | 331.7 |
| 10 | 344.5 | 440.1 | 262.3 | 351.1 | |
| 20 | 358.7 | 460.0 | 294.6 | 394.2 | |
| 40 | 386.2 | 479.0 | 318.9 | 430.1 | |
| PP×3 | 5 | 330.7 | 429.0 | 246.8 | 324.3 |
| 10 | 337.6 | 438.7 | 260.2 | 348.0 | |
| 20 | 354.7 | 456.2 | 288.1 | 385.0 | |
| 40 | 381.3 | 476.8 | 312.4 | 427.4 | |
| PP×5 | 5 | 327.4 | 423.5 | 238.5 | 308.7 |
| 10 | 334.7 | 436.7 | 257.9 | 338.6 | |
| 20 | 353.1 | 454.8 | 285.6 | 378.9 | |
| 40 | 371.6 | 474.3 | 309.7 | 426.2 s | |
| Sample | Heating Rate (°C/min) | Atmosphere | Tpeak (°C) | |||
|---|---|---|---|---|---|---|
| Hemicellulose | Cellulose | Lignin | PP | |||
| Wood | 5 °C/min | N2 | 290.3 | 335.96 | NA | - |
| Air | 267.9 | 308.5 | 442.2 | - | ||
| PPVW00 | N2 | - | - | - | 430.5 | |
| Air | - | - | - | 331.7 | ||
| PPVW40 | N2 | 291.2 | 342.8 | NA | 435.7 | |
| Air | 288.3 | 337.4 | 456.4 | 364.0 | ||
| PPVW60 | N2 | 291.4 | 344.5 | NA | 439.6 | |
| Air | 290.1 | 336.7 | 453.9 | 364.5 | ||
| Wood | 10 °C/min | N2 | 293.3 | 347.5 | NA | - |
| Air | 280.0 | 321.0 | 464.1 | - | ||
| PPVW00 | N2 | - | - | - | 440.1 | |
| Air | - | - | - | 351.1 | ||
| PPVW40 | N2 | 295.1 | 349.6 | NA | 441.9 | |
| Air | 288.9 | 347.2 | 471.1 | 380.3 | ||
| PPVW60 | N2 | 296.3 | 351.8 | NA | 449.6 | |
| Air | 288.7 | 342.4 | 468.4 | 374.4 | ||
| Wood | 20 °C/min | N2 | 311.5 | 359.5 | NA | - |
| Air | 292.5 | 335.7 | 482.2 | - | ||
| PPVW00 | N2 | - | - | - | 460.0 | |
| Air | - | - | - | 394.2 | ||
| PPVW40 | N2 | 317.6 | 368.0 | NA | 464.8 | |
| Air | 308.4 | 358.2 | 496.1 | 393.4 | ||
| PPVW60 | N2 | 319.5 | 370.5 | NA | 468.6 | |
| Air | 309.3 | 356.5 | 475.4 | 394.1 | ||
| Wood | 40 °C/min | N2 | 322.4 | 372.1 | NA | - |
| Air | 305.2 | 350.2 | 501.1 | - | ||
| PPVW00 | N2 | - | - | - | 479.0 | |
| Air | - | - | - | 430.1 | ||
| PPVW40 | N2 | 331.0 | 380.1 | NA | 485.2 | |
| Air | 311.0 | 370.4 | 536.4 | 425.7 | ||
| PPVW60 | N2 | 334.8 | 384.0 | NA | 486.1 | |
| Air | 310.8 | 369.3 | 535.7 | 422.5 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esmizadeh, E.; Tzoganakis, C.; Mekonnen, T.H. Degradation Behavior of Polypropylene during Reprocessing and Its Biocomposites: Thermal and Oxidative Degradation Kinetics. Polymers 2020, 12, 1627. https://doi.org/10.3390/polym12081627
Esmizadeh E, Tzoganakis C, Mekonnen TH. Degradation Behavior of Polypropylene during Reprocessing and Its Biocomposites: Thermal and Oxidative Degradation Kinetics. Polymers. 2020; 12(8):1627. https://doi.org/10.3390/polym12081627
Chicago/Turabian StyleEsmizadeh, Elnaz, Costas Tzoganakis, and Tizazu H. Mekonnen. 2020. "Degradation Behavior of Polypropylene during Reprocessing and Its Biocomposites: Thermal and Oxidative Degradation Kinetics" Polymers 12, no. 8: 1627. https://doi.org/10.3390/polym12081627
APA StyleEsmizadeh, E., Tzoganakis, C., & Mekonnen, T. H. (2020). Degradation Behavior of Polypropylene during Reprocessing and Its Biocomposites: Thermal and Oxidative Degradation Kinetics. Polymers, 12(8), 1627. https://doi.org/10.3390/polym12081627