Development and Performance of Bioactive Compounds-Loaded Cellulose/Collagen/Polyurethane Materials

 ,
,  , ,
, , 
 and
and
Abstract
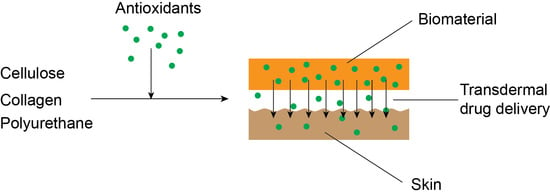
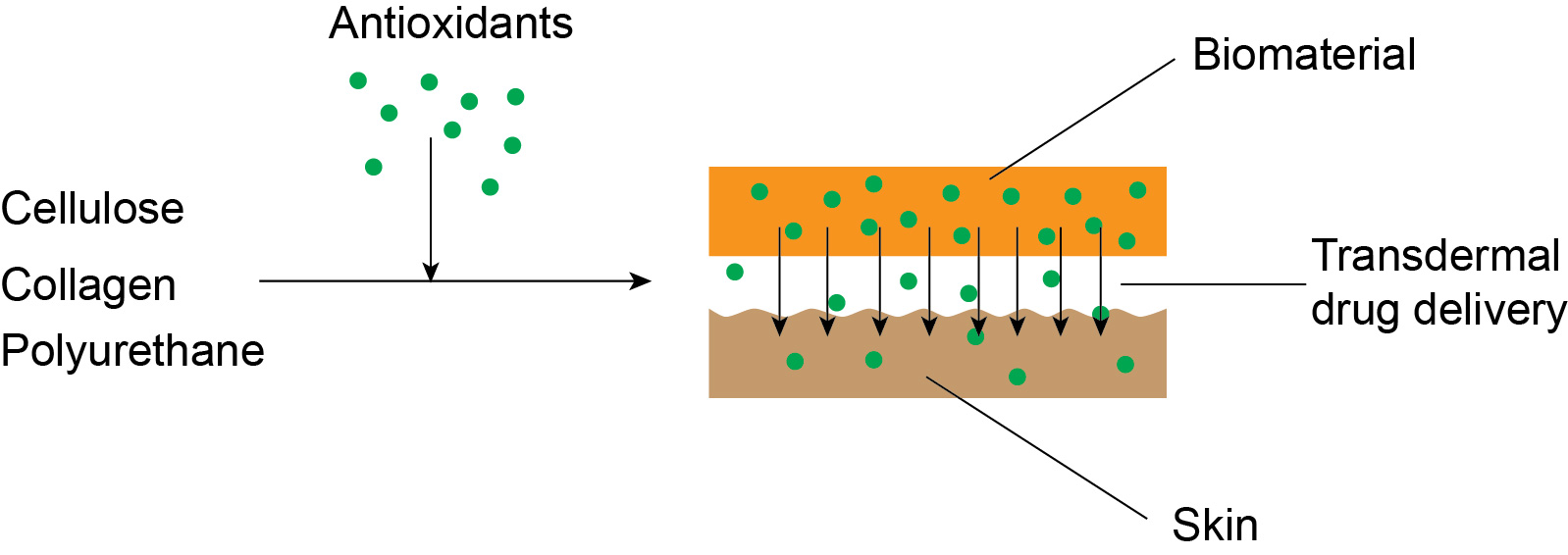
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
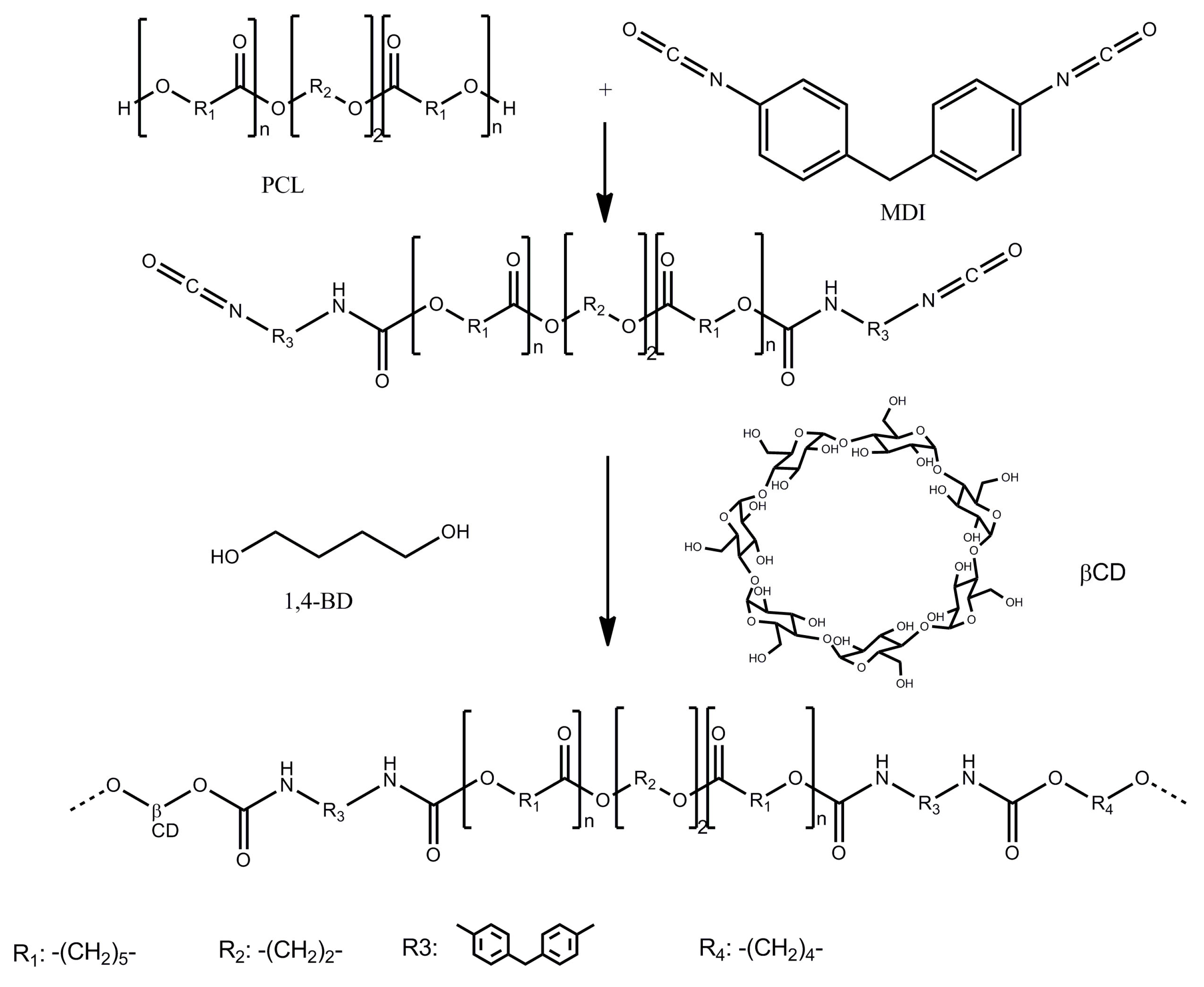
2.2.1. Polyurethane Synthesis
2.2.2. Tannins Extraction
2.2.3. Preparation of Biomaterials
2.2.4. Characterization
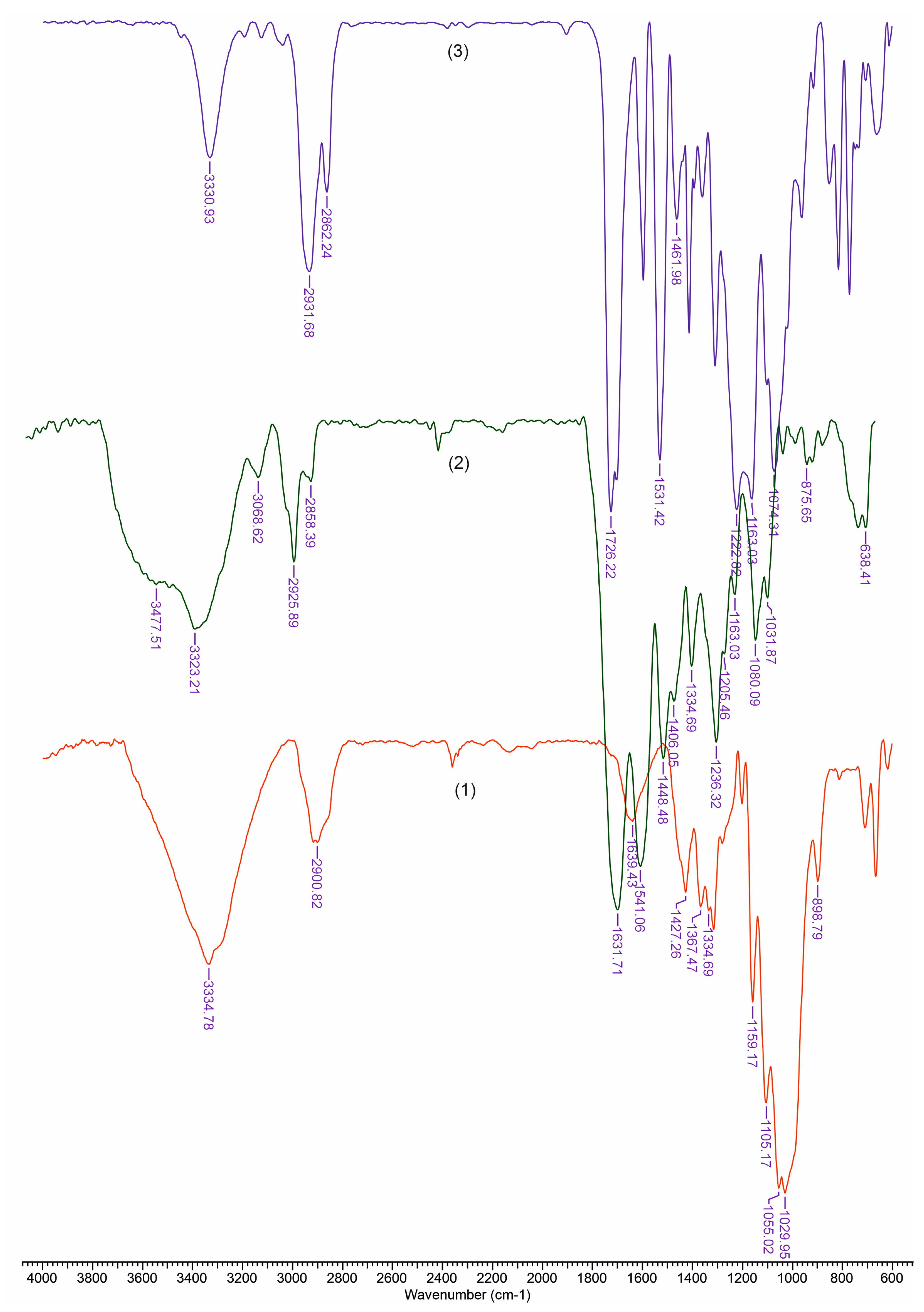
FTIR Spectroscopy
Scanning Electron Microscopy
2.2.5. Bioadhesivity Test
2.2.6. Compression Test
2.2.7. Dynamic Vapor Sorption (DVS)
2.2.8. In Vitro Release Studies
2.2.9. (DPPH—2,2 diphenyl-1-picrylhydrazyl) Assay
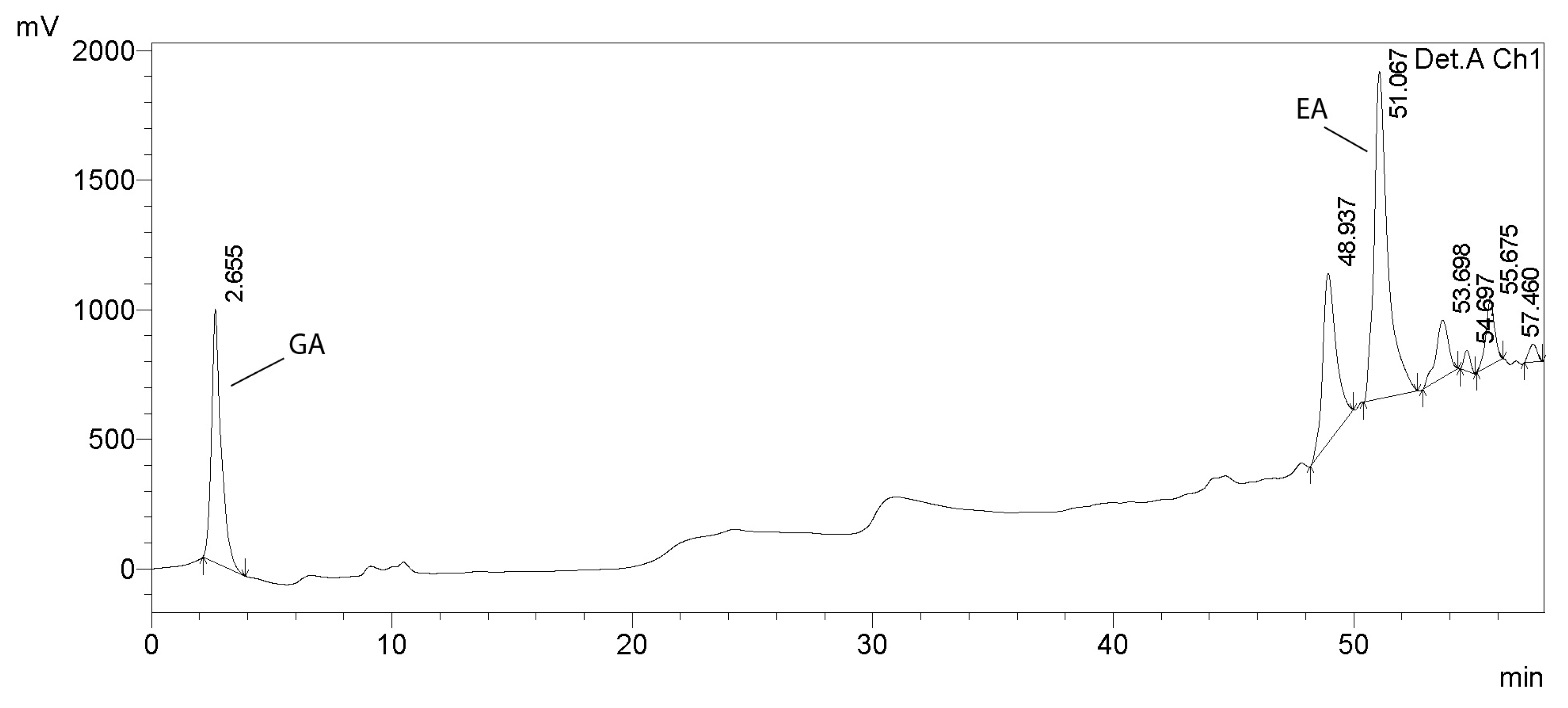
2.2.10. HPLC Analysis of Tannins
3. Results and Discussion
3.1. Analysis of Tannins in Oak Bark
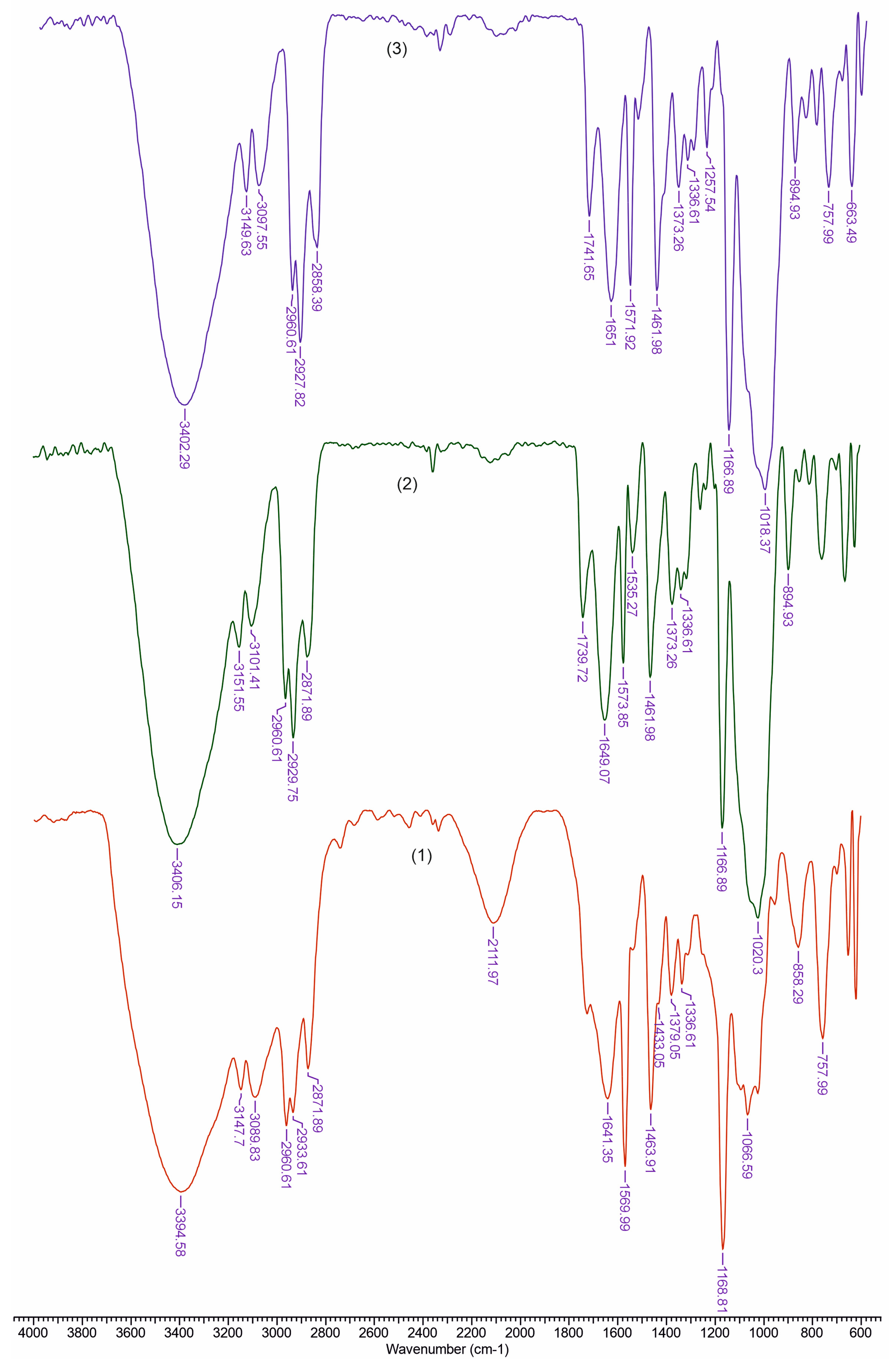
3.2. FTIR Analysis
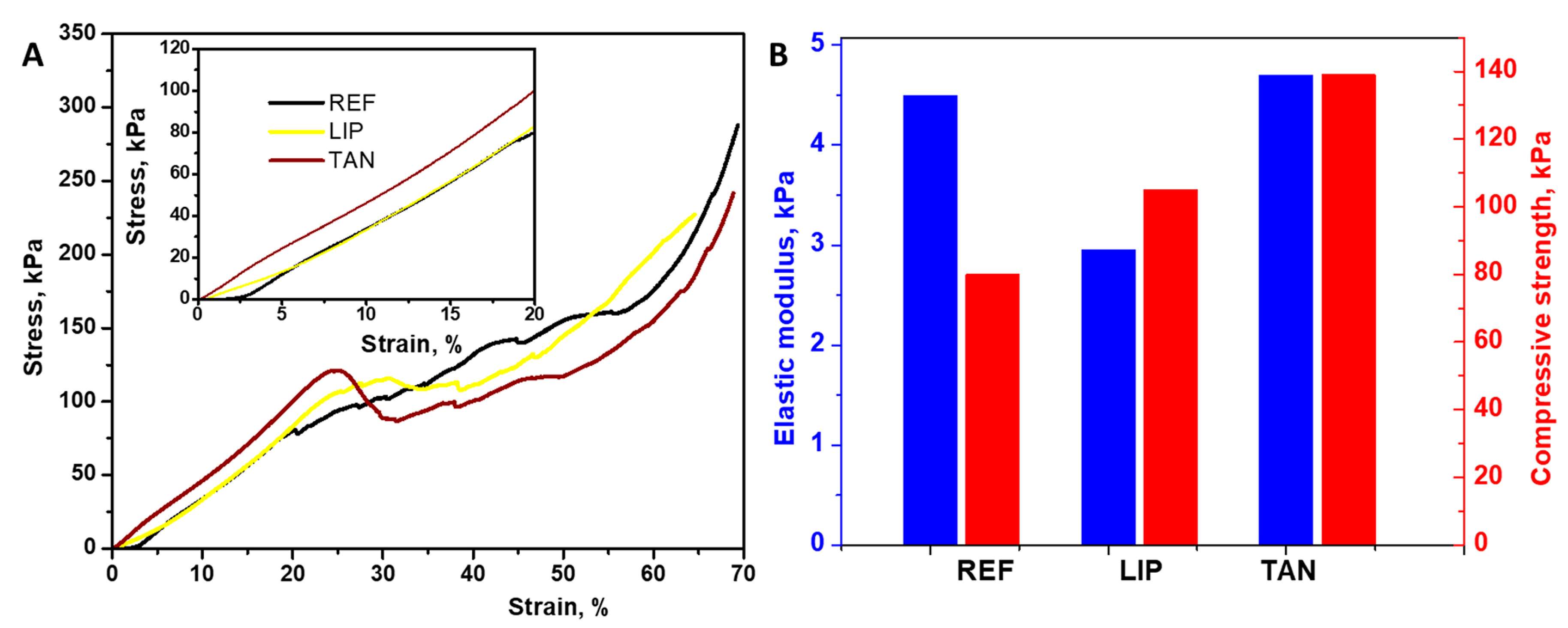
3.3. Mechanical Properties
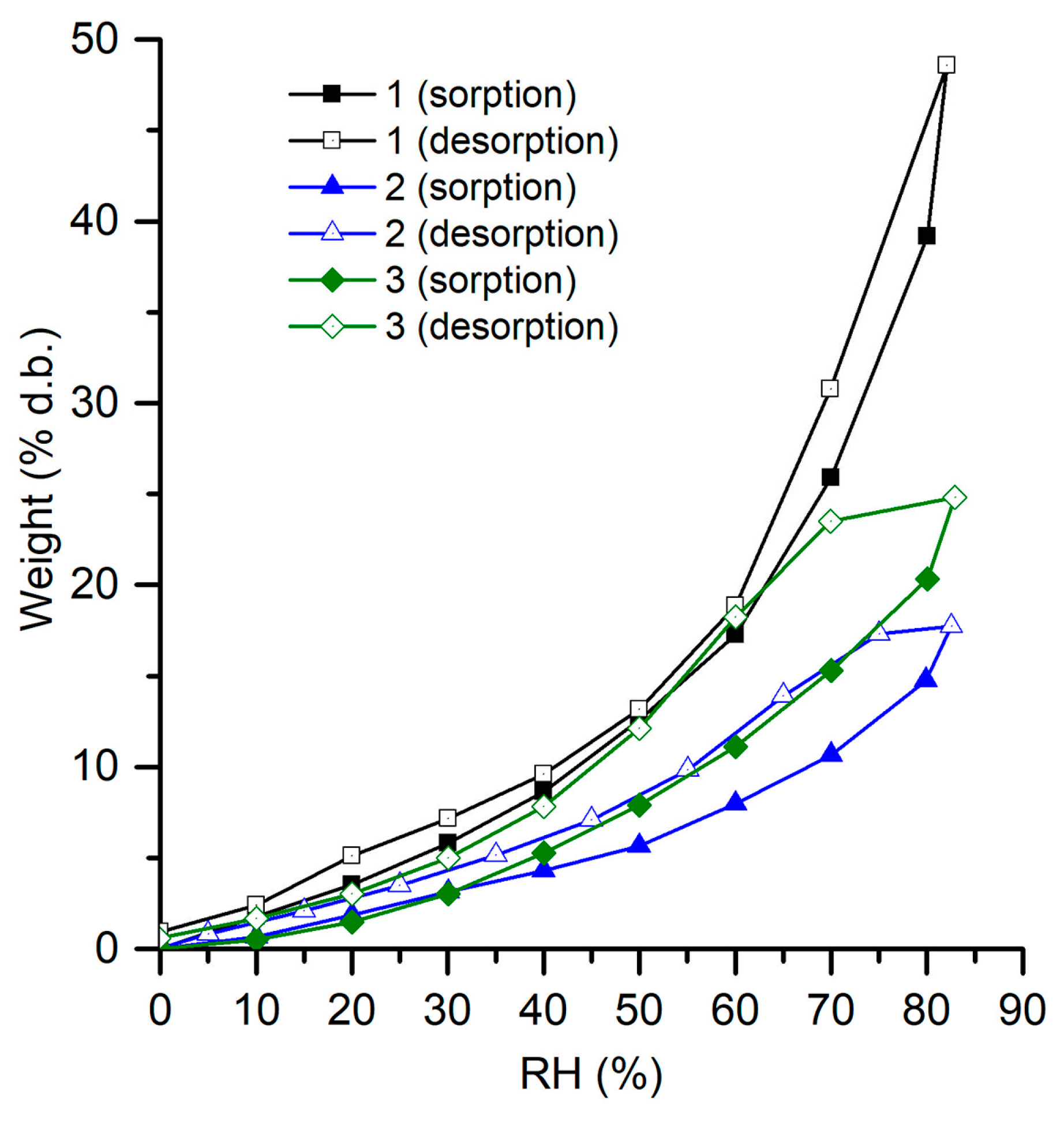
3.4. Water Adsorption Isotherms
3.5. Bio/Mucoadhesivity Properties
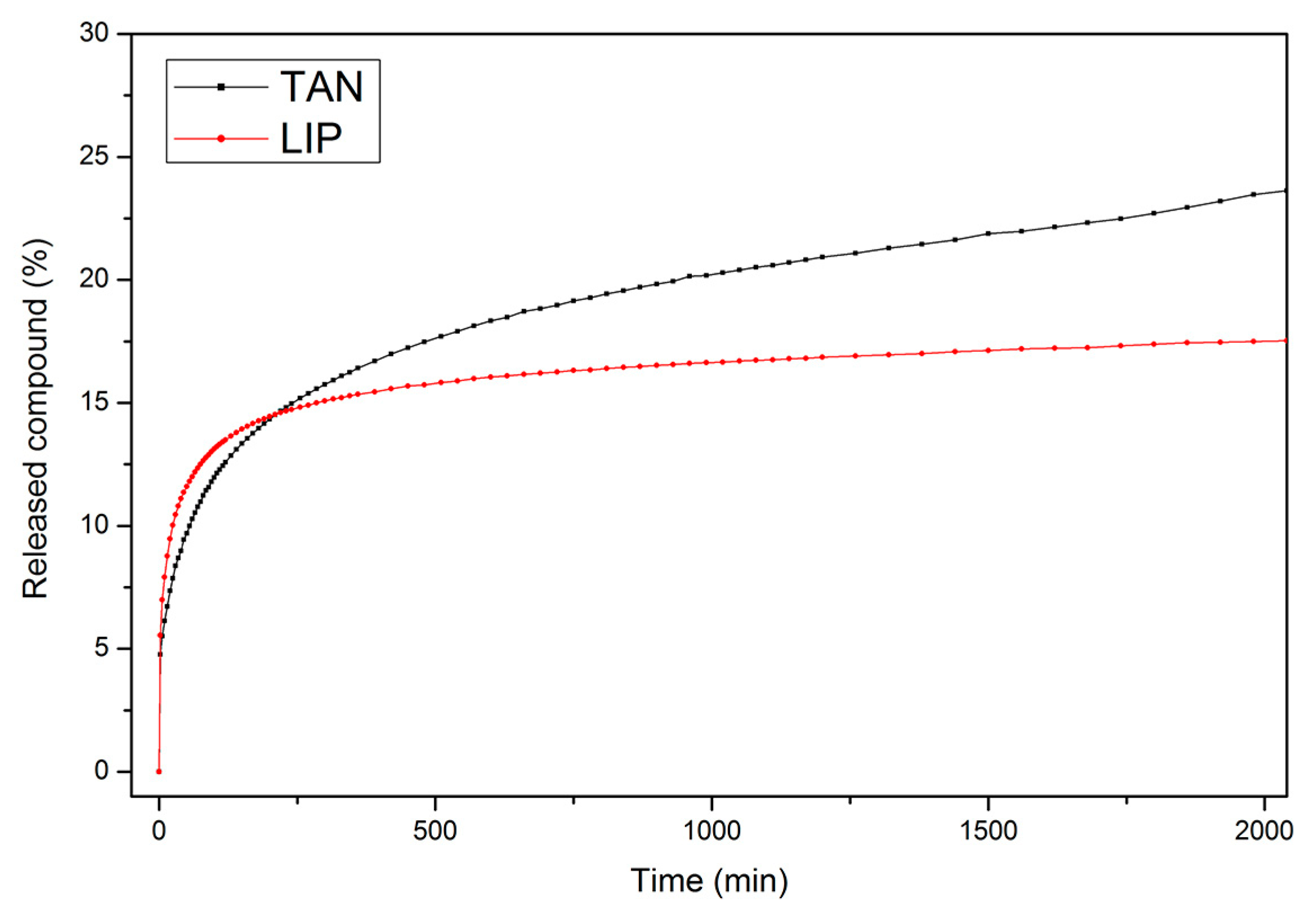
3.6. Controlled Release of Active Compounds from Biomaterials
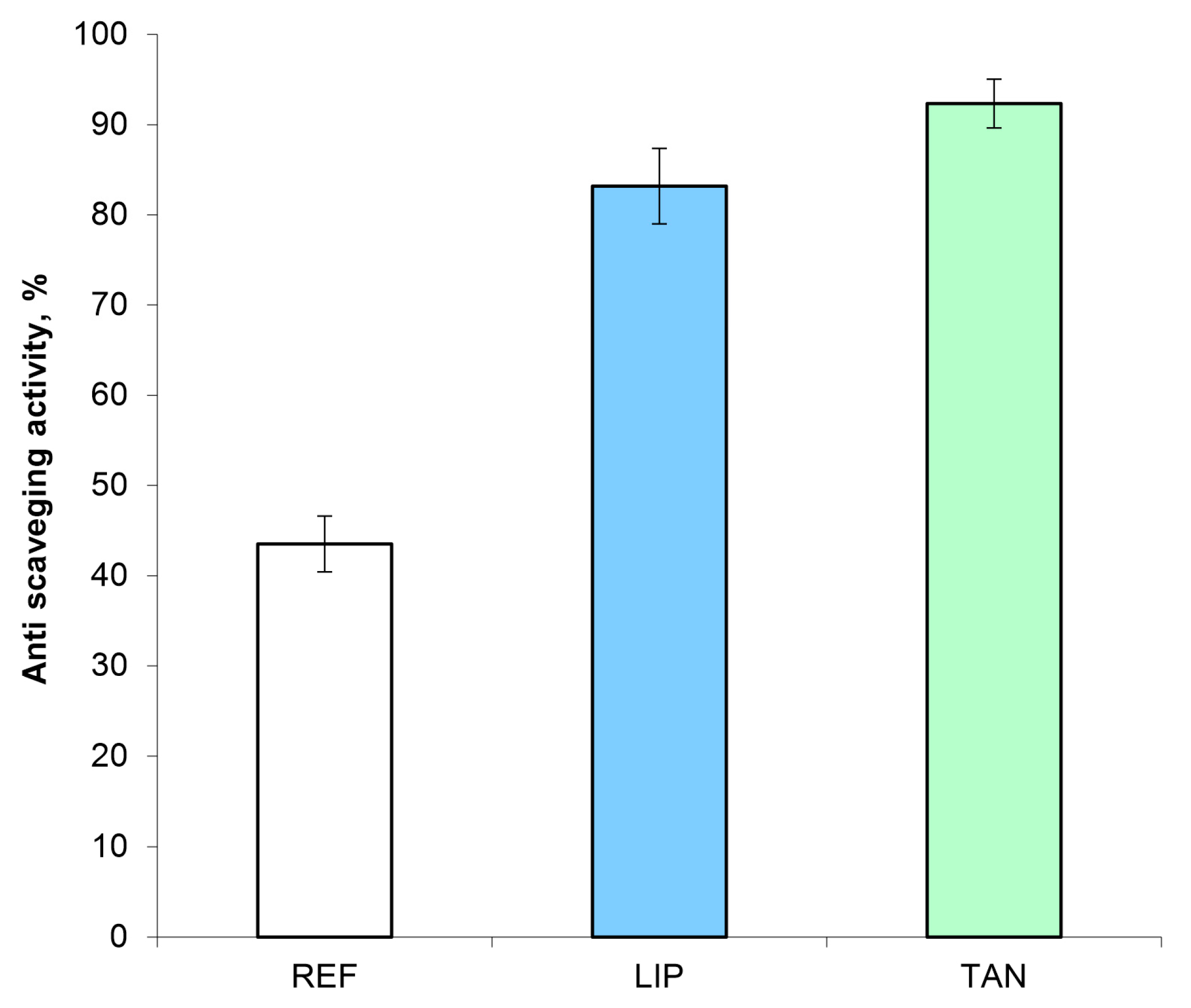
3.7. Antiradical Activity
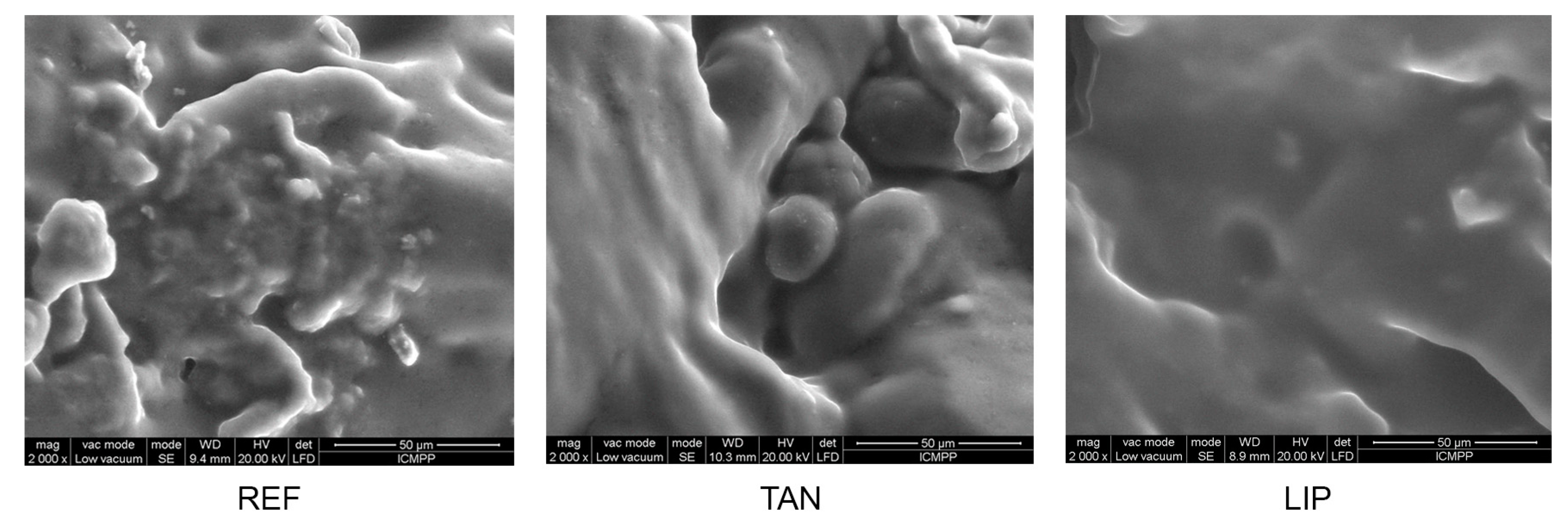
3.8. Materials’ Morphology
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stone, S.A.; Gosavi, P.; Athauda, T.J.; Ozer, R.R. In situ citric acid crosslinking of alginate/polyvinylalcohol electrospun nanofibers. Mater. Lett. 2013, 112, 32–35. [Google Scholar] [CrossRef]
- El Seoud, O.A.; Kostag, M.; Jedvert, K.; Malek, N.I. Cellulose in Ionic Liquids and Alkaline Solutions: Advances in the mechanisms of biopolymer dissolution and regeneration. Polymer 2019, 11, 1917. [Google Scholar] [CrossRef] [PubMed]
- Halayqa, M.; Pobudkowska, A.; Domańska, U.; Zawadzki, M. Studying of drug solubility in water and alcohols using drug—Ammonium ionic liquid-compounds. Eur. J. Pharm. Sci. 2018, 111, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Monti, D.; Egiziano, E.; Burgalassi, S.; Chetoni, P.; Chiappe, C.; Sanzone, A.; Tampucci, S. Ionic liquids as potential enhancers for transdermal drug delivery. Int. J. Pharm. 2017, 516, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, P.; Zhang, Y.; Lu, F.; Li, W.; Kang, H.; Xiang, J.; Huang, Y.; Liu, R. Hierarchical porous structures in cellulose: NMR relaxometry approach. Polymer 2016, 98, 237–243. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, H.J.; Kim, J.C. Nanocellulose applications for drug delivery: A review. J. Environ. Sci. 2019, 35, 141–149. [Google Scholar] [CrossRef]
- Sharma, P.R.; Joshi, R.; Sharma, S.K.; Hsiao, B.S. A Simple Approach to Prepare Carboxycellulose Nanofibers from Untreated Biomass. Biomacromolecules 2017, 18, 2333–2342. [Google Scholar] [CrossRef]
- Zhan, C.; Li, X.; Sharma, P.R.; He, H.; Sharma, S.K.; Wanga, R.; Hsiao, B.S. A study of TiO2 nanocrystal growth and environmental remediation capability of TiO2/CNC nanocomposites. RSC Adv 2019, 9, 40565–40576. [Google Scholar] [CrossRef]
- Chen, H.; Sharma, S.K.; Sharma, P.R.; Yeh, H.; Johnson, K.; Hsiao, B.S. Arsenic (III) Removal by Nanostructured Dialdehyde Cellulose−Cysteine Microscale and Nanoscale Fibers. ACS Omega 2019, 4, 22008–22020. [Google Scholar] [CrossRef]
- Sharma, P.R.; Chattopadhyay, A.; Sharma, S.K.; Geng, L.; Amiralian, N.; Martin, D.; Hsiao, B.S. Nanocellulose from Spinifex as an Effective Adsorbent to Remove Cadmium (II) from Water. ACS Sustain. Chem. Eng. 2018, 6, 3279–3290. [Google Scholar] [CrossRef]
- Yu, H.Y.; Zhang, D.Z.; Lu, F.F.; Yao, J. New Approach for Single-Step Extraction of Carboxylated Cellulose Nanocrystals for Their Use as Adsorbents and Flocculants. ACS Sustain. Chem. Eng. 2016, 4, 2632–2643. [Google Scholar] [CrossRef]
- Sharma, P.R.; Sharma, S.K.; Antoine, R.; Hsiao, B.S. Efficient Removal of Arsenic Using Zinc Oxide Nanocrystal-Decorated Regenerated Microfibrillated Cellulose Scaffolds. ACS Sustain. Chem. Eng. 2019, 7, 6140–6151. [Google Scholar] [CrossRef]
- Sharma, P.R.; Chattopadhyay, A.; Sharma, S.K.; Hsiao, B.S. Efficient Removal of UO22+ from Water Using Carboxycellulose Nanofibers Prepared by The Nitro-Oxidation Method. Ind. Eng. Chem. Res. 2017, 56, 13885–13893. [Google Scholar] [CrossRef]
- Klemm, D.; Heublein, B.; Fink, H.P.; Bohn, A. Cellulose: Fascinating Biopolymer and Sustainable Raw Material. Angew. Chem. Int. Ed. 2005, 44, 3358–3393. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, N.; Grishkewich, N.; Tam, K.C. Cellulose Nanomaterials: Promising Sustainable Nanomaterialsfor Application inWater/Wastewater Treatment Processes. Environ. Sci. Nano 2018, 5, 623–658. [Google Scholar] [CrossRef]
- Sharma, P.R.; Sharma, S.K.; Lindström, L.; Hsiao, B.S. Nanocellulose-Enabled Membranes for Water Purification: Perspectives. Adv. Sustain. Syst. 2020, 1900114. [Google Scholar] [CrossRef]
- Sharma, P.R.; Varma, A.J. Functional nanoparticles from cellulose: Engineering the shape and size of 6-carboxycellulose. Chem. Commun. 2013, 49, 8818–8820. [Google Scholar] [CrossRef]
- Klemm, D.; Cranston, E.D.; Fischer, D.; Gama, M.; Kedzior, S.A.; Kralisch, D.; Kramer, F.; Kondo, T.; Lindström, T.; Nietzsche, S.; et al. Nanocellulose as a natural source for ground breaking applications in materials science: Today’s. Mater. Today 2018, 21, 720–748. [Google Scholar] [CrossRef]
- Chang, S.W.; Flynn, B.P.; Ruberti, J.W.; Buehler, M.J. Molecular mechanism of force induced stabilization of collagen against enzymatic breakdown. Biomaterials 2012, 33, 3852–3859. [Google Scholar] [CrossRef]
- Salamanca, E.; Hsu, C.C.; Yao, W.L.; Choy, C.S.; Pan, Y.H.; Teng, N.C.; Chang, W.J. Porcine collagen–bone composite induced osteoblast diferentiation and bone regeneration in vitro and in vivo. Polymer 2020, 12, 93. [Google Scholar] [CrossRef]
- Lucarini, M.; Sciubba, F.; Capitani, D.; Di Cocco, M.E.; D’Evoli, L.; Durazzo, A.; Delfini, M.; Boccia, G.L. Role of catechin on collagen type I stability upon oxidation: A NMR approach. Nat. Prod. Res. 2020, 34, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Chenga, Y.; Lua, J.; Liub, S.; Zhaoc, P.; Luc, G.; Chen, J. The preparation, characterization and evaluation of regeneratedcellulose/collagen composite hydrogel films. Carbohydr. Polym. 2014, 107, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Yang, J.; Liu, P.; Xu, M.; Zhang, X.; Zhang, L. Fabrication, properties and bioapplications of cellulose/collagen hydrolysate composite films. Carbohydr. Polym. 2013, 92, 1752–1760. [Google Scholar] [CrossRef] [PubMed]
- Noha, Y.K.; Da Costaa, A.D.S.; Parkd, Y.S.; Due, P.; Kimb, I.K.; Park, K. Fabrication of bacterial cellulose-collagen composite scaffolds and their osteogenic effect on human mesenchymal stem cells. Carbohydr. Polym. 2019, 219, 210–218. [Google Scholar] [CrossRef]
- Brzeska, J.; Tercjak, A.; Sikorska, W.; Kowalczuk, M.; Rutkowska, M. Morphology and physicochemical properties of branched polyurethane/biopolymer blends. Polymer 2020, 12, 16. [Google Scholar] [CrossRef]
- Lei, W.; Fang, C.; Zhou, X.; Li, Y.; Pu, M. Polyurethane elastomer composites reinforced with waste natural cellulosic fibers from office paper in thermal properties. Carbohydr. Polym. 2018, 197, 385–394. [Google Scholar] [CrossRef]
- Stanzione, M.; Oliviero, M.; Cocca, M.; Errico, M.E.; Gentile, G.; Avella, M.; Lavorgna, M.; Buonocore, G.G.; Verdolotti, L. Tuning of polyurethane foam mechanical and thermal properties using ball-milled cellulose. Carbohydr. Polym. 2020, 231, 115772. [Google Scholar] [CrossRef]
- Pereira, R.; Carvalho, A.; Vaz, D.C.; Gil, M.H.; Mendes, A.; Bártolo, P. Development of novel alginate–based hydrogel films for wound healing applications. Int. J. Biol. Macromol. 2013, 52, 221–230. [Google Scholar] [CrossRef]
- Söhretog, D.; Sabuncuog, S.; Harput, U.S. Evaluation of antioxidative, protective effect against H2O2 induced cytotoxicity, and cytotoxic activities of three different Quercus species. Food Chem. Toxicol. 2012, 50, 141–146. [Google Scholar] [CrossRef]
- Lee, B.S.; Yuan, X.; Xu, Q.; McLafferty, F.S.; Petersen, B.A.; Collette, J.C.; Black, K.L.; Yu, J.S. Preparation and characterization of antioxidant nanospheres from multiplea-lipoic acid-containing compounds. Bioorg. Med. Chem. Lett. 2009, 19, 1678–1681. [Google Scholar] [CrossRef]
- Weerakody, R.; Fagan, P.; Kosaraju, S.L. Chitosan microspheres for encapsulation of alpha-lipoic acid. Int. J. Pharm. 2008, 357, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yu, J.; Feng, X.; Li, W.; Wang, Y.; Jin, H.; Huang, H.; Fan, D. Reduction-responsive core-crosslinked micelles based on a glycol chitosan–lipoic acid conjugate for triggered release of doxorubicin. RSC Adv. 2016, 6, 31391–31400. [Google Scholar] [CrossRef]
- Mândru, M.; Vlad, S.; Ciobanu, C.; Lebrun, L.; Popa, M. Polyurethane-hydroxypropyl cellulose membranes for sustained release of nystatin. Cellul. Chem. Technol. 2013, 47, 5–12. [Google Scholar]
- Sivakumar, V.; Ilanhtiraiyan, S.; Ilayaraja, K.; Ashly, A.; Hariharan, S. Influence of ultrasound on Avaram bark (Cassia auriculata) tannin extraction and tanning. Chem. Eng. Res. Des. 2014, 92, 1827–1833. [Google Scholar] [CrossRef]
- Anghel, N.; Lazar, S.; Ciubotariu, B.-I.; Verestiuc, L.; Spiridon, I. New cellulose-based materials as transdermal transfer systems for bioactive substances. Cellul. Chem. Technol. 2019, 53, 879–884. [Google Scholar] [CrossRef]
- Suflet, D.M.; Pelin, I.M.; Dinu, M.V.; Lupu, M.; Popescu, I. Hydrogels based on monobasic curdlan phosphate for biomedical applications. Cellul. Chem. Technol. 2019, 53, 897–906. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Lustig, S.R.; Peppas, N.A. Solute and penetrant diffusion in swellable polymers. I. Mathematical modeling. J. Polym. Sci. Part B Polym. Phys. 1986, 24, 395–408. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Sridhar, K.; Charles, A.L. In vitro antioxidant activity of Kyoho grape extracts in DPPH and ABTS assays: Estimation methods for EC50 using advanced statistical programs. Food Chem. 2019, 275, 41–49. [Google Scholar] [CrossRef]
- Mammela, P.; Savolainen, H.; Lindroos, L.; Kangas, J.; Vartiainen, T. Analysis of oak tannins by liquid chromatography-electrospray ionisation mass spectrometry. J. Chromatogr. A 2000, 891, 75–83. [Google Scholar] [CrossRef]
- Arinaa, M.Z.I.; Harisuna, Y. Effect of extraction temperatures on tannin content and antioxidant activity of Quercus infectoria (Manjakani). Biocatal. Agric. Biotechnol. 2019, 19, 101104. [Google Scholar] [CrossRef]
- Elansary, H.O.; Szopa, A.; Kubica, P.; Ekiert, H.; Mattar, M.A.; Al-Yafrasi, M.A.; El-Ansary, D.O.; El-Abedin, T.K.Z.; Yessoufou, K. Polyphenol Profile and Pharmaceutical Potential of Quercus spp. Bark Extracts. Plants 2019, 8, 486. [Google Scholar] [CrossRef] [PubMed]
- Rosa, M.F.; Medeiros, E.S.; Malmonge, J.A.; Gregorski, K.S.; Wood, D.F.; Mattoso, L.H.C.; Imam, S.H. Cellulose nanowhiskers from coconut husk fibers: Effect of preparation conditions on their thermal and morphological behavior. Carbohyd. Polym. 2010, 8, 83–92. [Google Scholar] [CrossRef]
- Poletto, M.; Pistor, V.; Zeni, M.; Attera, A.J. Crystalline properties and decomposition kinetics of cellulose fibers in wood pulp obtained by two pulping processes. Polym. Degrad. Stabil. 2011, 96, 679–685. [Google Scholar] [CrossRef]
- Xu, F.; Yu, J.; Tesso, T.; Dowell, F.; Wang, D. Qualitative and quantitative analysis of lignocellulosic biomass using Infrared Techniques: A mini-review. Appl. Energ. 2013, 104, 801–809. [Google Scholar] [CrossRef]
- Fackler, K.; Stevanic, J.S.; Ters, T.; Hinterstoisser, B.; Schwanninger, M.; Salmén, L. FTIR Imaging Spectroscopy to Localise and Characterise Simultaneous and Selective White-Rot Decay within Sprude Woodcell. Holzforschung 2011, 65, 411–420. [Google Scholar] [CrossRef]
- Bonakdar, S.; Emami, S.H.; Shokrgozar, M.A.; Farhadic, A.; Ahmadi, S.A.H.; Amanzadeh, A. Preparation and characterization of polyvinyl alcohol hydrogels crosslinked by biodegradable polyurethane for tissue engineering of cartilage. Mater. Sci. Eng. C Mater. Biol. Appl. 2010, 30, 636–643. [Google Scholar] [CrossRef]
- Riaz, T.; Zeeshan, R.; Zarif, F.; Ilyas, K.; Muhammad, N.; Safi, S.Z.; Rahim, A.; Rizvi, S.A.A.; Rehman, I.U. FTIR analysis of natural and synthetic collagen. Appl. Spectrosc. Rev. 2018, 53, 703–746. [Google Scholar] [CrossRef]
- Colom, X.; Carrillo, F. Crystallinity changes in lyocell and viscose-type fibres by caustic treatment. Eur. Polym. J. 2002, 38, 2225–2230. [Google Scholar] [CrossRef]
- Yue, Y.; Han, J.; Han, G.; French, A.D.; Qi, Y.; Wu, Q. Cellulose nanofibers reinforced sodium alginate-polyvinyl alcohol hydrogels: Core-shell structure formation and property characterization. Carbohydr. Polym. 2016, 147, 155–164. [Google Scholar] [CrossRef]
- Yue, Y.; Wang, X.; Han, J.; Yu, L.; Chen, J.; Wu, Q.; Jiang, J. Effects of nanocellulose on sodium alginate/polyacrylamide hydrogel: Mechanical properties and adsorption-desorption Capacities. Carbohydr. Polym. 2019, 206, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Raschip, I.E.; Fifere, N.; Varganici, C.D.; Dinu, M.V. Development of antioxidant and antimicrobial xanthan-based cryogels with tuned porous morphology and controlled swelling features. Int. J. Biol. Macromol. 2020, 156, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Kar, G.P.; Biswas, S.; Bose, S. Simultaneous enhancement in mechanical strength, electrical conductivity, and electromagnetic shielding properties in PVDF-ABS blends containing PMMA wrapped multiwall carbon nanotubes. Phys. Chem. Chem. Phys. 2015, 17, 14856–14865. [Google Scholar] [CrossRef] [PubMed]
- Felfel, R.M.; Gideon-Adeniyi, M.J.; Hossain, K.M.Z.; Roberts, G.A.F.; Grant, D.M. Structural, mechanical and swelling characteristics of 3D scaffolds from chitosan-agarose blends. Carbohydr. Polym. 2019, 204, 59–67. [Google Scholar] [CrossRef]
- Kalinoski, R.M.; Shi, J. Hydrogels derived from lignocellulosic compounds: Evaluation of the compositional, structural, mechanical and antimicrobial properties. Ind. Crop. Prod. 2019, 128, 323–330. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, T.; Feng, B.; Weng, J.; Duan, K.; Wang, J.; Lu, X. Biomimetic bacterial cellulose-enhanced double-network hydrogel with excellent mechanical properties applied for the osteochondral defect repair. ACS Biomater. Sci. Eng. 2018, 4, 3534–3544. [Google Scholar] [CrossRef]
- Rudzinski, W.E.; Dave, A.M.; Vaishnav, U.H.; Kumbar, S.G.; Kulkarni, A.R.; Aminabhavi, T.M. Hydrogels as controlled release devices in agriculture. Des. Monomers Polym. 2002, 5, 39–65. [Google Scholar] [CrossRef]
- Missio, A.L.; Mattos, B.D.; Ferreira, D.; Magalhaes, W.L.E.; Bertuol, D.A.; Gatto, D.A.; Petutschnigg, A.; Tondi, G. Nanocellulose-tannin films: From trees to sustainable active packaging. J. Clean Prod. 2018, 184, 143–151. [Google Scholar] [CrossRef]
- Zhang, Y.; Chan, J.W.; Moretti, A.; Uhrich, K.E. Designing polymers with sugar-based advantages for bioactive delivery applications. J. Control. Release 2015, 219, 355–368. [Google Scholar] [CrossRef]
- Hauptstein, S.; Bernkop-Schnurch, A. Thiomers and thiomer-based nanoparticles in protein and DNA drug delivery. Expert Opin. Drug Deliv. 2012, 9, 1069–1081. [Google Scholar] [CrossRef]
- Bernkop-Schnürch, A. Thiomers: A new generation of mucoadhesive polymers. Adv. Drug Deliv. Rev. 2005, 57, 1569–1582. [Google Scholar] [CrossRef] [PubMed]
- Mandapalli, P.K.; Venuganti, V.V.K. Layer-by-layer microcapsules for pH-controlled delivery of small molecules. J. Pharm. Investig. 2015, 45, 131–141. [Google Scholar] [CrossRef]
- Ma, M.; Dong, S.; Hussain, M.; Zhou, W. Effects of addition of condensed tannin on the structure and properties of silk fibroin film. Polym. Int. 2017, 66, 151–159. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCI (A1376/A2902) | LOI (A1437/A899) | HBI (A3336/A1336) | |
|---|---|---|---|
| REF | 0.492 | 1.533 | 3.459 |
| TAN | 0.447 | 2.132 | 4.368 |
| LIP | 0.406 | 2.250 | 4.177 |
| Sample | Sorption Capacity % d.b. | BET Data | |
|---|---|---|---|
| Area m2 × g−1 | Monolayer g × g−1 | ||
| REF | 48.5 | 331.062 | 0.094 |
| TAN | 17.7 | 151.100 | 0.043 |
| LIP | 24.8 | 758.750 | 0.216 |
| Sample | Bioadhesion Test | Muchoadhesion Test | ||
|---|---|---|---|---|
| Adhesion Force (n) | Total Work of Adhesion (n × s) | Adhesion Force (n) | Total Work of Adhesion (n × s) | |
| REF | 0.143 ± 0.00205 | 0.025 ± 0.00286 | 0.067 ± 0.00339 | 0.0099 ± 0.00033 |
| TAN | 0.095 ± 0.00205 | 0.020 ± 0.0017 | 0.087 ± 0.00449 | 0.0204 ± 0.00041 |
| LIP | 0.142 ± 0.00368 | 0.031 ± 0.00163 | 0.124 ± 0.0033 | 0.0317 ± 0.00057 |
| Samples | n | R2n | k, min−n | R2k |
|---|---|---|---|---|
| TAN | 0.294 | 0.998 | 0.0308 | 0.999 |
| LIP | 0.235 | 0.998 | 0.0500 | 0.999 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spiridon, I.; Anghel, N.; Dinu, M.V.; Vlad, S.; Bele, A.; Ciubotaru, B.I.; Verestiuc, L.; Pamfil, D. Development and Performance of Bioactive Compounds-Loaded Cellulose/Collagen/Polyurethane Materials. Polymers 2020, 12, 1191. https://doi.org/10.3390/polym12051191
Spiridon I, Anghel N, Dinu MV, Vlad S, Bele A, Ciubotaru BI, Verestiuc L, Pamfil D. Development and Performance of Bioactive Compounds-Loaded Cellulose/Collagen/Polyurethane Materials. Polymers. 2020; 12(5):1191. https://doi.org/10.3390/polym12051191
Chicago/Turabian StyleSpiridon, Iuliana, Narcis Anghel, Maria Valentina Dinu, Stelian Vlad, Adrian Bele, Bianca Iulia Ciubotaru, Liliana Verestiuc, and Daniela Pamfil. 2020. "Development and Performance of Bioactive Compounds-Loaded Cellulose/Collagen/Polyurethane Materials" Polymers 12, no. 5: 1191. https://doi.org/10.3390/polym12051191
APA StyleSpiridon, I., Anghel, N., Dinu, M. V., Vlad, S., Bele, A., Ciubotaru, B. I., Verestiuc, L., & Pamfil, D. (2020). Development and Performance of Bioactive Compounds-Loaded Cellulose/Collagen/Polyurethane Materials. Polymers, 12(5), 1191. https://doi.org/10.3390/polym12051191