Synthesis of Nixantphos Core-Functionalized Amphiphilic Nanoreactors and Application to Rhodium-Catalyzed Aqueous Biphasic 1-Octene Hydroformylation


 ,
,  , and
, and
Abstract
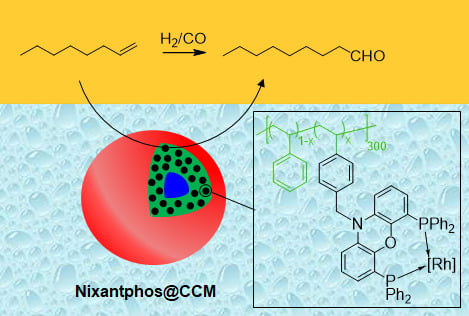
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instrumentation
2.3. Synthesis of the Ligand-Functionalized Styrene Monomer
2.4. Synthesis of the Nixantphos@CCM Latex
2.5. Hydroformylation Catalysis
3. Results and Discussion
3.1. Synthesis of the Ligand-Functionalized Monomer, 1
3.2. Synthesis and Characterization of the Nanoreactors Nixantphos@CCM
3.3. Biphasic Hydroformylation of 1-Octene
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cornils, B.; Herrmann, W.A. Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Cornils, B.; Herrmann, W.A. Concepts in homogeneous catalysis: The industrial view. J. Catal. 2003, 216, 23–31. [Google Scholar] [CrossRef]
- Wiese, K.-D.; Obst, D. Hydroformylation. In Catalytic Carbonylation Reactions; Beller, M., Ed.; Springer: Berlin, Germany, 2006; pp. 1–33. [Google Scholar]
- Van Leeuwen, P.W.N.M.; Claver, C. (Eds.) Rhodium Catalyzed Hydroformylation; Wiley: Hoboken, NJ, USA, 2005; Volume 22, pp. 231–269. [Google Scholar]
- Tudor, R.; Ashley, M. Enhancement of Industrial Hydroformylation Processes by the Adoption of Rhodium-Based Catalyst: Part II. Platin. Met. Rev. 2007, 51, 164–171. [Google Scholar] [CrossRef]
- Tudor, R.; Ashley, M. Enhancement of Industrial Hydroformylation Processes by the Adoption of Rhodium-Based Catalyst: Part I. Platin. Met. Rev. 2007, 51, 116–126. [Google Scholar] [CrossRef]
- Franke, R.; Selent, D.; Börner, A. Applied Hydroformylation. Chem. Rev. 2012, 112, 5675–5732. [Google Scholar] [CrossRef] [PubMed]
- Cornils, B.; Kuntz, E.G. Introducing TPPTS and related ligands for industrial biphasic processes. J. Organomet. Chem. 1995, 502, 177–186. [Google Scholar] [CrossRef]
- Bohnen, H.-W.; Cornils, B. Hydroformylation of alkenes: An industrial view of the status and importance. In Advances in Catalysis; Elsevier: Amsterdam, The Netherlands, 2002; Volume 47, pp. 1–64. [Google Scholar] [CrossRef]
- Cornils, B.; Herrmann, W.A. Aqueous-Phase Organometallic Catalysis. Chem. Ing. Tech. 1999, 71, 168–169. [Google Scholar] [CrossRef]
- De, C.; Saha, R.; Ghosh, S.K.; Ghosh, A.; Mukherjee, K.; Bhattacharyya, S.S.; Saha, B. A review of biphasic hydroformylation for long chain substrates. In Research on Chemical Intermediates; Springer: Berlin/Heidelberg, Germany, 2012; Volume 39, pp. 3463–3474. [Google Scholar] [CrossRef]
- Sharma, S.K.; Jasra, R.V. Aqueous phase catalytic hydroformylation reactions of alkenes. Catal. Today 2015, 247, 70–81. [Google Scholar] [CrossRef]
- Börner, A.; Franke, R. Hydroformylation: Fundamentals, Processes, and Applications in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2016. [Google Scholar]
- Matsinha, L.C.; Siangwata, S.; Smith, G.S.; Makhubela, B.C.E. Aqueous biphasic hydroformylation of olefins: From classical phosphine-containing systems to emerging strategies based on water-soluble nonphosphine ligands. Catal. Rev. 2018, 61, 111–133. [Google Scholar] [CrossRef]
- Obrecht, L.; Kamer, P.C.J.; Laan, W. Alternative approaches for the aqueous–organic biphasic hydroformylation of higher alkenes. Catal. Sci. Technol. 2013, 3, 541–551. [Google Scholar] [CrossRef]
- Purwanto, P.; Delmas, H. Gas-liquid-liquid reaction engineering: Hydroformylation of 1-octene using a water soluble rhodium complex catalyst. Catal. Today 1995, 24, 135–140. [Google Scholar] [CrossRef]
- Purwanto; Deshpande, R.M.; Chaudhari, R.V.; Delmas, H. Solubility of Hydrogen, Carbon Monoxide, and 1-Octene in Various Solvents and Solvent Mixtures. J. Chem. Eng. Data 1996, 41, 1414–1417. [Google Scholar] [CrossRef]
- Deshpande, R.M.; Purwanto; Delmas, H.; Chaudhari, R.V. Kinetics of Hydroformylation of 1-Octene Using [Rh(COD)Cl]2−TPPTS Complex Catalyst in a Two-Phase System in the Presence of a Cosolvent. Ind. Eng. Chem. Res. 1996, 35, 3927–3933. [Google Scholar] [CrossRef]
- Deshpande, R. Effect of pH on rate and selectivity behavior in biphasic hydroformylation of 1-octene. J. Mol. Catal. A: Chem. 1997, 126, 133–140. [Google Scholar] [CrossRef][Green Version]
- Lekhal, A.; Chaudhari, R.; Wilhelm, A.; Delmas, H. Mass transfer effects on hydroformylation catalyzed by a water soluble complex. Catal. Today 1999, 48, 265–272. [Google Scholar] [CrossRef]
- Dabbawala, A.A.; Parmar, J.N.; Jasra, R.V.; Bajaj, H.; Monflier, E. Cobalt catalyzed hydroformylation of higher olefins in the presence of chemically modified cyclodextrins. Catal. Commun. 2009, 10, 1808–1812. [Google Scholar] [CrossRef]
- Legrand, F.X.; Hapiot, F.; Tilloy, S.; Guerriero, A.; Peruzzini, M.; Gonsalvi, L.; Monflier, E. Aqueous rhodium-catalyzed hydroformylation of 1-decene in the presence of randomly methylated beta-cyclodextrin and 1,3,5-triaza-7-phosphaadamantane derivatives. Appl. Catal. A 2009, 362, 62–66. [Google Scholar] [CrossRef]
- Leclercq, L.; Hapiot, F.; Tilloy, S.; Ramkisoensing, K.; Reek, J.N.H.; Van Leeuwen, P.W.N.M.; Monflier, E. Sulfonated Xantphos Ligand and Methylated Cyclodextrin: A Winning Combination for Rhodium-Catalyzed Hydroformylation of Higher Olefins in Aqueous Medium. Organometallics 2005, 24, 2070–2075. [Google Scholar] [CrossRef]
- Tilloy, S.; Crowyn, G.; Monflier, E.; Van Leeuwen, P.W.N.M.; Reek, J.N.H. Hydroformylation of 1-decene in aqueous medium catalysed by rhodium–alkyl sulfonated diphosphines system in the presence of methylated cyclodextrins. How the flexibility of the diphosphine backbone influences the regioselectivity. New J. Chem. 2006, 30, 377–383. [Google Scholar] [CrossRef]
- Hapiot, F.; Menuel, S.; Ferreira, M.; Leger, B.; Bricout, H.; Tilloy, S.; Monflier, E. Catalysis in Cyclodextrin-Based Unconventional Reaction Media: Recent Developments and Future Opportunities. ACS Sustain. Chem. Eng. 2017, 5, 3598–3606. [Google Scholar] [CrossRef]
- Chen, H.; Li, Y.; Chen, J.; Cheng, P.; He, Y.-E.; Li, X. Micellar effect in high olefin hydroformylation catalyzed by water-soluble rhodium complex. J. Mol. Catal. A: Chem. 1999, 149, 1–6. [Google Scholar] [CrossRef]
- Riisager, A. CTAB micelles and the hydroformylation of octene with rhodium/TPPTS catalysts Evidence for the interaction of TPPTS with micelle surfaces. J. Mol. Catal. A: Chem. 2002, 189, 195–202. [Google Scholar] [CrossRef]
- Haumann, M.; Yıldız, H.; Koch, H.; Schomäcker, R.; Yildiz, H.; Schomäcker, R. Hydroformylation of 7-tetradecene using Rh-TPPTS in a microemulsion. Appl. Catal. A: Gen. 2002, 236, 173–178. [Google Scholar] [CrossRef]
- Peng, Q. Micelle effect of disulfonated cetyldiphenyl phosphine in biphasic hydroformylation of higher olefins. Catal. Commun. 2004, 5, 447–451. [Google Scholar] [CrossRef]
- Miyagawa, C.C.; Kupka, J.; Schumpe, A. Rhodium-catalyzed hydroformylation of 1-octene in micro-emulsions and micellar media. J. Mol. Catal. A: Chem. 2005, 234, 9–17. [Google Scholar] [CrossRef]
- Ünveren, H.H.Y.; Schomäcker, R.; Schomäcker, R. Rhodium catalyzed hydroformylation of 1-octene in microemulsion: Comparison with various catalytic systems. Catal. Lett. 2006, 110, 195–201. [Google Scholar] [CrossRef]
- Desset, S.L.; Cole-Hamilton, D.J.; Foster, D.F. Aqueous-biphasic hydroformylation of higher alkenes promoted by alkylimidazolium salts. Chem. Commun. 2007, 1933–1935. Available online: https://pubs.rsc.org/en/content/articlelanding/2007/cc/b618785d/unauth#!divAbstract (accessed on 12 May 2020).
- Desset, S.L.; Reader, S.W.; Cole-Hamilton, D.J. Aqueous-biphasic hydroformylation of alkenes promoted by “weak” surfactants. Green Chem. 2009, 11, 630. [Google Scholar] [CrossRef]
- Nowothnick, H.; Rost, A.; Hamerla, T.; Schomäcker, R.; Müller, C.; Vogt, D. Comparison of phase transfer agents in the aqueous biphasic hydroformylation of higher alkenes. Catal. Sci. Technol. 2013, 3, 600–605. [Google Scholar] [CrossRef]
- Potier, J.; Menuel, S.; Chambrier, M.-H.; Burylo, L.; Blach, J.-F.; Woisel, P.; Monflier, E.; Hapiot, F. Pickering Emulsions Based on Supramolecular Hydrogels: Application to Higher Olefins’ Hydroformylation. ACS Catal. 2013, 3, 1618–1621. [Google Scholar] [CrossRef]
- Vanbésien, T.; Sayede, A.; Monflier, E.; Hapiot, F. A self-emulsifying catalytic system for the aqueous biphasic hydroformylation of triglycerides. Catal. Sci. Technol. 2016, 6, 3064–3073. [Google Scholar] [CrossRef]
- Pogrzeba, T.; Schmidt, M.; Hohl, L.; Weber, A.; Buchner, G.; Schulz, J.; Schwarze, M.; Kraume, M.; Schomäcker, R. Catalytic Reactions in Aqueous Surfactant-Free Multiphase Emulsions. Ind. Eng. Chem. Res. 2016, 55, 12765–12775. [Google Scholar] [CrossRef]
- Behr, A.; Roll, R. Hydroaminomethylation in thermomorphic solvent systems. J. Mol. Catal. A: Chem. 2005, 239, 180–184. [Google Scholar] [CrossRef]
- Hamamoto, H.; Suzuki, Y.; Yamada, Y.M.A.; Tabata, H.; Takahashi, H.; Ikegami, S. A recyclable catalytic system based on a temperature-responsive catalyst. Angew. Chem. Int. Ed. 2005, 44, 4536–4538. [Google Scholar] [CrossRef] [PubMed]
- Tijani, J.; El Ali, B. Selective thermomorphic biphasic hydroformylation of higher olefins catalyzed by HRhCO(PPh3)3/P(OPh)3. Appl. Catal. A 2006, 303, 158–165. [Google Scholar] [CrossRef]
- Hermanns, E.; Hasenjäger, J.; Drießen-Hölscher, B. PEG-Modified Ligands for Catalysis and Catalyst Recycling in Thermoregulated Systems. In Organometallics in Process Chemistry; Springer: Berlin/Heidelberg, Germany, 2007; Volume 23, pp. 53–66. [Google Scholar] [CrossRef]
- Behr, A.; Henze, G.; Schomaecker, R. Thermoregulated liquid/liquid catalyst separation and recycling. Adv. Synth. Catal. 2006, 348, 1485–1495. [Google Scholar] [CrossRef]
- Shaharun, M.S.; Dutta, B.K.; Mukhtar, H.; Maitra, S. Hydroformylation of 1-octene using rhodium–phosphite catalyst in a thermomorphic solvent system. Chem. Eng. Sci. 2010, 65, 273–281. [Google Scholar] [CrossRef]
- Ji, Z.J.; Jiang, J.Y.; Wang, Y.H. A novel thermoregulated phosphine ligand used for the Rh-catalyzed hydroformylation of mixed C11–12 olefins in aqueous/organic biphasic system. Chin. Chem. Lett. 2010, 21, 515–518. [Google Scholar] [CrossRef]
- Li, K.; Wang, Y.; Jiang, J.; Jin, Z. Hydroformylation of Higher Olefins by Thermoregulated Phase-Transfer Catalysis with Rhodium Nanoparticles. Chin. J. Catal. 2010, 31, 1191–1194. [Google Scholar] [CrossRef]
- Liu, X.; Kong, F.; Zheng, X.; Jin, Z. Polyether triaryl phosphine oxides for hydroformylation of oleyl alcohol in micellar catalysis. Catal. Commun. 2003, 4, 129–133. [Google Scholar] [CrossRef]
- Zarka, M.T.; Bortenschlager, M.; Wurst, K.; Nuyken, O.; Weberskirch, R. Immobilization of a Rhodium Carbene Complex to an Amphiphilic Block Copolymer for Hydroformylation of 1-Octene under Aqueous Two-Phase Conditions. Organometallics 2004, 23, 4817–4820. [Google Scholar] [CrossRef]
- Pawar, G.; Weckesser, J.; Blechert, S.; Buchmeiser, M.R. Ring opening metathesis polymerization-derived block copolymers bearing chelating ligands: Synthesis, metal immobilization and use in hydroformylation under micellar conditions. Beilstein J. Org. Chem. 2010, 6. [Google Scholar] [CrossRef]
- Oehme, G. Micellar catalysis. A Comprehensive Handbook. In Applied Homogeneous Catalysis with Organometallic Compounds, 2nd ed.; Cornils, B., Herrmann, W.A., Eds.; Wiley-VCH: Weinheim, Germany, 2002; Volume 2, pp. 835–841. [Google Scholar]
- Reinsborough, V.C. Micellar Catalysis. In Interfacial Catalysis; Marcel Dekker: New York, NY, USA, 2002; pp. 377–390. [Google Scholar] [CrossRef]
- Oehme, G. Micellar systems. In Aqueous-Phase Organometallic Catalysis: Concepts and Applications, 2nd ed.; Cornils, B., Herrmann, W.A., Eds.; Wiley-VCH: Weinheim, Germany, 2004; pp. 256–271. [Google Scholar]
- Khan, M.N. Micellar Catalysis; CRC Press: Boca Raton, FL, USA, 2006; p. 464. [Google Scholar]
- Zhang, X.; Cardozo, A.F.; Chen, S.; Zhang, W.; Julcour, C.; Lansalot, M.; Blanco, J.-F.; Gayet, F.; Delmas, H.; Charleux, B.; et al. Core-Shell Nanoreactors for Efficient Aqueous Biphasic Catalysis. Chem. Eur. J. 2014, 20, 15505–15517. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, A.F.; Julcour, C.; Barthe, L.; Blanco, J.-F.; Chen, S.; Gayet, F.; Manoury, E.; Zhang, X.; Lansalot, M.; Charleux, B.; et al. Aqueous phase homogeneous catalysis using core-shell nano-reactors: Application to rhodium catalyzed hydroformylation of 1-octene. J. Catal. 2015, 324, 1–8. [Google Scholar] [CrossRef]
- Chen, S.; Cardozo, A.F.; Julcour, C.; Blanco, J.-F.; Barthe, L.; Gayet, F.; Lansalot, M.; D’Agosto, F.; Delmas, H.; Manoury, E.; et al. Amphiphilic core-cross-linked micelles functionalized with bis (4-methoxyphenyl) phenylphosphine as catalytic nanoreactors for biphasic hydroformylation. Polymer 2015, 72, 327–335. [Google Scholar] [CrossRef]
- Poli, R.; Chen, S.; Zhang, X.; Cardozo, A.; Lansalot, M.; D’Agosto, F.; Charleux, B.; Manoury, E.; Gayet, F.; Julcour, C.; et al. One-Pot RAFT Synthesis of Triphenylphosphine-Functionalized Amphiphilic Core-Shell Polymers and Application as Catalytic Nanoreactors in Aqueous Biphasic Hydroformylation. ACS Symp. Ser. 2015, 1188, 203–220. [Google Scholar] [CrossRef]
- Lobry, E.; Cardozo, A.F.; Barthe, L.; Blanco, J.-F.; Delmas, H.; Chen, S.; Gayet, F.; Zhang, X.; Lansalot, M.; D’Agosto, F.; et al. Core phosphine-functionalized amphiphilic nanogels as catalytic nanoreactors for aqueous biphasic hydroformylation. J. Catal. 2016, 342, 164–172. [Google Scholar] [CrossRef]
- Manoury, E.; Gayet, F.; D’Agosto, F.; Lansalot, M.; Delmas, H.; Julcour, C.; Blanco, J.-F.; Barthe, L.; Poli, R. Core-Cross-Linked Micelles and Amphiphilic Nanogels as Unimolecular Nanoreactors for Micellar-Type, Metal-Based Aqueous Biphasic Catalysis. In Effects of Nanoconfinement on Catalysis; Poli, R., Ed.; Springer: New York, NY, USA, 2017; pp. 147–172. [Google Scholar]
- Kamer, P.C.J.; Van Leeuwen, P.W.N.M.; Reek, J.N.H. Wide bite angle diphosphines: Xantphos ligands in transition metal complexes and catalysis. Acc. Chem. Res. 2001, 34, 895–904. [Google Scholar] [CrossRef]
- Van Der Veen, L.A.; Keeven, P.H.; Schoemaker, G.C.; Reek, J.N.H.; Kamer, P.C.J.; Van Leeuwen, P.W.N.M.; Lutz, M.; Spek, A.L. Origin of the Bite Angle Effect on Rhodium Diphosphine Catalyzed Hydroformylation. Organometallics 2000, 19, 872–883. [Google Scholar] [CrossRef]
- Van Leeuwen, P.W.N.M.; Kamer, P.C.J. Featuring Xantphos. Catal. Sci. Technol. 2018, 8, 26–113. [Google Scholar] [CrossRef]
- Deprele, S.; Montchamp, J.-L. Environmentally Benign Synthesis of H-Phosphinic Acids Using a Water-Tolerant, Recyclable Polymer-Supported Catalyst. Org. Lett. 2004, 6, 3805–3808. [Google Scholar] [CrossRef]
- Sandee, A.J.; Reek, J.N.H.; Kamer, P.C.J.; Van Leeuwen, P.W.N.M. A Silica-Supported, Switchable, and Recyclable Hydroformylation−Hydrogenation Catalyst. J. Am. Chem. Soc. 2001, 123, 8468–8476. [Google Scholar] [CrossRef]
- Kluwer, A.M.; Simons, C.; Knijnenburg, Q.; Van Der Vlugt, J.I.; De Bruin, B.; Reek, J.N.H. Catalyst recycling via specific non-covalent adsorption on modified silicas. Dalton Trans. 2013, 42, 3609. [Google Scholar] [CrossRef] [PubMed]
- Ricken, S.; Osinski, P.W.; Eilbracht, P.; Haag, R. A new approach to dendritic supported NIXANTPHOS-based hydroformylation catalysts. J. Mol. Catal. A: Chem. 2006, 257, 78–88. [Google Scholar] [CrossRef]
- Xu, X.; Smith, A.E.; Kirkland, S.E.; McCormick, C.L. Aqueous RAFT Synthesis of pH-Responsive Triblock Copolymer mPEO−PAPMA−PDPAEMA and Formation of Shell Cross-Linked Micelles†. Macromolecules 2008, 41, 8429–8435. [Google Scholar] [CrossRef]
- Boursier, T.; Chaduc, I.; Rieger, J.; D’Agosto, F.; Lansalot, M.; Charleux, B. Controlled radical polymerization of styrene in miniemulsion mediated by PEO-based trithiocarbonate macromolecular RAFT agents. Polym. Chem. 2011, 2, 355–362. [Google Scholar] [CrossRef]
- Mann, B.E.; Taylor, B.F. 13-C NMR Data for Organometallic Compounds; Academic Press: London, UK, 1981; pp. 200–211. [Google Scholar]
- Chiefari, J.; Chong, Y.K. (Bill); Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.T.; Mayadunne, R.T.A.; Meijs, G.F.; Moad, C.L.; Moad, G.; et al. Living Free-Radical Polymerization by Reversible Addition−Fragmentation Chain Transfer: The RAFT Process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Barner-Kowollik, C. (Ed.) Handbook of RAFT Polymerization; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Keddie, D.J.; Moad, G.; Rizzardo, E.; Thang, S.H. RAFT Agent Design and Synthesis. Macromolecules 2012, 45, 5321–5342. [Google Scholar] [CrossRef]
- Hill, M.R.; Carmean, R.N.; Sumerlin, B.S. Expanding the Scope of RAFT Polymerization: Recent Advances and New Horizons. Macromolecules 2015, 48, 5459–5469. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S. Radical Addition–Fragmentation Chemistry and RAFT Polymerization. In Reference Module in Materials Science and Materials Engineering; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Jenkins, A.D.; Jones, R.G.; Moad, G. Terminology for reversible-deactivation radical polymerization previously called “controlled” radical or “living” radical polymerization (IUPAC Recommendations 2010). Pure Appl. Chem. 2009, 82, 483–491. [Google Scholar] [CrossRef]
- Charleux, B.; Delaittre, G.; Rieger, J.; D’Agosto, F. Polymerization-Induced Self-Assembly: From Soluble Macromolecules to Block Copolymer Nano-Objects in One Step. Macromolecules 2012, 45, 6753–6765. [Google Scholar] [CrossRef]
- Warren, N.J.; Armes, S.P. Polymerization-Induced Self-Assembly of Block Copolymer Nano-objects via RAFT Aqueous Dispersion Polymerization. J. Am. Chem. Soc. 2014, 136, 10174–10185. [Google Scholar] [CrossRef]
- Canning, S.L.; Smith, G.N.; Armes, S.P. A Critical Appraisal of RAFT-Mediated Polymerization-Induced Self-Assembly. Macromolecules 2016, 49, 1985–2001. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Gayet, F.; Manoury, E.; Joumaa, A.; Lansalot, M.; D’Agosto, F.; Poli, R. Coordination Chemistry Inside Polymeric Nanoreactors: Interparticle Metal Exchange and Ionic Compound Vectorization in Phosphine-Functionalized Amphiphilic Polymer Latexes. Chem. Eur. J. 2016, 22, 6302–6313. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Rh Loading b (mol%) | P/Rh Ratio (mol/mol) | Time (h) | Pressure (bar) | Yield n-nonanal c (%) | Isomers d |
|---|---|---|---|---|---|---|---|
| 1 | decanal | 0.18 | 4 | 3 | 20 | 7 | yes |
| 2 | heptanal | 0.18 | 4 | 3 | 20 | 5 | yes |
| 3 | dodecane | 0.18 | 4 | 3 | 20 | 0 | no |
| 4 | decanal | 0.36 | 2 | 3 | 20 | 12 | yes |
| 5 | dodecane | 0.36 | 2 | 20 | 20 | 13 | traces |
| 6 | dodecane | 0.36 | 2 | 72 | 20 | 14 | yes |
| 7 | dodecane | 0.36 | 2 | 20 | 30 | 28 | traces |
| 8 e | dodecane | 0.36 | 2 | 20 | 20 | <1 | traces |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joumaa, A.; Gayet, F.; Garcia-Suarez, E.J.; Himmelstrup, J.; Riisager, A.; Poli, R.; Manoury, E. Synthesis of Nixantphos Core-Functionalized Amphiphilic Nanoreactors and Application to Rhodium-Catalyzed Aqueous Biphasic 1-Octene Hydroformylation. Polymers 2020, 12, 1107. https://doi.org/10.3390/polym12051107
Joumaa A, Gayet F, Garcia-Suarez EJ, Himmelstrup J, Riisager A, Poli R, Manoury E. Synthesis of Nixantphos Core-Functionalized Amphiphilic Nanoreactors and Application to Rhodium-Catalyzed Aqueous Biphasic 1-Octene Hydroformylation. Polymers. 2020; 12(5):1107. https://doi.org/10.3390/polym12051107
Chicago/Turabian StyleJoumaa, Ahmad, Florence Gayet, Eduardo J. Garcia-Suarez, Jonas Himmelstrup, Anders Riisager, Rinaldo Poli, and Eric Manoury. 2020. "Synthesis of Nixantphos Core-Functionalized Amphiphilic Nanoreactors and Application to Rhodium-Catalyzed Aqueous Biphasic 1-Octene Hydroformylation" Polymers 12, no. 5: 1107. https://doi.org/10.3390/polym12051107
APA StyleJoumaa, A., Gayet, F., Garcia-Suarez, E. J., Himmelstrup, J., Riisager, A., Poli, R., & Manoury, E. (2020). Synthesis of Nixantphos Core-Functionalized Amphiphilic Nanoreactors and Application to Rhodium-Catalyzed Aqueous Biphasic 1-Octene Hydroformylation. Polymers, 12(5), 1107. https://doi.org/10.3390/polym12051107