Improvement of Peptide Affinity and Stability by Complexing to Cyclodextrin-Grafted Ammonium Chitosan

 ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Analytical Methods
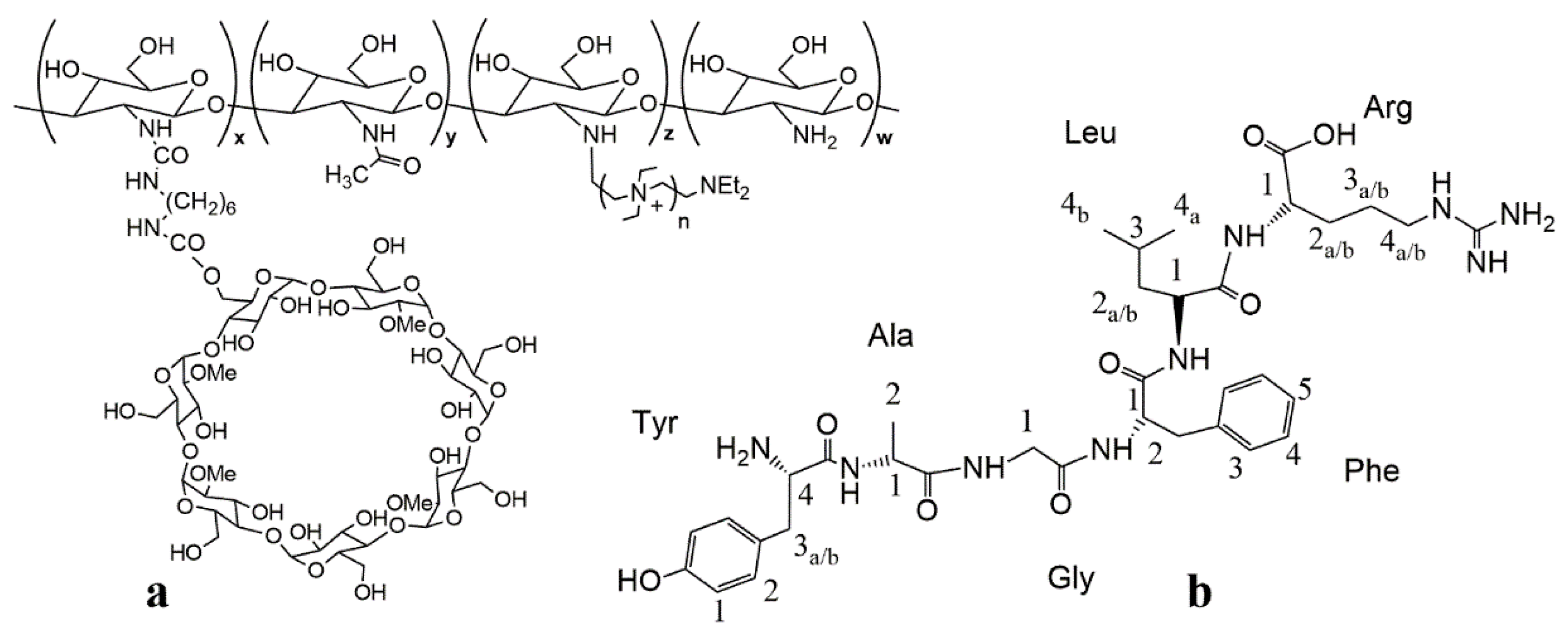
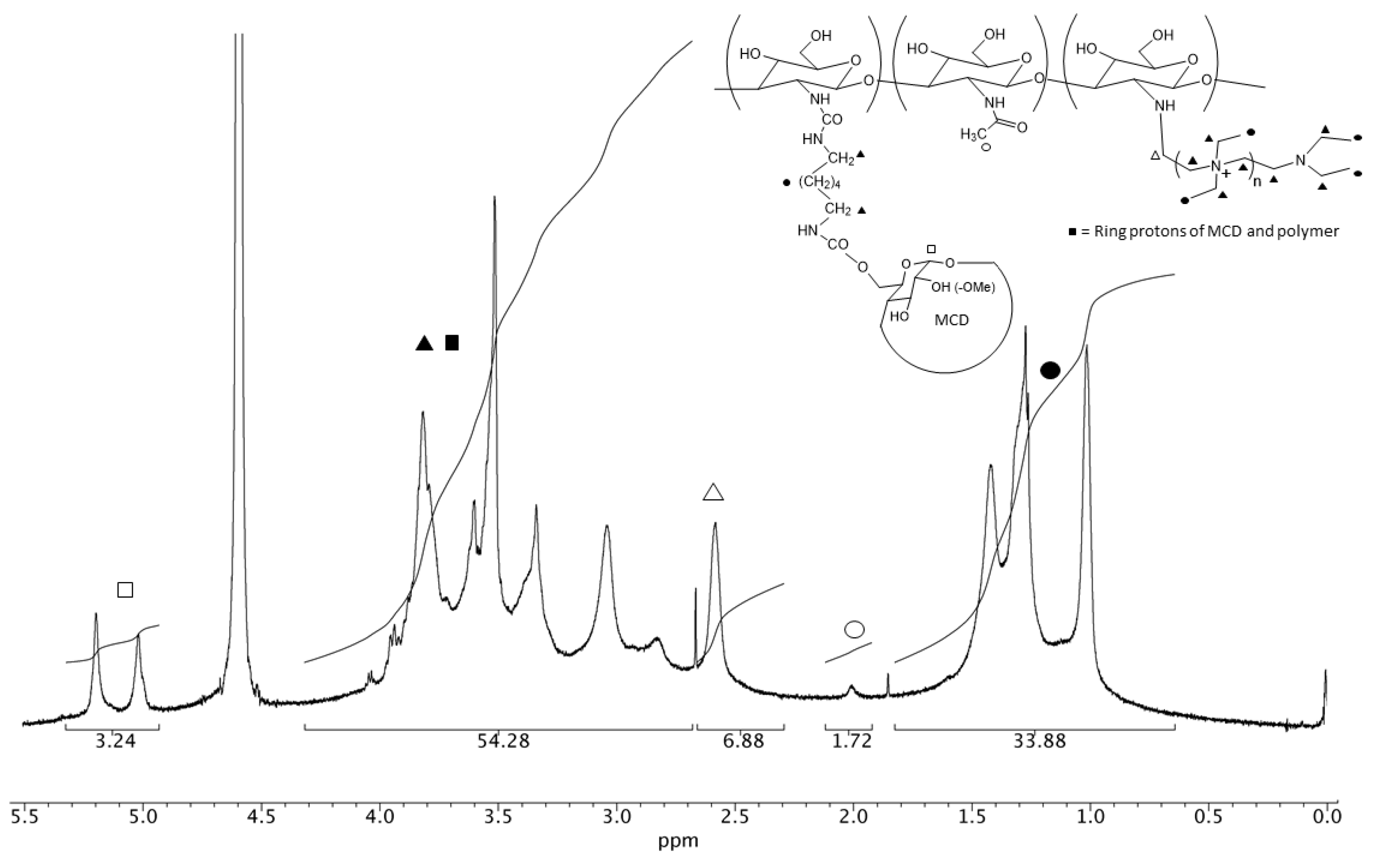
2.3. Preparation of Quaternary Methyl-β-Cyclodextrin Ammonium–Chitosan Conjugate
2.4. Characterization of DAL Inclusion Complexes
2.4.1. Complexes Stoichiometry (Job’s Plot)
2.4.2. Evaluation of Complex Association Constant (Benesi–Hildebrand Method)
2.5. Stability under Enzymatic Hydrolyses
2.6. Sample Preparation for NMR Studies
2.6.1. Affinity Studies
2.6.2. Enzymatic Hydrolyses
2.7. Biological Evaluation
2.7.1. Cell Viability
2.7.2. In Vitro Evaluation of Protection from Enzymatic Degradation
3. Results and Discussion
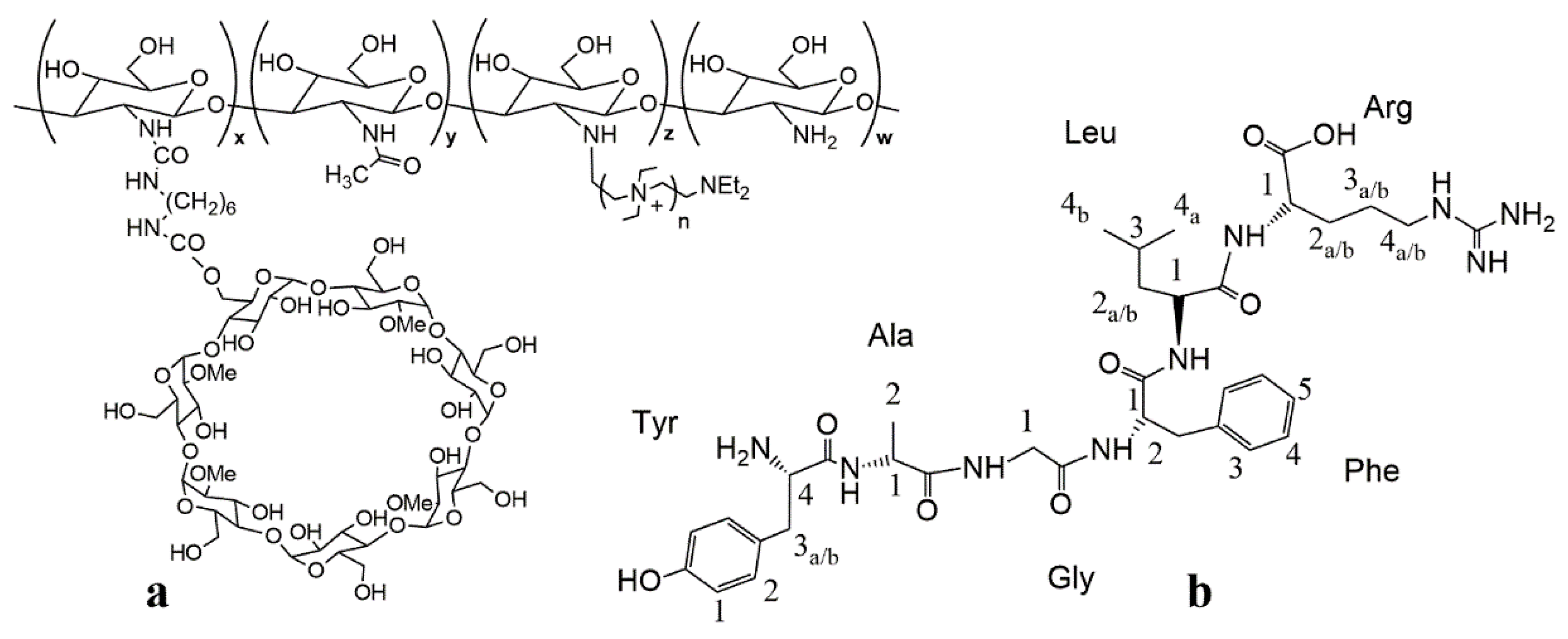
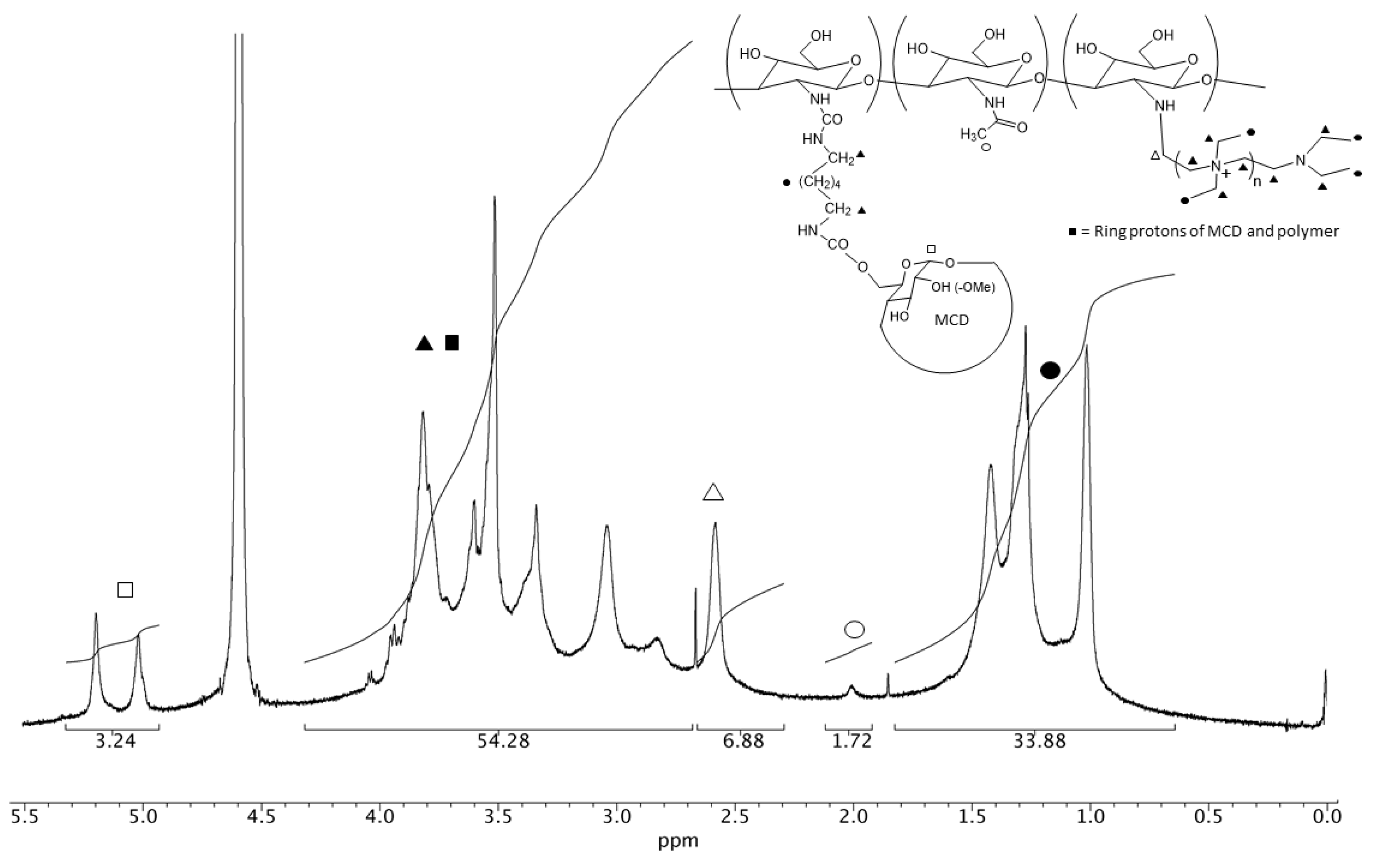
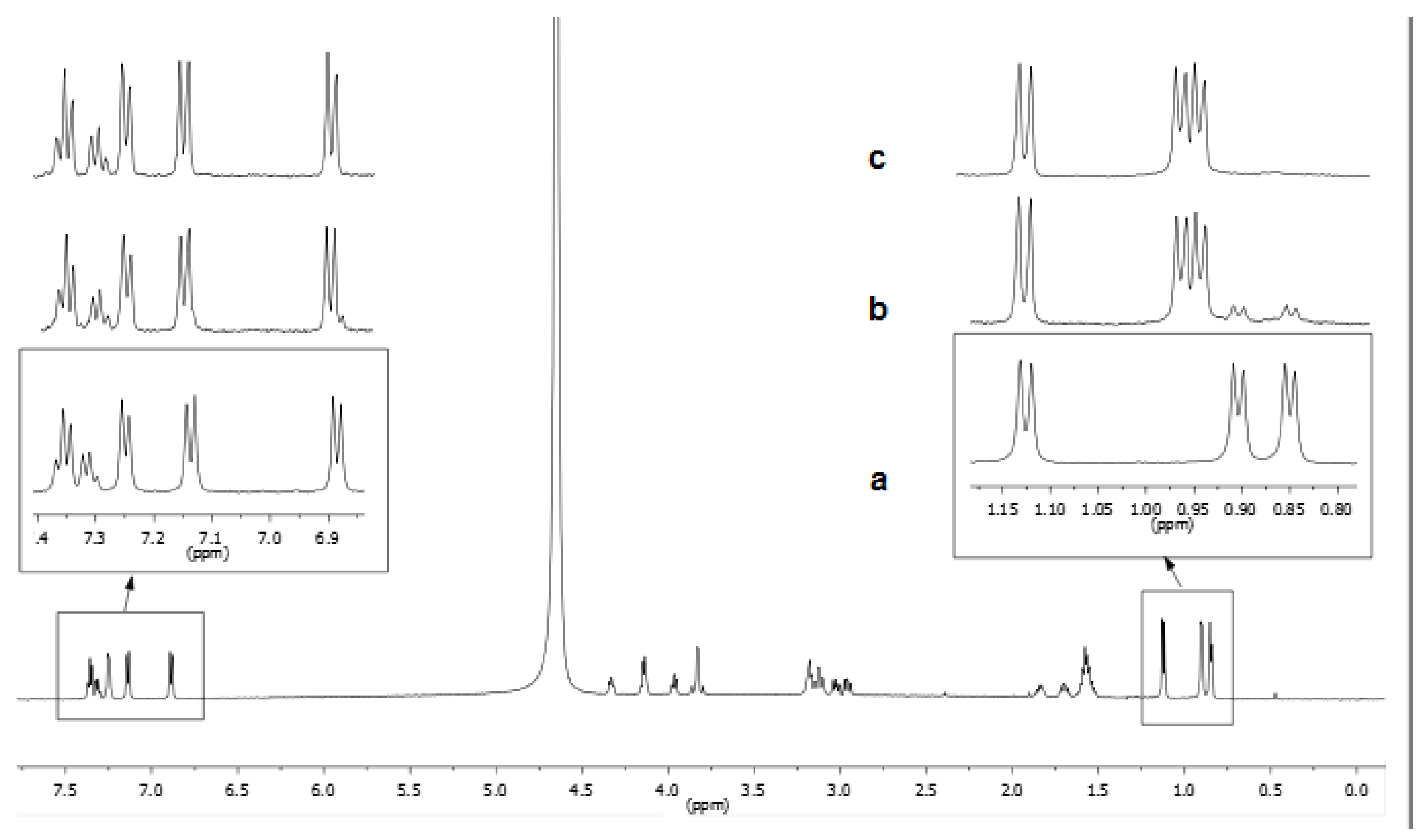
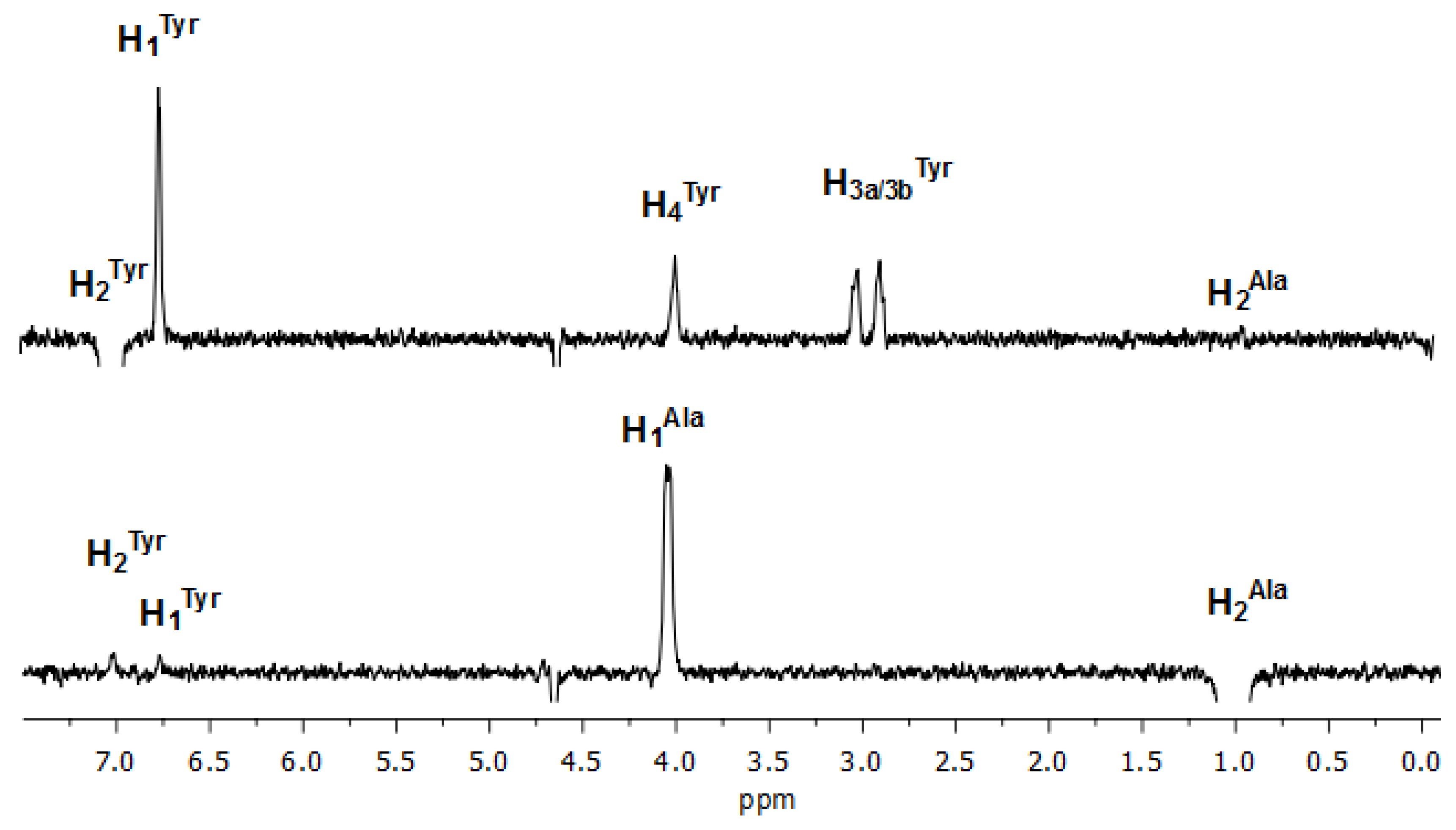
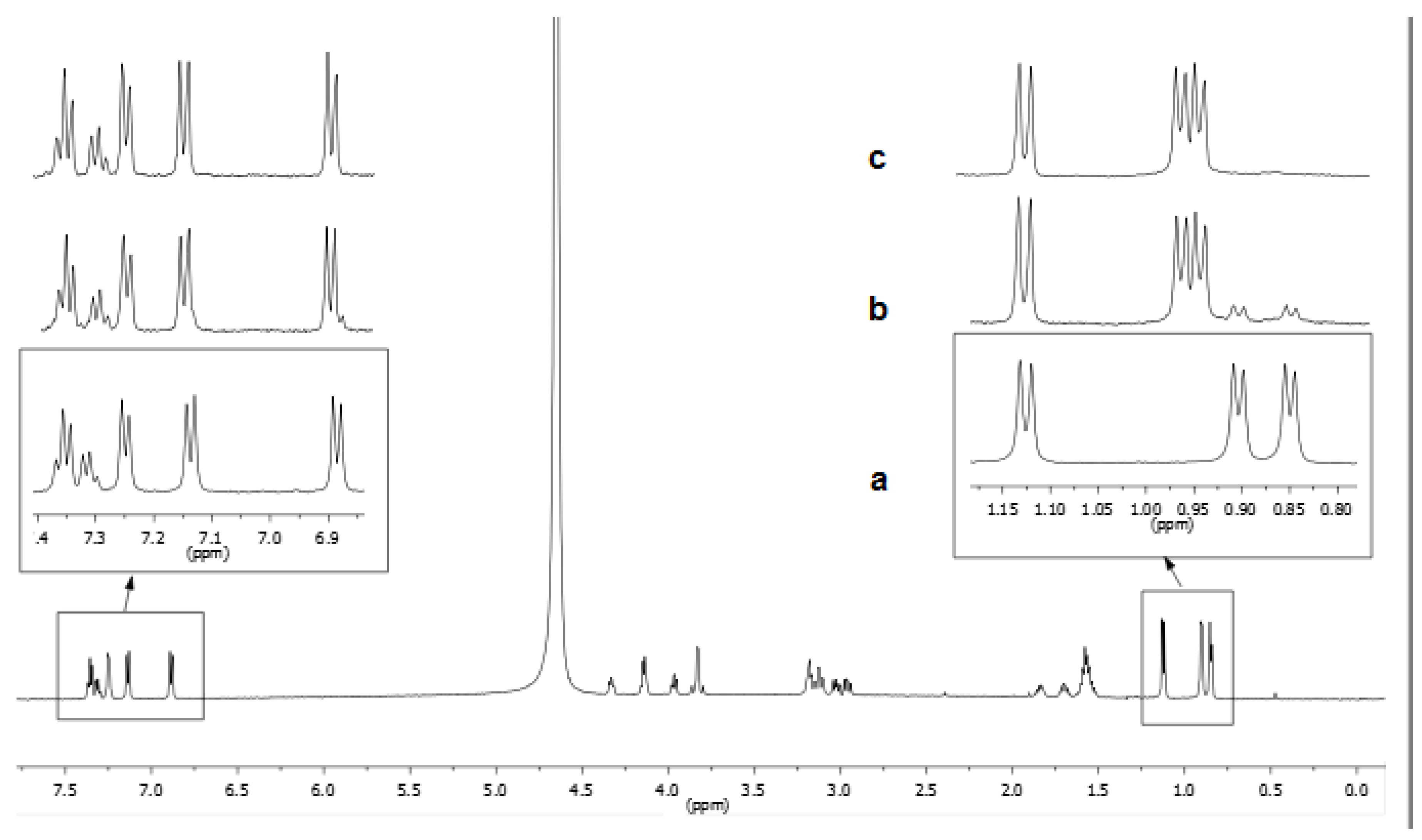
3.1. NMR Characterization
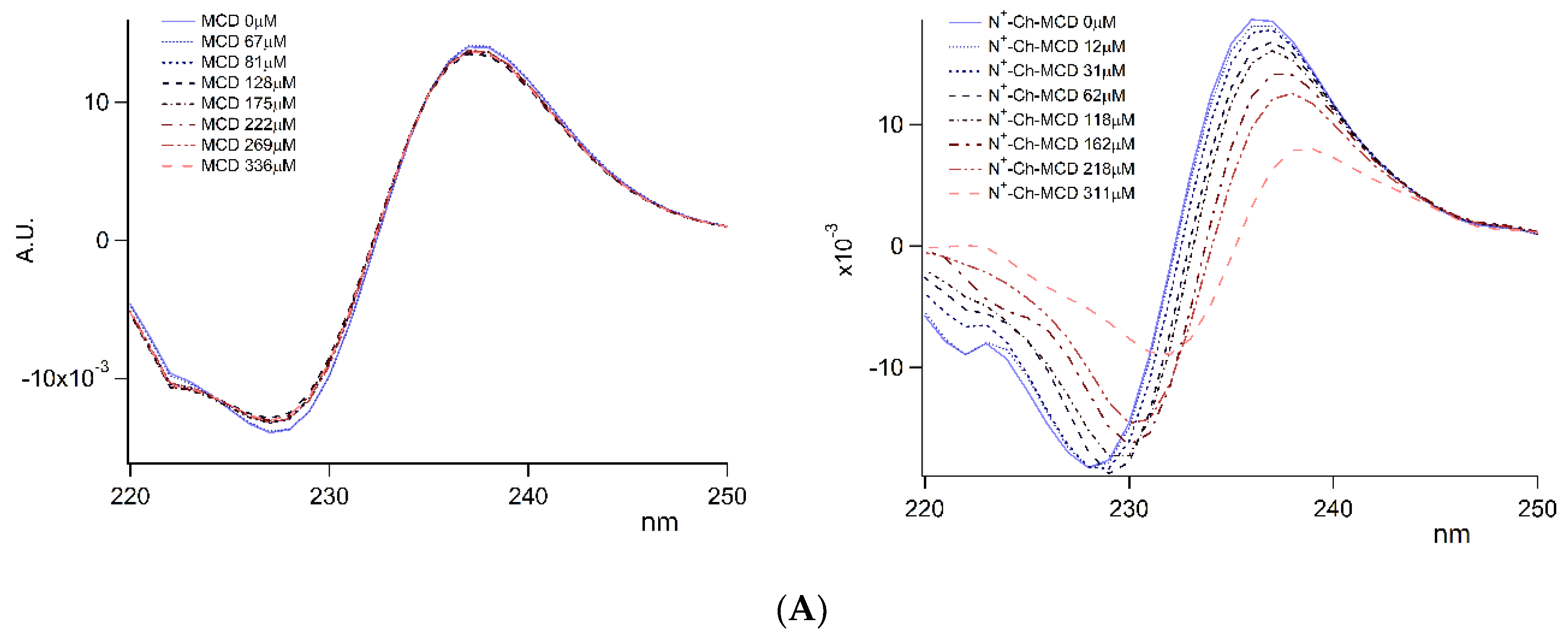
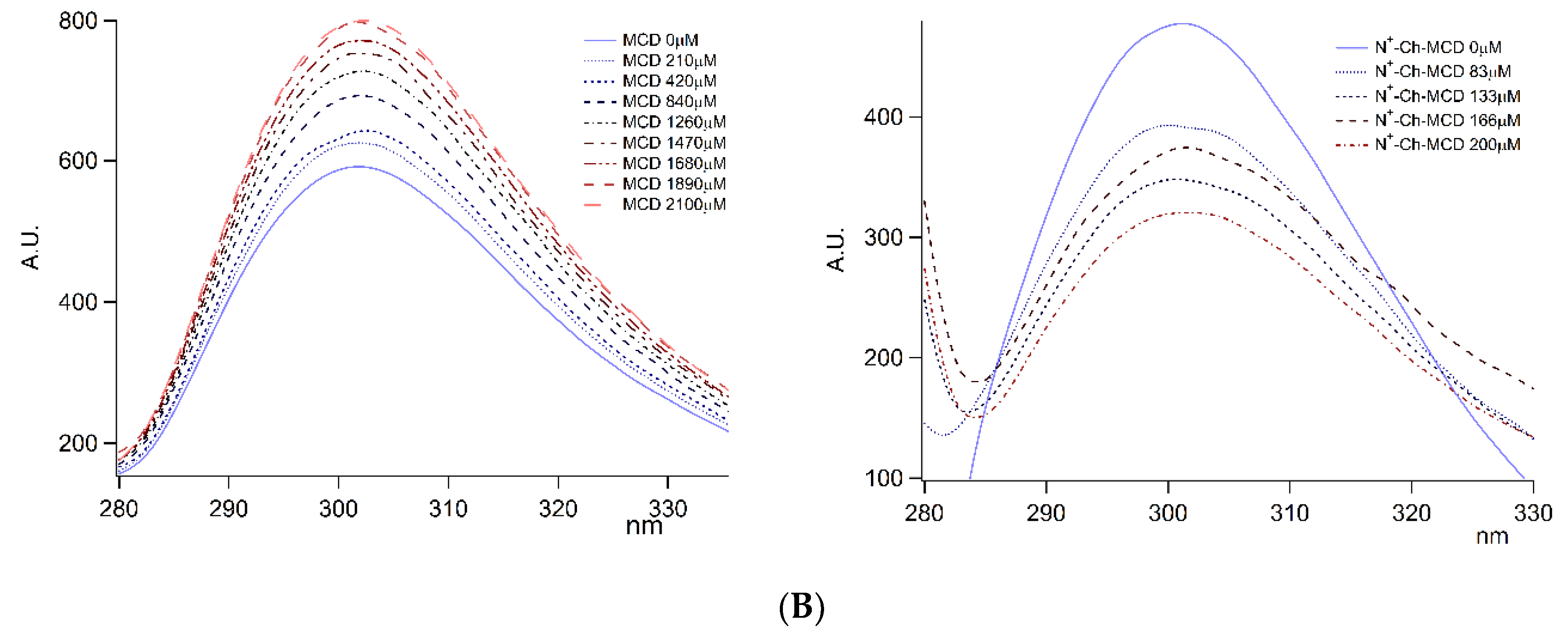
3.2. Inclusion Complexes
3.2.1. Complexes Stoichiometry and Association Constant
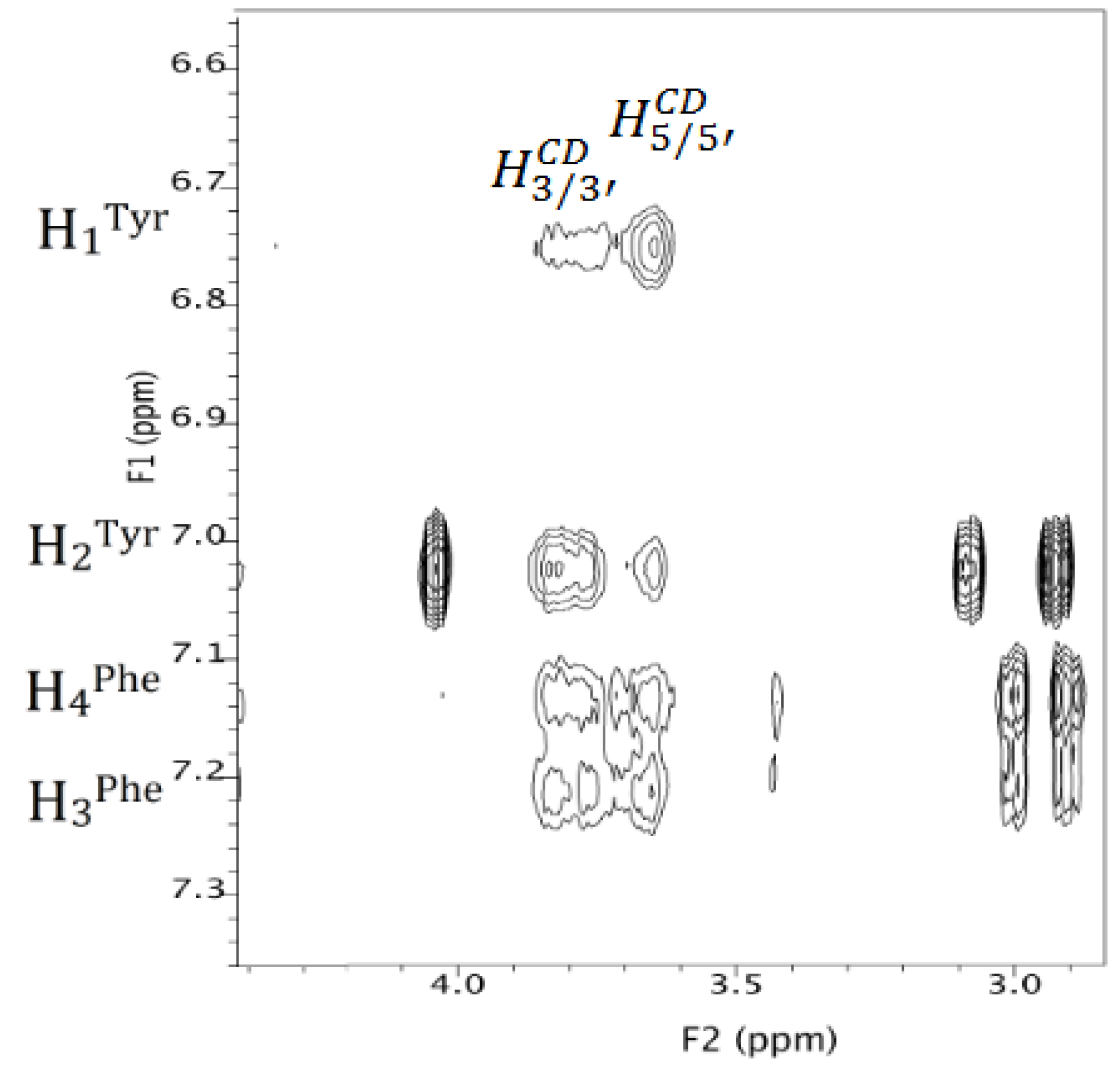
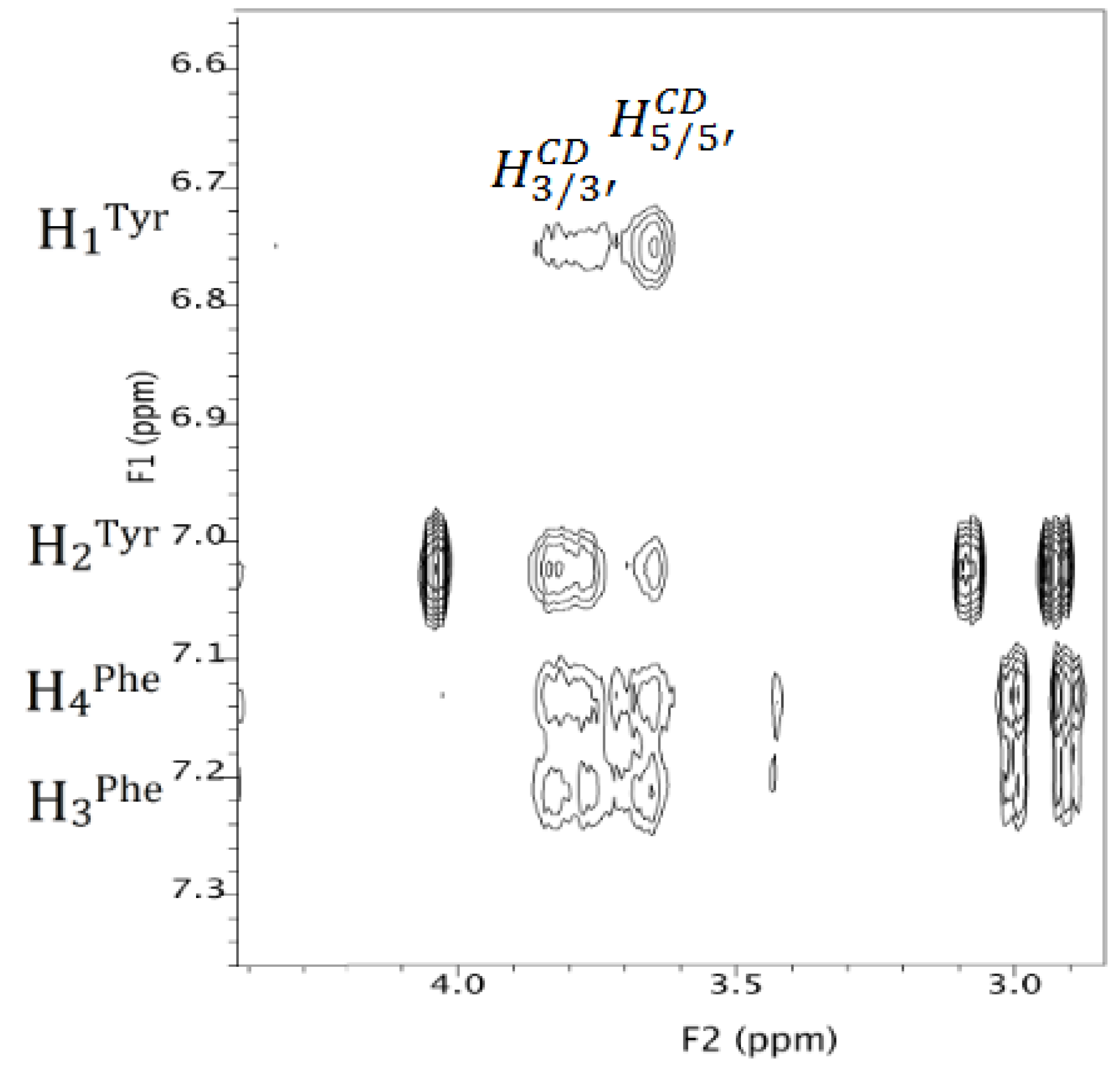
3.2.2. NMR Interaction Studies and Complex Stereochemistry
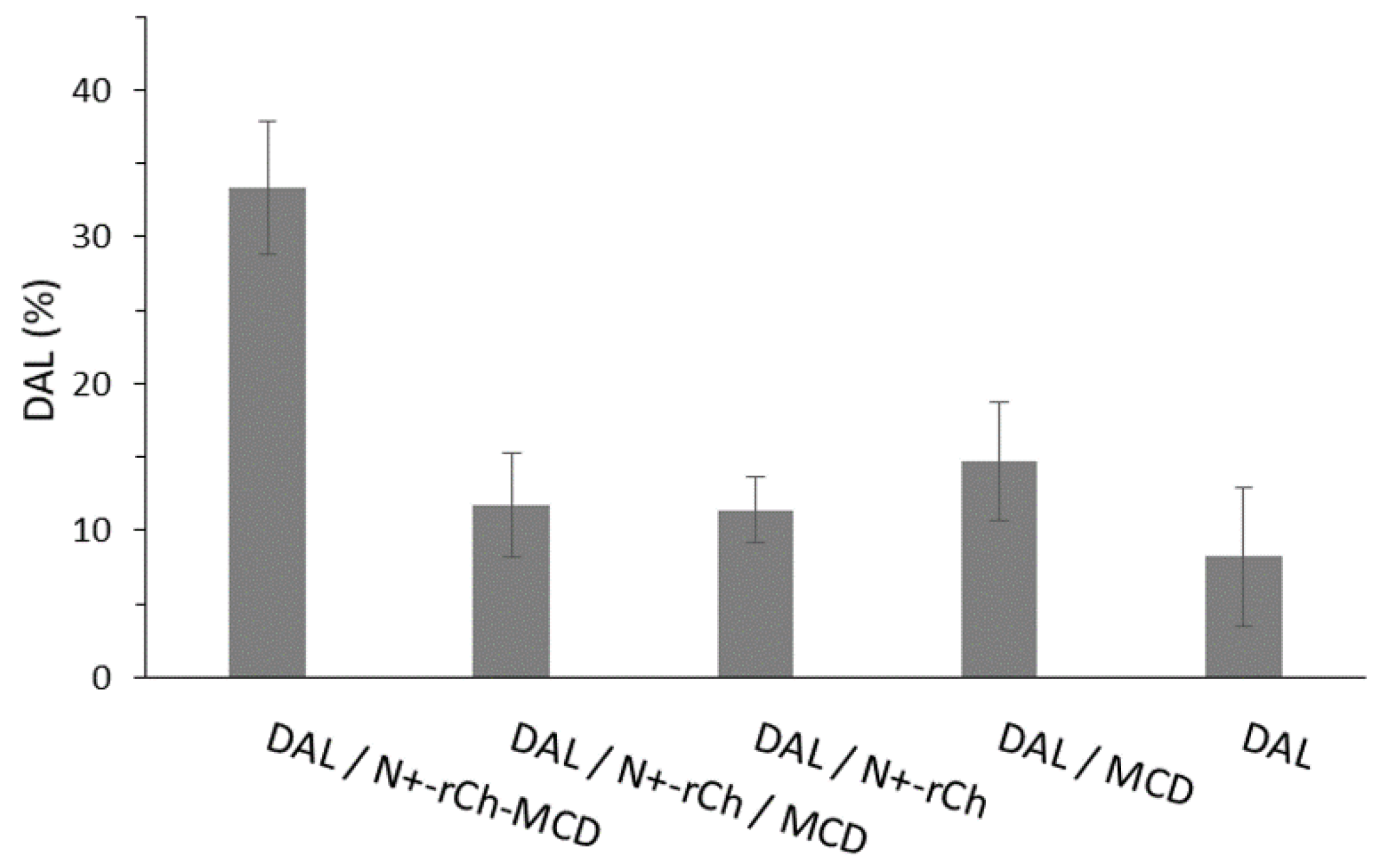
3.3. Kinetic Studies of Dalargin Enzymatic Hydrolysis
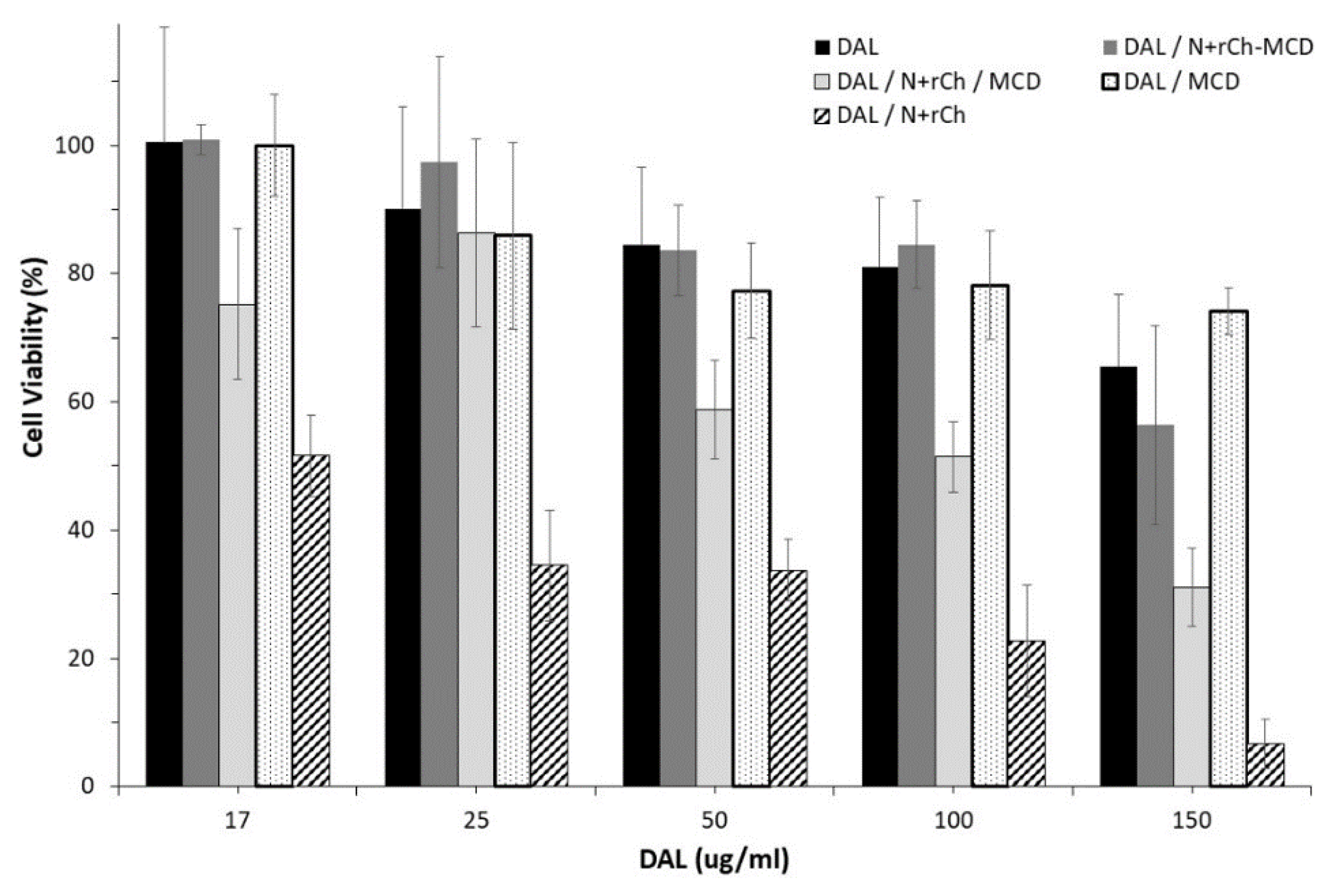
3.4. Biological Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BCS | bovine fetal serum |
| Ch | chitosan |
| CHT | α-chymotrypsin |
| DAL | dalargin |
| DEAE-Cl HCl | 2 diethylaminoethyl chloride hydrochloride |
| DMSO | dimethyl sulfoxide |
| DPBS | Dulbecco’s phosphate buffer |
| HMDI | 1,6-hexamethylene diisocyanate |
| MCD | 2-methyl-β-cyclodextrin |
| MEM | minimum essential medium |
| N+-rCh | reduced molecular weight ammonium chitosan |
| N+-rCh-MCD | ammonium-chitosan grafted with 2-methyl-β-cyclodextrin |
| rCh | reduced molecular weight chitosan |
| TEA | triethylamine |
References
- Kosaraju, S.L. Colon targeted delivery systems: Review of polysaccharides for encapsulation and delivery. Crit. Rev. Food Sci. Nutr. 2005, 45, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Zambito, Y.; Uccello-Barretta, G.; Zaino, C.; Balzano, F.; Di Colo, G. Novel transmucosal absorption enhancers obtained by aminoalkylation of chitosan. Eur. J. Pharm. Sci. 2006, 29, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for drug delivery systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, M.; Goswami, P.; Paritosh, K.; Kumar, M.; Pareek, N.; Vivekanand, V. Seafood waste: A source for preparation of commercially employable chitin/chitosan materials. Bioresour. Bioprocess. 2019, 6. [Google Scholar] [CrossRef]
- Dash, M.; Chiellini, F.; Ottenbrite, R.M.; Chiellini, E. Chitosan—A versatile semi-synthetic polymer in biomedical applications. Prog. Polym. Sci. (Oxford) 2011, 36, 981–1014. [Google Scholar] [CrossRef]
- Zhang, J.; Xia, W.; Liu, P.; Cheng, Q.; Tahirou, T.; Gu, W.; Li, B. Chitosan modification and pharmaceutical/biomedical applications. Mar. Drugs 2010, 8, 1962–1987. [Google Scholar] [CrossRef] [Green Version]
- Harish Prashanth, K.V.; Tharanathan, R.N. Chitin/chitosan: Modifications and their unlimited application potential-an overview. Trends Food Sci. Technol. 2007, 18, 117–131. [Google Scholar] [CrossRef]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. (Oxford) 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Aiello, F.; Balzano, F.; Carpita, L.; Fabiano, A.; Zambito, Y.; Uccello Barretta, G. Role of nanostructured aggregation of chitosan derivatives on [5-methionine]enkephalin affinity. Carbohydr. Polym. 2017, 157, 321–324. [Google Scholar] [CrossRef]
- Uccello-Barretta, G.; Balzano, F.; Aiello, F.; Senatore, A.; Fabiano, A.; Zambito, Y. Mucoadhesivity and release properties of quaternary ammonium-chitosan conjugates and their nanoparticulate supramolecular aggregates: An NMR investigation. Int. J. Pharm. 2014, 461, 489–494. [Google Scholar] [CrossRef]
- Zambito, Y.; Zaino, C.; Uccello-Barretta, G.; Balzano, F.; Di Colo, G. Improved synthesis of quaternary ammonium-chitosan conjugates (N+-Ch) for enhanced intestinal drug permeation. Eur. J. Pharm. Sci. 2008, 33, 343–350. [Google Scholar] [CrossRef]
- Cesari, A.; Fabiano, A.; Piras, A.M.; Zambito, Y.; Uccello-Barretta, G.; Balzano, F. Binding and mucoadhesion of sulfurated derivatives of quaternary ammonium-chitosans and their nanoaggregates: An NMR investigation. J. Pharm. Biomed. Anal. 2020, 177. [Google Scholar] [CrossRef] [PubMed]
- Fabiano, A.; Piras, A.M.; Uccello-Barretta, G.; Balzano, F.; Cesari, A.; Testai, L.; Citi, V.; Zambito, Y. Impact of mucoadhesive polymeric nanoparticulate systems on oral bioavailability of a macromolecular model drug. Eur. J. Pharm. Biopharm. 2018, 130, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Dünnhaupt, S.; Barthelmes, J.; Thurner, C.C.; Waldner, C.; Sakloetsakun, D.; Bernkop-Schnürch, A. S-protected thiolated chitosan: Synthesis and in vitro characterization. Carbohydr. Polym. 2012, 90, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambito, Y.; Felice, F.; Fabiano, A.; Di Stefano, R.; Di Colo, G. Mucoadhesive nanoparticles made of thiolated quaternary chitosan crosslinked with hyaluronan. Carbohydr. Polym. 2013, 92, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Zambito, Y.; Fogli, S.; Zaino, C.; Stefanelli, F.; Breschi, M.C.; Di Colo, G. Synthesis, characterization and evaluation of thiolated quaternary ammonium-chitosan conjugates for enhanced intestinal drug permeation. Eur. J. Pharm. Sci. 2009, 38, 112–120. [Google Scholar] [CrossRef]
- Auzély-Velty, R.; Rinaudo, M. Chitosan derivatives bearing pendant cyclodextrin cavities: Synthesis and inclusion performance. Macromolecules 2001, 34, 3574–3580. [Google Scholar] [CrossRef]
- Song, M.; Li, L.; Zhang, Y.; Chen, K.; Wang, H.; Gong, R. Carboxymethyl-β-cyclodextrin grafted chitosan nanoparticles as oral delivery carrier of protein drugs. React. Funct. Polym. 2017, 117, 10–15. [Google Scholar] [CrossRef]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J. Pharm. Sci. 1996, 85, 1017–1025. [Google Scholar] [CrossRef]
- Loftsson, T.; Duchêne, D. Cyclodextrins and their pharmaceutical applications. Int. J. Pharm. 2007, 329, 1–11. [Google Scholar] [CrossRef]
- Piras, A.M.; Zambito, Y.; Burgalassi, S.; Monti, D.; Tampucci, S.; Terreni, E.; Fabiano, A.; Balzano, F.; Uccello-Barretta, G.; Chetoni, P. A water-soluble, mucoadhesive quaternary ammonium chitosan-methyl-β-cyclodextrin conjugate forming inclusion complexes with dexamethasone. J. Mater. Sci. Mater. Med. 2018, 29. [Google Scholar] [CrossRef] [PubMed]
- Fleishman, M.U.; Zhivotova, E.U.; Lebedko, O.A.; Deigin, V.I.; Timoshin, S.S. Mechanisms for the effect of arginine-containing dermorphin analogue on proliferative processes in the gastric mucosa of albino rats. Bull. Exp. Biol. Med. 2007, 144, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Maslov, L.N.; Lishmanov, Y.B. The anti-arrhythmic effect of d-Ala 2, Leu 5, Arg 6-enkephalin and its possible mechanism. Int. J. Cardiol. 1993, 40, 89–94. [Google Scholar] [CrossRef]
- Zhivotova, E.Y.; Fleishman, M.Y.; Sazonova, E.N.; Lebed’ko, O.A.; Timoshin, S.S. Gastroprotective effect of dalargin in gastropathy due to treatment with nonsteroid antiinflammatory drugs. Bull. Exp. Biol. Med. 2009, 147, 441–443. [Google Scholar] [CrossRef]
- Zivotova, E.U.; Fleishman, M.U.; Lebedko, O.A.; Sazonova, E.N.; Timoshin, S.S. Effect of dalargin on DNA synthesis in the gastric mucosa of albino rats. Bull. Exp. Biol. Med. 2007, 144, 314–316. [Google Scholar] [CrossRef]
- Aliautdin, R.N.; Petrov, V.E.; Ivanov, A.A.; Kreuter, J.; Kharkevich, D.A. Transport of the hexapeptide dalargin across the hemato-encephalic barrier into the brain using polymer nanoparticles. Eksperimental’naia Klin. Farmakol. 1996, 59, 57–60. [Google Scholar]
- AboulFotouh, K.; Allam, A.A.; El-Badry, M.; El-Sayed, A.M. Role of self-emulsifying drug delivery systems in optimizing the oral delivery of hydrophilic macromolecules and reducing interindividual variability. Colloids Surf. B Biointerfaces 2018, 167, 82–92. [Google Scholar] [CrossRef]
- Chen, Y.; Siddalingappa, B.; Chan, P.H.H.; Benson, H.A.E. Development of a chitosan-based nanoparticle formulation for delivery of a hydrophilic hexapeptide, dalargin. Biopolym. Pept. Sci. Sect. 2008, 90, 663–670. [Google Scholar] [CrossRef]
- Martin-Serrano, Á.; Gómez, R.; Ortega, P.; Mata, F.J.D.L. Nanosystems as vehicles for the delivery of antimicrobial peptides (Amps). Pharmaceutics 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Pan, D.; Yi, J.; Hao, L.; Kang, Q.; Liu, X.; Lu, L.; Lu, J. Protective effect of β-cyclodextrin on stability of nisin and corresponding interactions involved. Carbohydr. Polym. 2019, 223. [Google Scholar] [CrossRef] [PubMed]
- Freeman, R.; Wittekoek, S. Selective determination of relaxation times in high resolution NMR. J. Magn. Reson. (1969) 1969, 1, 238–276. [Google Scholar] [CrossRef]
- Mao, S.; Shuai, X.; Unger, F.; Simon, M.; Bi, D.; Kissel, T. The depolymerization of chitosan: Effects on physicochemical and biological properties. Int. J. Pharm. 2004, 281, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Benesi, H.A.; Hildebrand, J.H. A Spectrophotometric Investigation of the Interaction of Iodine with Aromatic Hydrocarbons. J. Am. Chem. Soc. 1949, 71, 2703–2707. [Google Scholar] [CrossRef]
- Barman, S.; Barman, B.K.; Roy, M.N. Preparation, characterization and binding behaviors of host-guest inclusion complexes of metoclopramide hydrochloride with α- and β-cyclodextrin molecules. J. Mol. Struct. 2018, 1155, 503–512. [Google Scholar] [CrossRef]
- Zupančič, O.; Rohrer, J.; Thanh Lam, H.; Grießinger, J.A.; Bernkop-Schnürch, A. Development and in vitro characterization of self-emulsifying drug delivery system (SEDDS) for oral opioid peptide delivery. Drug Dev. Ind. Pharm. 2017, 43, 1694–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubatsch, I.; Ragnarsson, E.G.E.; Artursson, P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Schmid, F.-X. Biological Macromolecules: UV-visible Spectrophotometry. In eLS; Wiley: Hoboken, NJ, USA, 2001. [Google Scholar]
- Yang, H.; Xiao, X.; Zhao, X.; Wu, Y. Intrinsic fluorescence spectra of tryptophan, tyrosine and phenyloalanine. In Proceedings of the SPIE—The International Society for Optical Engineering, Jinhua, Suzhou, Chengdu, Xi’an, Wuxi, China, October and November 2016. [Google Scholar]
- Valensin, G.; Kushnir, T.; Navon, G. Selective and nonselective proton spin-lattice relaxation studies of enzyme-substrate interactions. J. Magn. Reson. (1969) 1982, 46, 23–29. [Google Scholar] [CrossRef]
- Gráf, L.A.; Szilágyi, L.A.; Venekei, I.A. Chymotrypsin. In Handbook of Proteolytic Enzymes; Academic Press, Elsevier: San Diego, CA, USA, 2013; Volume 3, pp. 2626–2633. [Google Scholar]
- Loftsson, T.; Brewster, M.E. Cyclodextrins as functional excipients: Methods to enhance complexation efficiency. J. Pharm. Sci. 2012, 101, 3019–3032. [Google Scholar] [CrossRef]
- Piras, A.M.; Fabiano, A.; Chiellini, F.; Zambito, Y. Methyl-β-cyclodextrin quaternary ammonium chitosan conjugate: Nanoparticles vs. macromolecular soluble complex. Int. J. Nanomed. 2018, 13, 2531–2541. [Google Scholar] [CrossRef] [Green Version]
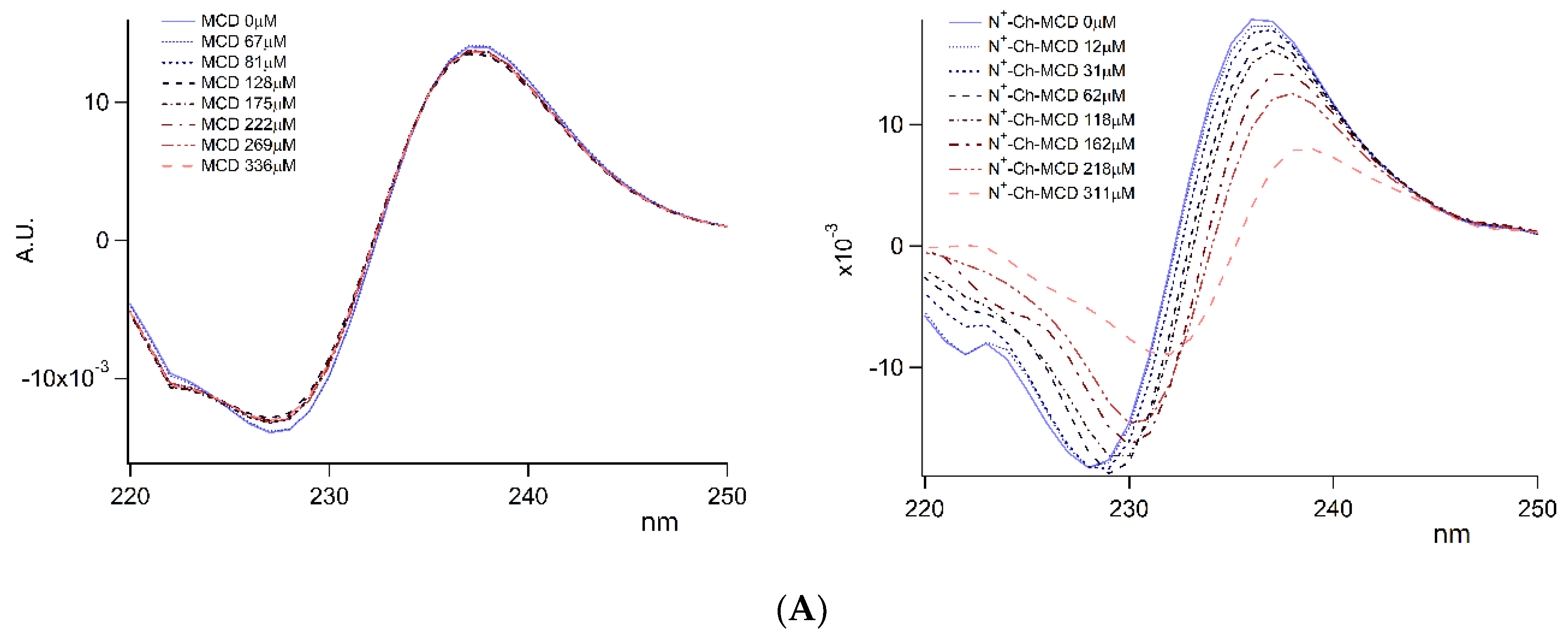
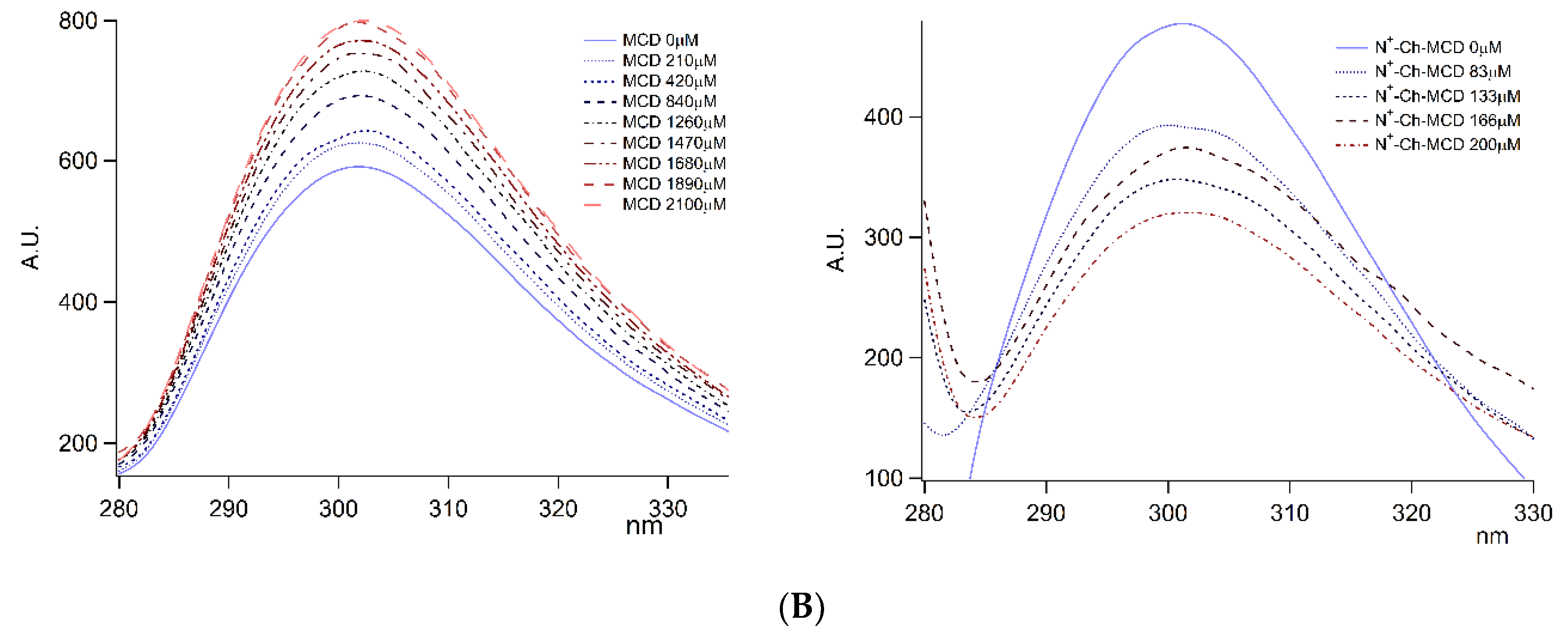

 ); on lyophilized complexes (
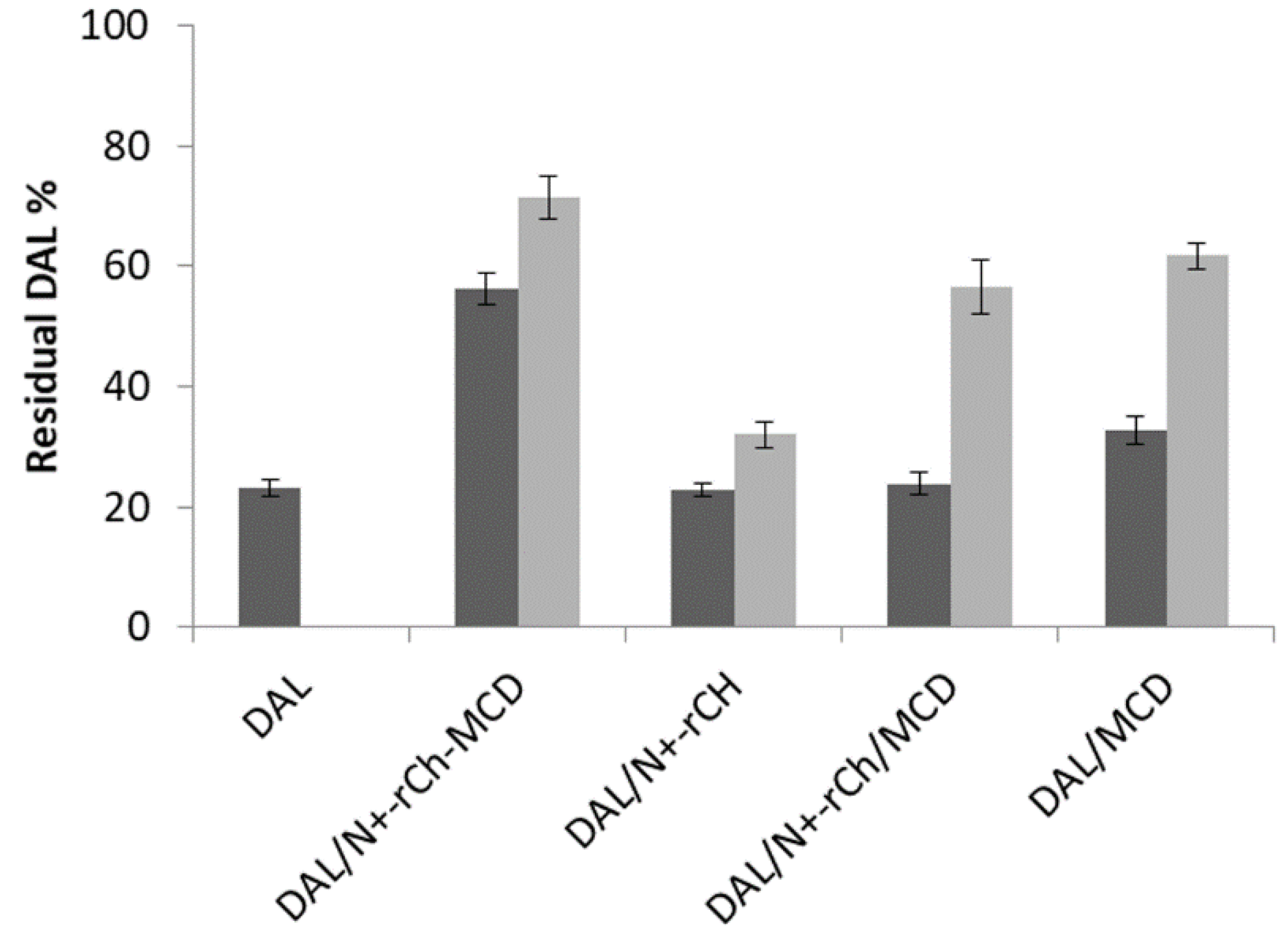
); on lyophilized complexes (  ). Error bars indicate the SD values of three independent experiments.
); on lyophilized complexes ( ). Error bars indicate the SD values of three independent experiments.
). Error bars indicate the SD values of three independent experiments.
); on lyophilized complexes ( ). Error bars indicate the SD values of three independent experiments.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Absorbance | Fluorescence | |||||
|---|---|---|---|---|---|---|
| Ka (M−1) | SD | R2 | Ka (M−1) | SD | R2 | |
| DAL/MCD | - | - | - | 120 | 10 | 0.999 |
| DAL/N+-rCh-MCD | 2326 | 406 | 0.999 | 2617 | 307 | 0.990 |
| δ (ppm) | R1ms (s−1) | |||||
|---|---|---|---|---|---|---|
| DAL | DAL/MCD | DAL/N+-rCh | DAL/N+-rCh-MCD | DAL/MCD/N+-rCh | ||
| H3Phe | 7.11 | 0.62 | 0.59 | 0.61 | 4.54 | 0.60 |
| H4Phe | 7.22 | 0.54 | 0.54 | 0.52 | 4.35 | 0.50 |
| H5Phe | 7.18 | 0.66 | 0.57 | 0.61 | 5.00 | 0.53 |
| H1Tyr | 6.77 | 0.36 | 0.32 | 0.34 | 4.54 | 0.34 |
| H2Tyr | 7.02 | 0.58 | 0.57 | 0.55 | 4.54 | 0.54 |
| H4aLeu | 0.78 | 1.69 | 1.65 | 1.67 | 4.17 | 1.67 |
| H4bLeu | 0.72 | 1.59 | 1.58 | 1.59 | 4.17 | 1.59 |
| R1bs (s−1) | ||||||
| H3Phe | 7.11 | 0.63 | 0.60 | 0.62 | 3.66 | 0.62 |
| H4Phe | 7.22 | 0.55 | 0.55 | 0.54 | 3.47 | 0.53 |
| H1Tyr | 6.77 | 0.39 | 0.35 | 0.36 | 3.92 | 0.37 |
| H2Tyr | 7.02 | 0.61 | 0.60 | 0.57 | 3.92 | 0.57 |
| σ (s−1) | ||||||
| H3/H4Phe | - | 0.01 | 0.01 | 0.01 | −0.88 | 0.02 |
| H1/H2Tyr | - | 0.03 | 0.03 | 0.02 | −0.62 | 0.03 |
| DAL/N+-rCh-MCD | |||||||
|---|---|---|---|---|---|---|---|
| H3Phe | H4Phe | H5Phe | H1Tyr | H2Tyr | H4aLeu | H4bLeu | |
| |R| | 6.32 | 7.06 | 6.58 | 11.6 | 6.83 | 1.47 | 1.62 |
| MCD | |Δδ| (Hz) | MCD | |Δδ| (Hz) |
|---|---|---|---|
| H1 | 9.5 | H4 | 4.2 |
| H1′ | 4.1 | H4′ | 1.2 |
| H2 | 4.5 | H5 | 41.8 |
| H2′ | 6.1 | H5′ | 49.2 |
| H3 | 27.6 | H6/H6′ | 3.2 |
| H3′ | 25.3 |
| Mixture | tf (min) |
|---|---|
| DAL/CHT/MCD | 16 |
| DAL/CHT/N+-rCh | 18 |
| DAL/CHT/N+-rCh/MCD | 17 |
| DAL/CHT/N+-rCh-MCD | 61 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cesari, A.; Recchimurzo, A.; Fabiano, A.; Balzano, F.; Rossi, N.; Migone, C.; Uccello-Barretta, G.; Zambito, Y.; Piras, A.M. Improvement of Peptide Affinity and Stability by Complexing to Cyclodextrin-Grafted Ammonium Chitosan. Polymers 2020, 12, 474. https://doi.org/10.3390/polym12020474
Cesari A, Recchimurzo A, Fabiano A, Balzano F, Rossi N, Migone C, Uccello-Barretta G, Zambito Y, Piras AM. Improvement of Peptide Affinity and Stability by Complexing to Cyclodextrin-Grafted Ammonium Chitosan. Polymers. 2020; 12(2):474. https://doi.org/10.3390/polym12020474
Chicago/Turabian StyleCesari, Andrea, Alessandra Recchimurzo, Angela Fabiano, Federica Balzano, Nicolò Rossi, Chiara Migone, Gloria Uccello-Barretta, Ylenia Zambito, and Anna Maria Piras. 2020. "Improvement of Peptide Affinity and Stability by Complexing to Cyclodextrin-Grafted Ammonium Chitosan" Polymers 12, no. 2: 474. https://doi.org/10.3390/polym12020474
APA StyleCesari, A., Recchimurzo, A., Fabiano, A., Balzano, F., Rossi, N., Migone, C., Uccello-Barretta, G., Zambito, Y., & Piras, A. M. (2020). Improvement of Peptide Affinity and Stability by Complexing to Cyclodextrin-Grafted Ammonium Chitosan. Polymers, 12(2), 474. https://doi.org/10.3390/polym12020474