Triple-Shape Memory Behavior of Modified Lactide/Glycolide Copolymers

 , ,
, , 
Abstract
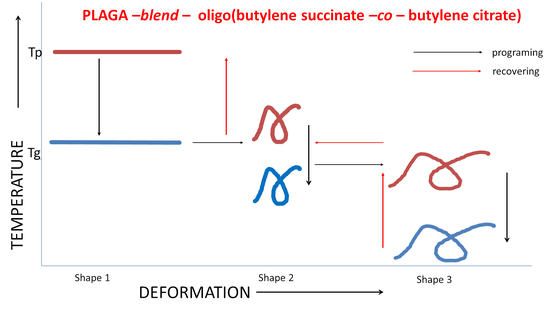
:
1. Introduction
2. Materials and Methods
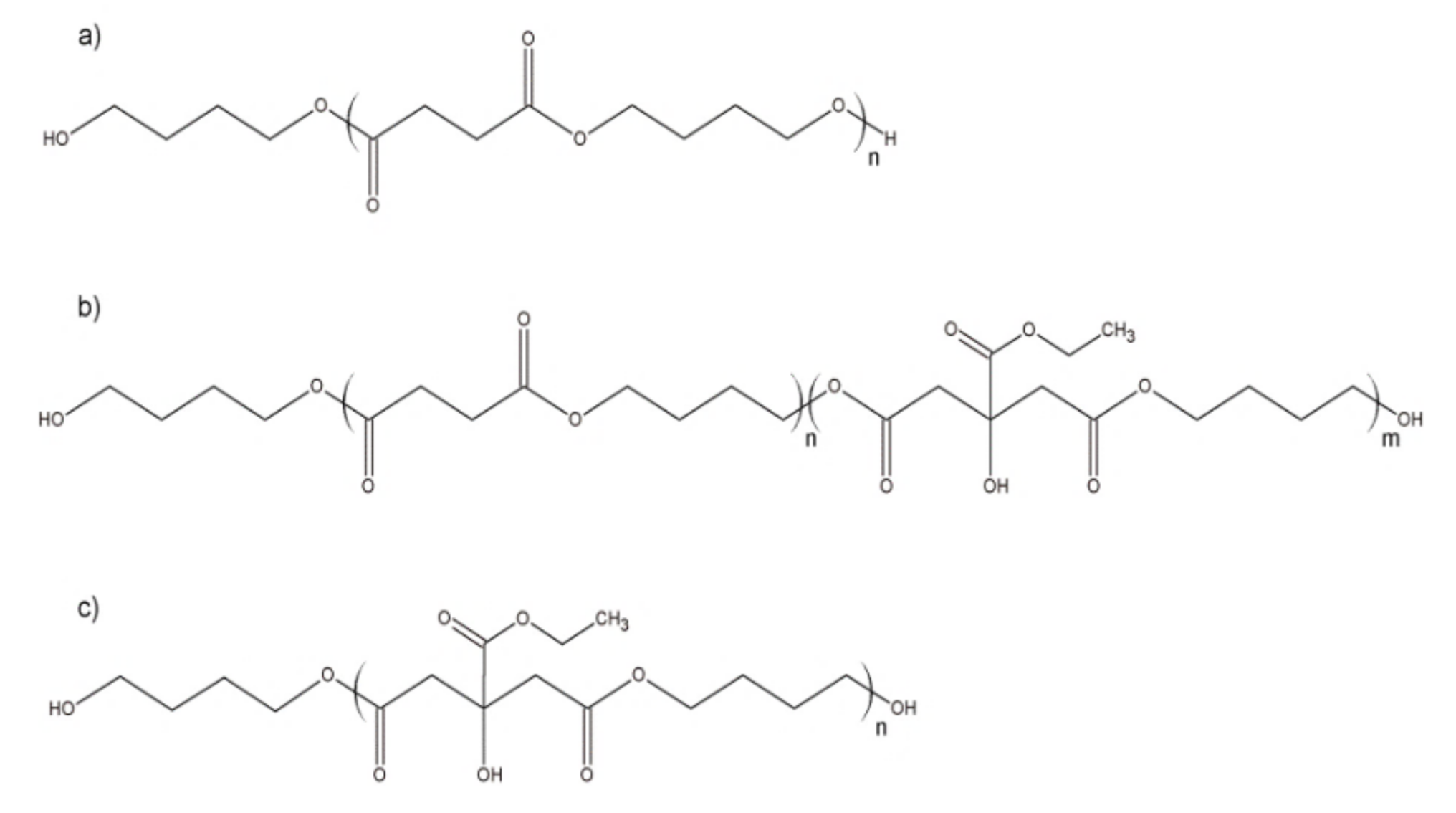
2.1. Materials
Copolymers and Blends Preparation
2.2. Characterizations
- (a)
- Each dog-bone shaped sample, firstly, was heated to a temperature a few degrees below the glass transition or at glass transition temperature, after fixing the sample in both jaws, the sample was maintained for 5 min in the chamber of the testing machine.
- (b)
- The samples were stretched to an elongation equal to 100% at a strain rate of 0.01 s−1 and then fast cooled. When the temperature reached 0 °C, the value of tension was measured and then reset.
- (c)
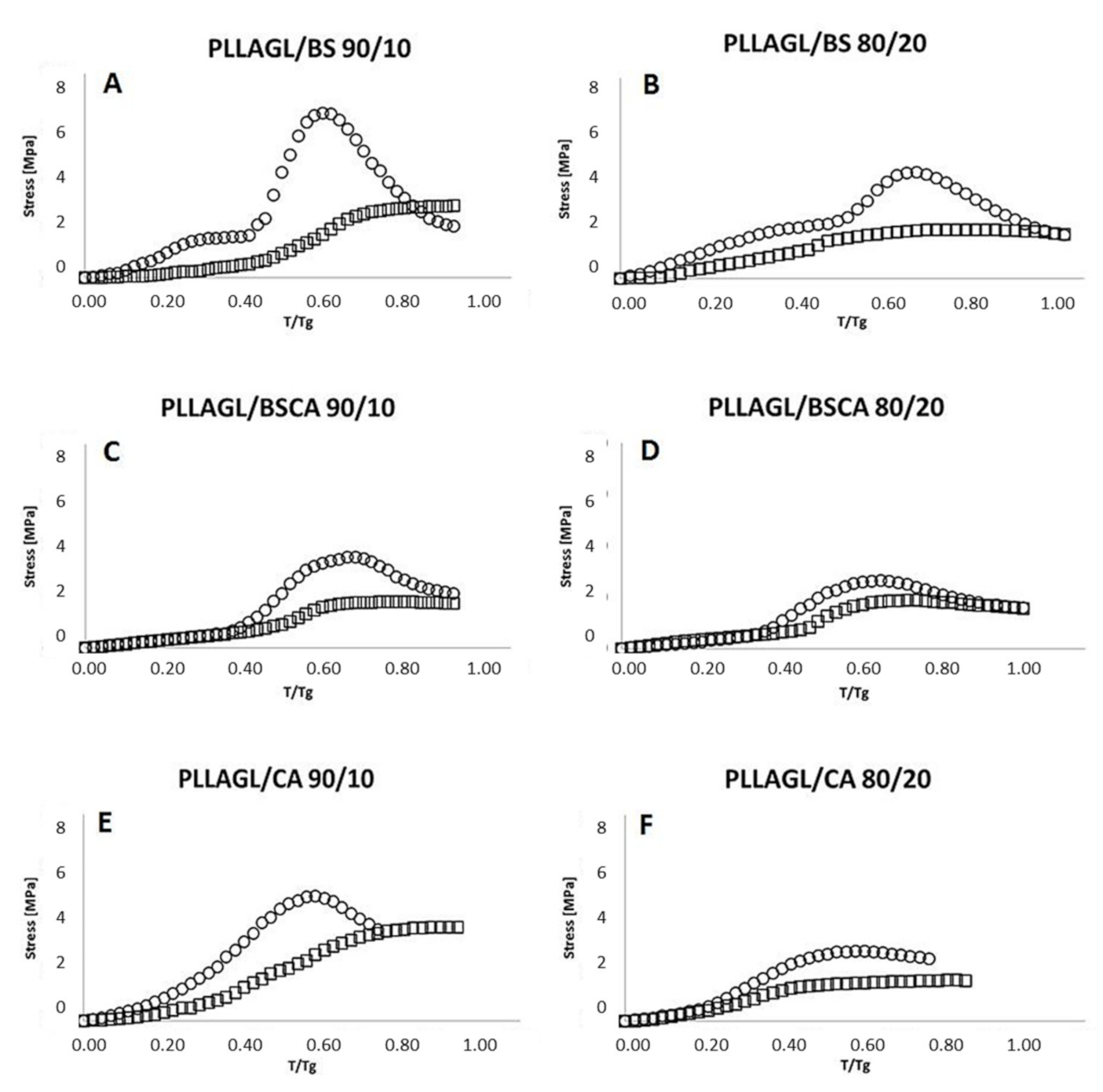
- Then, the sample was gradually heated (with the jaws of the handles still closed) with temperature rising rate 2 °C/min and continued observation of the gradual increasing of the stress values, determining on this basis the initial recovery temperature (IRT). The obtained relationship between the temperature change and the value of the observed stress is illustrated in Figure 4, additionally.
- (d)
- The other samples, after shaping into a temporary shape using the method described above, were put into the water bath heated to the correct temperature to measure the time of the shape return (tR) and the change of sample length. Since the samples, after giving a temporary shape fastened in the jaws of the testing machine, were quickly cooled and did not show any shrinkage, their fixing ratio was 1, and they also did not show dimensional changes after the period of storage at room temperature; the shape recovery ratio (RR) [37] was calculated according to the following simplified Equation (1).where em—engineering strain of the temporary shape after unloading, er—engineering strain of the recovered permanent shape.
- (a)
- Samples in the dog-bone shape with a gauge length of L0 = 35 mm (permanent shape obtained by injection molding at 180 °C were heated in the chamber to 56–57 °C and maintained for 5 min fastened in the jaws;
- (b)
- Keeping the temperature around 57 °C, the specimen was stretched at a strain rate 0.01 s−1 to about 20% strain, reaching a length of L1 = 42 mm; then, it was cooled to 0 °C with stress loaded without opening the jaws;
- (c)
- Then, the temperature in the chamber was increased from 0 °C to just below the glass transition temperature Tg or Tg2 of the tested sample, while the next temporary shape with a length of L2 = 49 mm was obtained by re-stretching the sample at the same rate and cooled to 0 °C. After removal, the sample was conditioned for one day at room temperature;
- (d)
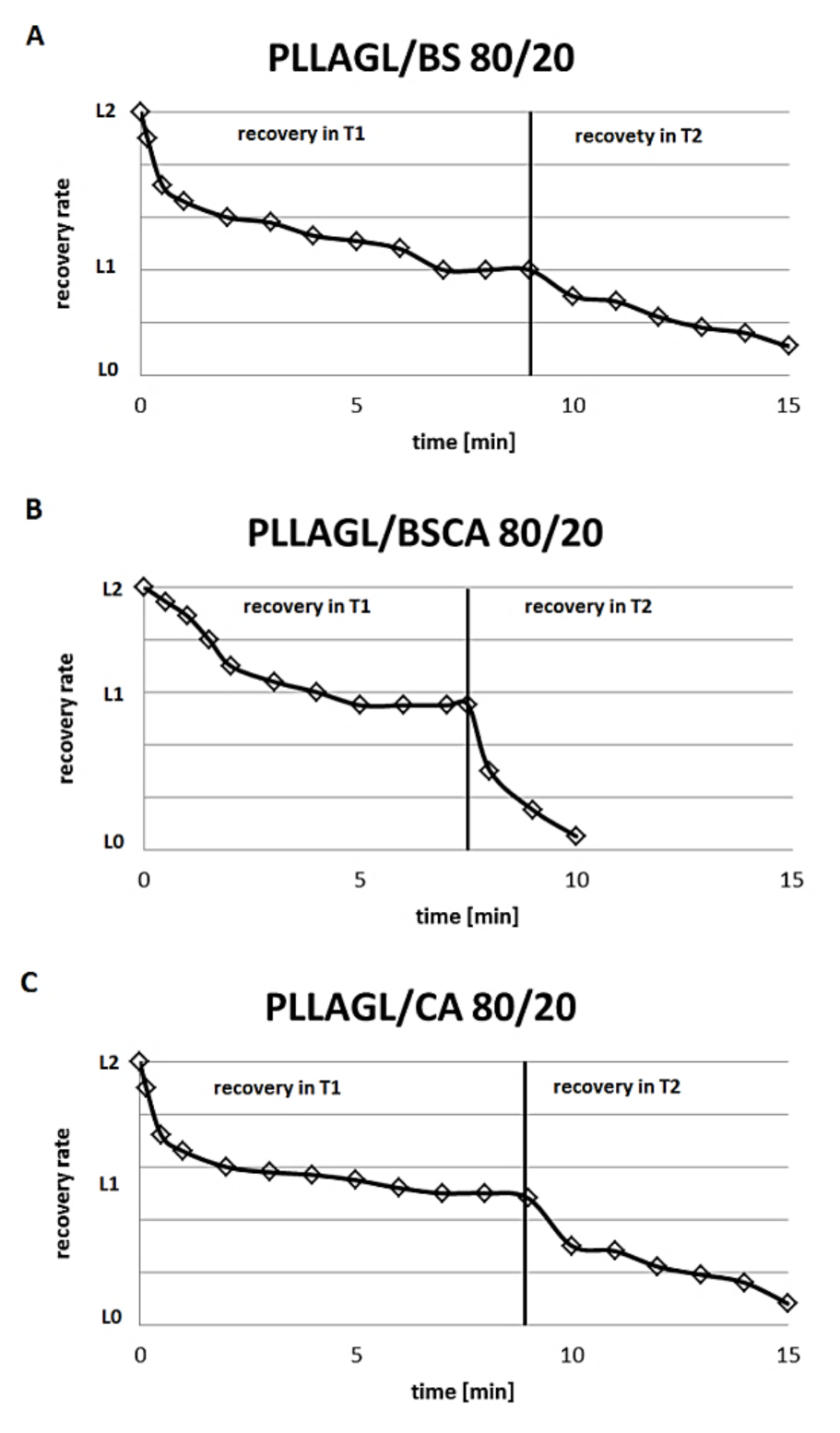
- The next day, the samples obtained previously were introduced to a water bath at a temperature Tg. The gradual changes of sample length have been noted. When the length of the sample stopped the change, the temperature of the bath was raised up to 57 °C. At this temperature, we noted the start of a further gradual change of the length (results are pictured in Figure 6).
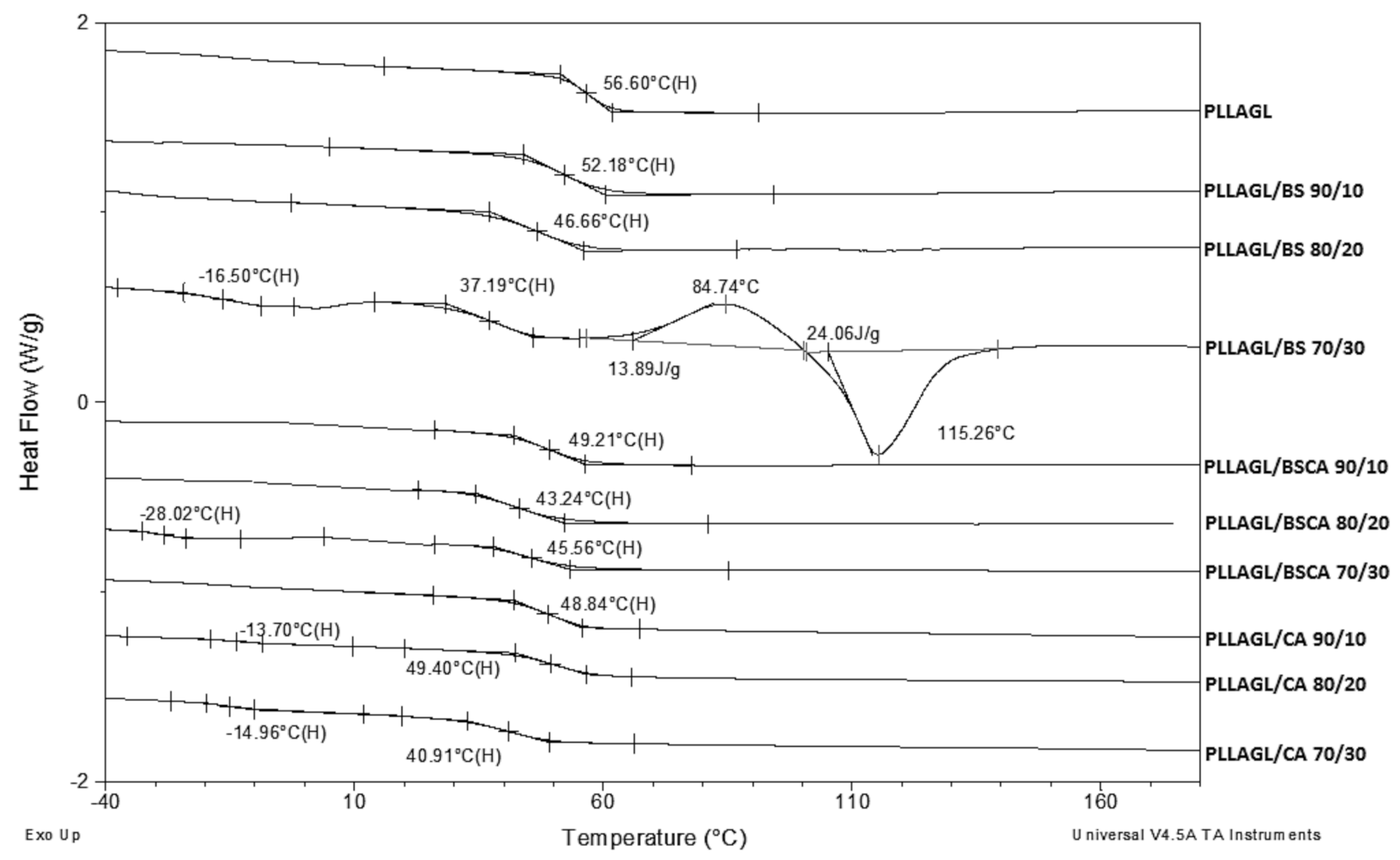
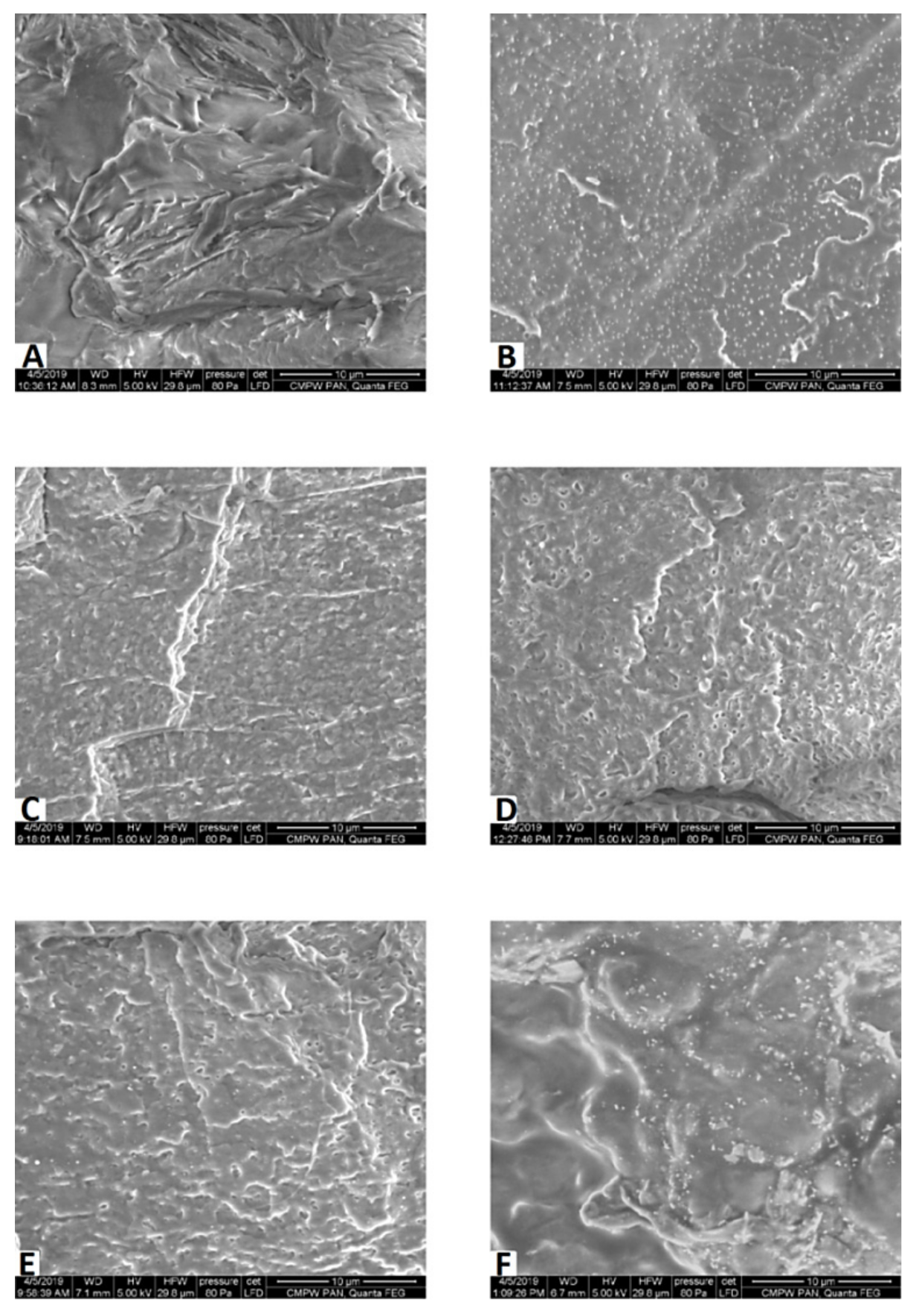
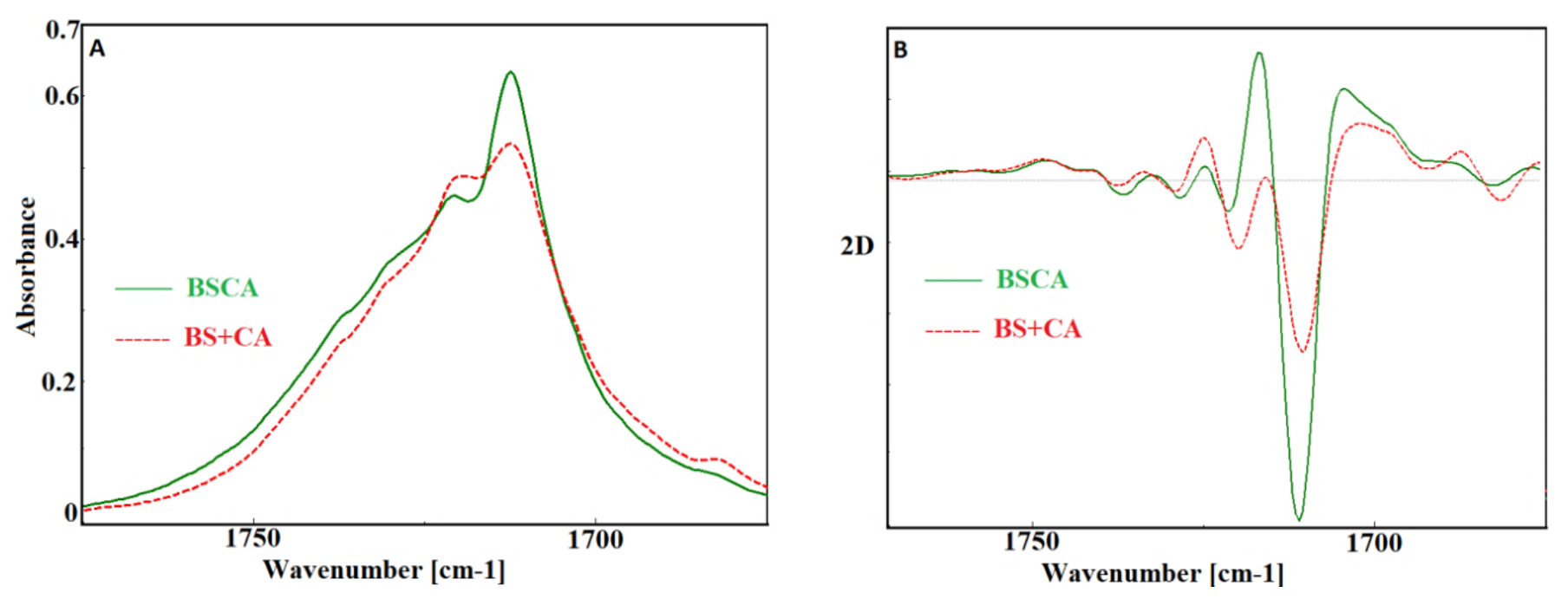
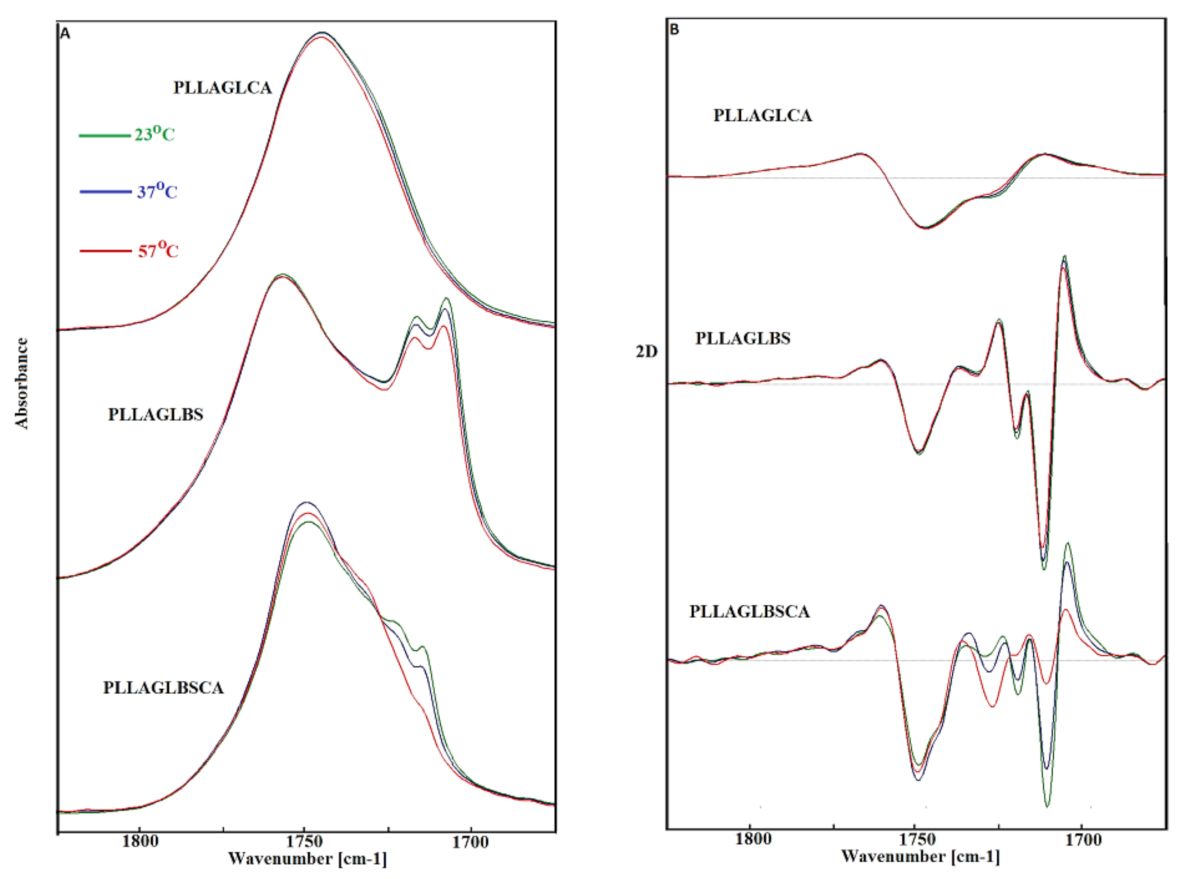
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ratna, D.; Karger-Kocsis, J. Recent Advances in Shape Memory Polymers and Composites: A Review. J. Mater. Sci. 2008, 43, 254–269. [Google Scholar] [CrossRef]
- Liu, C.; Qin, H.; Mather, P.T. Review of Progress in Shape-Memory Polymers. J. Mater. Chem. 2007, 17, 1543–1558. [Google Scholar] [CrossRef]
- Wu, X.; Huang, W.M.; Zhao, Y.; Ding, Z.; Tang, C.; Zhang, J. Mechanisms of the Shape Memory Effect in Polymeric Materials. Polymers 2013, 5, 1169–1202. [Google Scholar] [CrossRef]
- Behl, M.; Lendlein, A. Shape-Memory Polymers. Mater. Today 2007, 10, 20–28. [Google Scholar] [CrossRef]
- Lendlein, A. (Ed.) Shape-Memory Polymers Series: Advances in Polymer Science; Shape-Memory Polymers and Shape-Changing Polymers; Springer: Berlin/Herdelberg, Germany, 2010; Chapter 1; pp. 6–19. [Google Scholar]
- Kashif, M.; Chang, Y.-W. Triple-shape Memory Effects of Modified Semicrystalline Ethylene–propylene–diene rubber/poly(ε-caprolactone) Blends. Eur. Poly. J. 2015, 70, 306–316. [Google Scholar] [CrossRef]
- Ware, T.; Hearon, K.; Lonnecker, A.; Wooley, K.L.; Maitland, D.J.; Voit, W. Triple-Shape Memory Polymers Based on Self-Complementary Hydrogen Bonding. Macromolecules 2012, 45, 1062–1069. [Google Scholar] [CrossRef] [Green Version]
- Samuel, C.; Barrau, S.; Raquez, J.-M.; Dubois, P. Designing Multiple-Shape Memory Polymers with Miscible Polymer Blends: Evidence and Origins of a Triple-Shape Memory Effect for Miscible PLLA/PMMA Blends. Macromolecules 2014, 47, 6791–6803. [Google Scholar] [CrossRef]
- Wang, K.; Jia, Y.G.; Zhu, X.X. Biocompound-Based Multiple Shape Memory Polymers Reinforced by Photo-Cross-Linking. ACS Biomater. Sci. Eng. 2015, 1, 855–863. [Google Scholar] [CrossRef]
- Sun, L.; Huang, W.M. Mechanisms of the multi-shape memory effect and temperature memory effect in shape memory polymers. Soft Matter 2010, 6, 4403–4406. [Google Scholar] [CrossRef]
- Gu, S.Y.; Liu, L.L.; Gao, X.F. Triple-shape Memory Properties of Polyurethane/Polylactide–Polytetramethylene Ether Blends. Polym. Int. 2015, 64, 1155–1162. [Google Scholar] [CrossRef]
- Li, H.; Luo, Y.; Gao, X. Core–shell nano-latex blending method to prepare multi-shape memory polymers. Soft Matter 2017, 13, 5324–5331. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Gao, W.; Hu, J.; Xu, X.; Ning, N.; Yu, B.; Zhang, L. Multidirectional Triple-Shape-Memory Polymer by Tunable Cross-linking and Crystallization. ACS Appl. Mater. Interfaces 2020, 12, 6426–6435. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Zhang, Z.; Wang, C.; Ma, S.; Li, T.; Zhao, X.; Wang, D.; Zhoua, H.; Chen, C. Multi-shape memory effect of polyimides with extremely high strain. RSC Adv. 2017, 7, 53492–53496. [Google Scholar] [CrossRef] [Green Version]
- Duarah, R.; Singh, Y.P.; Gupta, P.; Mandal, B.B.; Karak, N. Smart self-tightening surgical suture from a tough bio-based hyperbranched polyurethane/reduced carbon dot nanocomposite. Biomed. Mater. 2018, 13, 045004. [Google Scholar] [CrossRef] [PubMed]
- Yeazel, T.R.; Becker, M.L. Advancing Toward 3D Printing of Bioresorbable Shape Memory Polymer Stents. Biomacromolecules 2020, 21, 3957–3965. [Google Scholar] [CrossRef] [PubMed]
- Dalton, E.; Chai, Q.; Shaw, M.W.; McKenzie, T.J.; Mullins, E.S.; Ayres, N. Hydrogel-Coated Polyurethane/Urea Shape Memory Polymer Foams. J. Polym. Sci. Part A 2019, 57, 1389–1395. [Google Scholar] [CrossRef]
- Rychter, P.; Pamula, E.; Orchel, A.; Posadowska, U.; Krok-Borkowicz, M.; Kaps, A.; Smigiel-Gac, N.; Smola, A.; Kasperczyk, J.; Prochwicz, W.; et al. Scaffolds with shape memory behavior for the treatment of large bone defects. J. Biomed. Mater. Res. Part A 2015, 103, 3503. [Google Scholar] [CrossRef]
- Davidson-Rozenfeld, G.; Stricker, L.; Simke, J.; Fadeev, M.; Vázquez-González, M.; Ravoo, B.J.; Willner, I. Light-responsive arylazopyrazole-based hydrogels: Their applications as shape-memory materials, self-healing matrices and controlled drug release systems. Polym. Chem. 2019, 10, 4106–4115. [Google Scholar] [CrossRef]
- Athanasiou, K.A.; Niederauer, G.G.; Agrawal, C. Sterilization, Toxicity, Biocompatibility and Clinical Applications of Polylactic Acid/Polyglycolic Acid Copolymers. Biomaterials 1996, 172, 93–102. [Google Scholar] [CrossRef]
- Song, J.J.; Chang, H.H.; Naguib, H.E. Biocompatible shape memory polymer actuators with high force capabilities. Eur. Polym. J. 2015, 67, 186–198. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, H.; Zhong, W.; Du, Q. A Novel Type of Shape Memory Polymer Blend and the Shape Memory Mechanism. Polymer 2009, 50, 1596–1601. [Google Scholar] [CrossRef]
- Lai, S.M.; Lan, Y.C. Shape Memory Properties of Melt-blended Polylactic Acid (PLA)/Thermoplastic Polyurethane (TPU) Bio-based Blends. J. Polym. Res. 2013, 20, 140. [Google Scholar] [CrossRef]
- Martin, O.; Averous, L. Poly(lactic acid): Plasticization and Properties of Biodegradable Multiphase Systems. Polymer 2001, 42, 6209–6219. [Google Scholar] [CrossRef]
- Mainardes, R.M.; Gremiao, M.P.; Brunetti, I.L.; Fonseca, L.M.; Khalil, N.M. Zidovudine-loaded PLA and PLA–PEG Blend Nanoparticles: Influence of Polymer Type on Phagocytic Uptake by Polymorphonuclear Cells. J. Pham. Sci. 2009, 98, 257–267. [Google Scholar] [CrossRef]
- Sonseca, A.; Madani, S.; Muñoz-Bonilla, A.; Fernández-García, M.; Peponi, L.; Leonés, A.; Rodríguez, G.; Echeverría, C.; López, D. Biodegradable and Antimicrobial PLA–OLA Blends Containing Chitosan-Mediated Silver Nanoparticles with Shape Memory Properties for Potential Medical Applications. Nanomaterials 2020, 10, 1065. [Google Scholar] [CrossRef]
- Chen, H.; Wang, L.; Zhou, S. Recent Progress in Shape Memory Polymers for Biomedical Applications, Chinese. J. Polym. Sci. 2018, 36, 905–917. [Google Scholar]
- Han, J.; Lai, S.; Chiu, Y. Two-way multi-shape memory properties of peroxide crosslinked ethylene vinyl-acetate copolymer (EVA)/polycaprolactone (PCL) blends. Polym. Adv. Technol. 2018, 29, 2010–2024. [Google Scholar] [CrossRef]
- Ba, C.; Yang, J.; Hao, Q.; Liu, X.; Cao, A. Syntheses and Physical Characterization of New Aliphatic Triblock Poly(l-lactide-b-butylene succinate-b-l-lactide)s Bearing Soft and Hard Biodegradable Building Blocks. Biomacromolecules 2003, 4, 1827–1834. [Google Scholar] [CrossRef]
- Shibata, M.; Inoue, Y.; Miyoshi, M. Mechanical Properties, Morphology, and Crystallization Behavior of Blends of Poly(l-lactide) with Poly(butylene succinate-co-l-lactate) and Poly(butylene succinate). Polymer 2006, 47, 3557–3564. [Google Scholar] [CrossRef]
- Park, J.W.; Im, S.S. Phase Behavior and Morphology in Blends of Poly(L-lactic acid) and Poly(butylene succinate). J. Appl. Polym. Sci. 2002, 86, 647–655. [Google Scholar] [CrossRef]
- Li, H.; Chang, J.; Cao, A.; Wang, J. In Vitro Evaluation of Biodegradable Poly(butylene succinate) as a Novel Biomaterial. Macromol. Biosci. 2005, 5, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, T.; Tsujisaka, T.; Masuda, T.; Nakayama, K. Plasma Surface Treatments and Biodegradation of Poly(butylene succinate) Sheets. J. Appl. Polym. Sci. 2000, 78, 1121–1129. [Google Scholar] [CrossRef]
- Yang, J.; Webb, A.R.; Pickerill, S.J.; Hageman, G.; Ameer, G.A. Synthesis and Evaluation of Poly(diol citrate) Biodegradable Elastomers. Biomaterials 2006, 27, 1889–1898. [Google Scholar] [CrossRef] [PubMed]
- Dobrzynski, P.; Kasperczyk, J.; Janeczek, H.; Bero, M. Synthesis of Biodegradable Copolymers with the Use of Low Toxic Zirconium Compounds. 1. Copolymerization of Glycolide with l-Lactide Initiated by Zr(Acac)4. Macromolecules 2001, 34, 5090–5098. [Google Scholar] [CrossRef]
- Śmigiel-Gac, N.; Pamuła, E.; Krok-Borkowicz, M.; Smola-Dmochowska, A.; Dobrzyński, P. Synthesis and Properties of Bioresorbable Block Copolymers of l-Lactide, Glycolide, Butyl Succinate and Butyl Citrate. Polymers 2020, 12, 214. [Google Scholar] [CrossRef] [Green Version]
- Lendlein, A.; Kelch, S. Shape-Memory Polymers. Angew. Chem. 2002, 41, 2034–2057. [Google Scholar] [CrossRef]
- Qiu, J.; Xing, C.; Cao, X.; Wang, H.; Wang, L.; Zhao, L.; Li, Y. Miscibility and Double Glass Transition Temperature Depression of Poly(l-lactic acid) (PLLA)/Poly(oxymethylene) (POM) Blends. Macromolecules 2013, 46, 5806–5814. [Google Scholar] [CrossRef]
- Kalogeras, I.M.; Brostow, W. Glass Transition Temperatures in Binary Polymer Blends. J. Polym. Sci. B Polym. Phys. 2009, 47, 80–95. [Google Scholar] [CrossRef]
- Deng, Y.; Thomas, N.L. Blending Poly(butylene succinate) with Poly(lactic acid): Ductility and Phase Inversion Effects. Eur. Polym. J. 2015, 71, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Huang, W.M.; Wang, C.C.; Zhao, Y.; Ding, Z.; Purnawali, H. Optimization of the shape memory effect in shape memory polymers. J. Polym. Sci. Part A 2011, 49, 3574–3581. [Google Scholar] [CrossRef]
- Smola, A.; Dobrzynski, P.; Cristea, M.; Kasperczyk, J.; Sobota, M.; Gebarowska, K.; Janeczek, H. Bioresorbable terpolymers based on l-lactide, glycolide and trimethylene carbonate with shape memory behavior. Polym. Chem. 2014, 5, 2442–2452. [Google Scholar] [CrossRef]
- Yao, S.F.; Chen, X.T.; Ye, H.M. Investigation of Structure and Crystallization Behavior of Poly(butylene succinate) by Fourier Transform Infrared Spectroscopy. J. Phys. Chem. B 2017, 121, 9476–9485. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Tg [°C] | Tm [°C] | ΔH [J/g] | Mn [g/mol] |
|---|---|---|---|---|
| BS | −18 | 114 | 81 | 4500 |
| BSCA | −33 | 77 | 52 | 4800 |
| CA | −29 | - | - | 4000 |
| Sample | Tg1 [°C] | Tg2 [°C] | TgT [°C] | E25 [MPa] | σ25 [MPa] | E37 [MPa] | σ37 [MPa] |
|---|---|---|---|---|---|---|---|
| PLLAGL | 56.6 | - | 2480 ± 160 | 77 ± 1 | 2020 ± 30 | 58 ± 0.1 | |
| PLLAGL/BS 90/10 | 52.2 | - | 47.2 | 1968 ± 117 | 72 ± 0.9 | 1464 ± 205 | 42 ± 0.6 |
| PLLAGL/BS 80/20 | 46.7 | - | 38.3 | 1645 ± 328 | 52 ± 7.7 | 460 ± 200 | 14 ± 1.2 |
| PLLAGL/BS 70/30 | −16.5 | 37.2 | 29.9 | 1504 ± 11 | 49 ± 2.3 | 255 ± 30 | 14 ± 0.9 |
| PLLAGL/BSCA 90/10 | 49.2 | - | 45.3 | 2010 ± 107 | 71 ± 1.3 | 613 ± 19 | 13 ± 0.5 |
| PLLAGL/BSCA 80/20 | 43.2 | - | 34.8 | 1809 ± 52 | 62 ± 1.7 | 310 ± 12 | 7 ± 0.8 |
| PLLAGL/BSCA 70/30 | −28.0 | 45.6 | 24.9 | 1620 ± 87 | 39 ± 4.5 | 233 ± 12 | 7 ± 0.9 |
| PLLAGL/CA 90/10 | 48.8 | - | 46.7 | 2110 ± 181 | 29 ± 1.1 | 757 ± 21 | 14 ± 0.9 |
| PLLAGL/CA 80/20 | −13.7 | 49.4 | 37.4 | 1410 ± 45 | 16 ± 0.2 | 64 ± 19 | 6 ± 0.3 |
| PLLAGL/CA 70/30 | −15.0 | 40.9 | 28.7 | 628 ± 16 | 13 ± 0.2 | 64 ± 23 | 5 ± 0.9 |
| Sample | TR [°C] | tR [s] | Rr [%] | VR [%/s] | IRT [°C] |
|---|---|---|---|---|---|
| PLLAGL | 48 | 2760 | 97.8 | 0.03 | 45 |
| 52 | 420 | 98.8 | 0.23 | ||
| 56 | 30 | 99.8 | 3.33 | ||
| PLLAGL/BS 90/10 | 42 | 3000 | 95.8 | 0.03 | 38 |
| 47 | 1320 | 96.0 | 0.07 | ||
| 52 | 120 | 99.9 | 0.83 | ||
| PLLAGL/BS 80/20 | 39 | 1800 | 54.0 | 0.03 | 36 |
| 42 | 900 | 77.8 | 0.09 | ||
| 47 | 540 | 86.7 | 0.16 | ||
| PLLAGL/BS 70/30 | 28 | 3000 | 21.5 | 0.01 | 27 |
| 33 | 1680 | 26.6 | 0.02 | ||
| 37 | 1200 | 77.7 | 0.06 | ||
| PLLAGL/BSCA90/10 | 39 | 2400 | 83.2 | 0.03 | 35 |
| 44 | 480 | 86.4 | 0.18 | ||
| 49 | 80 | 99.2 | 1.24 | ||
| PLLAGL/BSCA 80/20 | 33 | 1860 | 79.2 | 0.04 | 32 |
| 37 | 600 | 81.2 | 0.13 | ||
| 43 | 160 | 99.3 | 1.60 | ||
| PLLAGL/BSCA 70/30 | 37 | 2040 | 86.2 | 0.04 | 36 |
| 42 | 480 | 92.1 | 0.19 | ||
| 47 | 210 | 93.7 | 0.45 | ||
| PLLAGL/CA 90/10 | 37 | 3000 | 72.0 | 0.02 | 36 |
| 49 | 60 | 99.3 | 1.65 | ||
| PLLAGL/CA 80/20 | 37 | 1800 | 93.3 | 0.05 | 35 |
| 50 | 55 | 99.1 | 1.80 | ||
| PLLAGL/CA 70/30 | 37 | 360 | 91.1 | 0.25 | 32 |
| 41 | 180 | 95.5 | 0.53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smola-Dmochowska, A.; Śmigiel-Gac, N.; Kaczmarczyk, B.; Sobota, M.; Janeczek, H.; Karpeta-Jarząbek, P.; Kasperczyk, J.; Dobrzyński, P. Triple-Shape Memory Behavior of Modified Lactide/Glycolide Copolymers. Polymers 2020, 12, 2984. https://doi.org/10.3390/polym12122984
Smola-Dmochowska A, Śmigiel-Gac N, Kaczmarczyk B, Sobota M, Janeczek H, Karpeta-Jarząbek P, Kasperczyk J, Dobrzyński P. Triple-Shape Memory Behavior of Modified Lactide/Glycolide Copolymers. Polymers. 2020; 12(12):2984. https://doi.org/10.3390/polym12122984
Chicago/Turabian StyleSmola-Dmochowska, Anna, Natalia Śmigiel-Gac, Bożena Kaczmarczyk, Michał Sobota, Henryk Janeczek, Paulina Karpeta-Jarząbek, Janusz Kasperczyk, and Piotr Dobrzyński. 2020. "Triple-Shape Memory Behavior of Modified Lactide/Glycolide Copolymers" Polymers 12, no. 12: 2984. https://doi.org/10.3390/polym12122984
APA StyleSmola-Dmochowska, A., Śmigiel-Gac, N., Kaczmarczyk, B., Sobota, M., Janeczek, H., Karpeta-Jarząbek, P., Kasperczyk, J., & Dobrzyński, P. (2020). Triple-Shape Memory Behavior of Modified Lactide/Glycolide Copolymers. Polymers, 12(12), 2984. https://doi.org/10.3390/polym12122984