Introduction of Stilbene Derivatives and Cinnamate Ester Derivatives at the ω-End Groups of Poly(Methyl Methacrylate) Prepared via RAFT Polymerization


Abstract

1. Introduction

2. Materials and Methods
2.1. Materials
2.2. Conventional Free Radical Polymerization
2.3. Standard Method for RAFT Free Radical Polymerization
2.4. Chain Extension of the “Living” Polymers with the Non-Homopolymerizable Monomers: “Capping”
2.5. Modified Method for Capping the “Living” Polymers
2.6. Size Exclusion Chromatography (SEC)
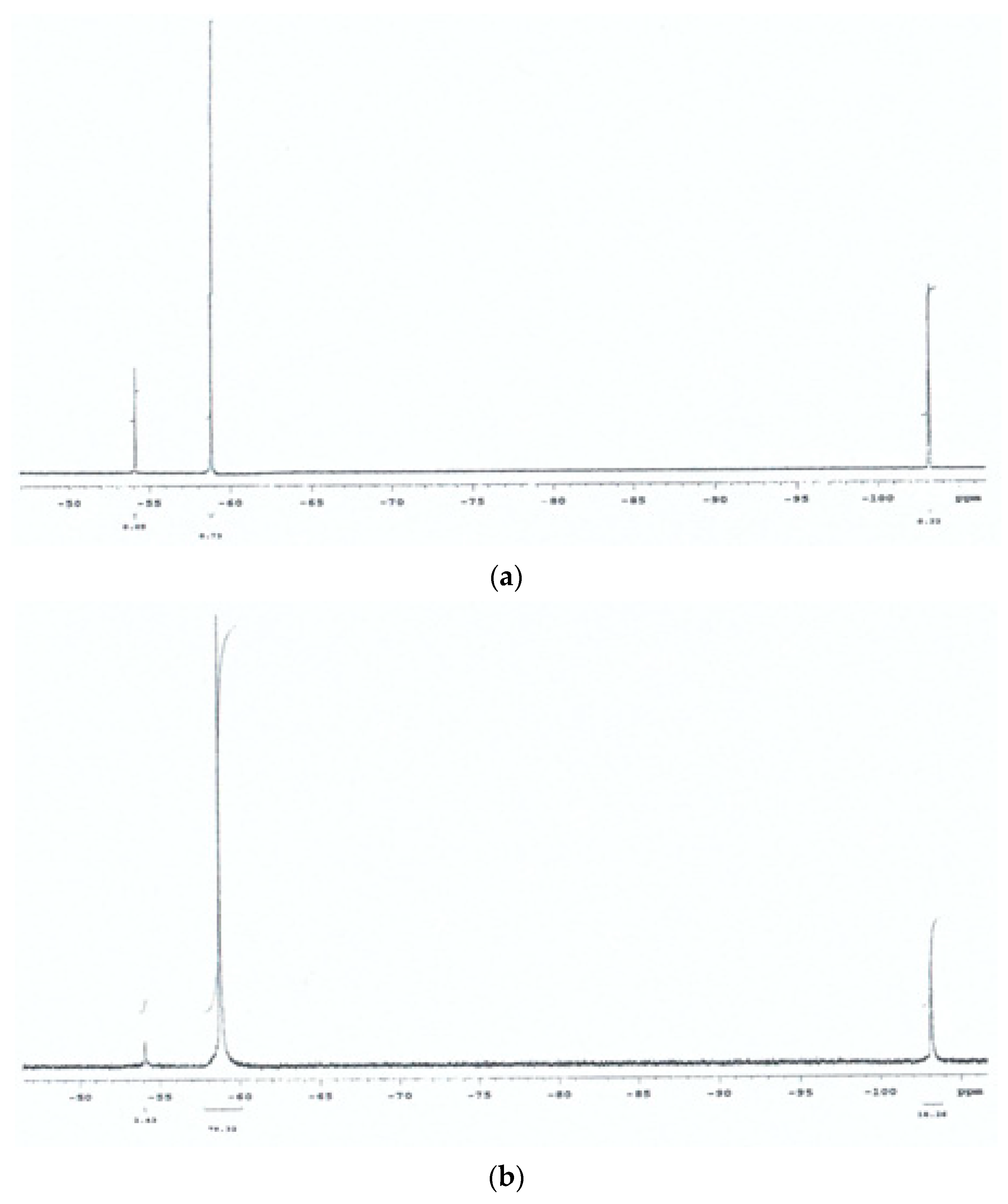
2.7. 19F-Nuclear Magnetic Resonance Spectroscopy
2.8. MALDI-ToF Mass Spectrometry (MALDI-ToF MS)
3. Results
3.1. Preliminary 19F NMR Spectroscopy Studies
3.2. Syntheses of “Living” PMMAs (13) and (15) and “Living” PS (14)
3.3. Capping Reactions
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A
Appendix A.1. General Experimental Procedures
Appendix A.2. Syntheses of Fluorocompounds (4)–(12)
Appendix A.2.1. Synthesis of Bis-(4-trifluoromethoxybenzoyl) Peroxide (4)
Appendix A.2.2. Synthesis of CTA (5)
Appendix A.2.3. Synthesis of E-4,4′-Bis(Trifluoromethyl)stilbene (6)
Appendix A.2.4. Synthesis of 1,1-Bis-(4-Trifluoromethylphenyl)ethene (7)
Appendix A.2.5. 1,1-Bis-(4-(4,4,4-Trifluorobutoxy)phenyl)ethane (8)
Appendix A.2.6. Methyl E-3-Trifluoromethylcinnamate (9)
Appendix A.2.7. Synthesis of 4,4,4-Trifluorobutyl E-Cinnamates (10)–(12)
References
- Bethell, D.; Brust, M.; Hasan, M. The Fate of Sulfur-Bound Hydrogen on Formation of Self-Assembled Thiol Monolayers on Gold: 1H NMR Spectroscopic Evidence from Solutions of Gold Clusters. J. Amer. Chem. Soc. 2002, 124, 1132–1133. [Google Scholar]
- Sasso, B.; Dobinson, M.; Hodge, P.; Wear, T. Synthesis of ω-end group functionalized poly(methyl methacrylate)s via RAFT polymerization. Macromolecules 2010, 43, 7453–7464. [Google Scholar] [CrossRef]
- Feng, X.-S.; Pan, C.-Y. Synthesis of Amphiphilic Miktoarm ABC Star Copolymers by RAFT Mechanism Using Maleic Anhydride as Linking Agent. Macromolecules 2002, 35, 4888–4893. [Google Scholar] [CrossRef]
- Bevington, J.C.; Huckerby, T.N. The involvement of stilbene in radical polymerizations of methyl methacrylate and styrene. Macromolecules 1985, 18, 176–178. [Google Scholar] [CrossRef]
- Bevington, J.C.; Breuer, S.W.; Huckerby, T.N. End-groups in poly(methyl methacrylate) and polystyrene prepared by radical polymerization in the presence of derivatives of stilbene. Die Angew. Makromol. Chem. 1986, 140, 145–152. [Google Scholar] [CrossRef]
- Bevington, J.C.; Huckerby, T.N.; Vickerstaff, N. Incorporation of stilbene and p-fluoro-stilbene in polystyrene during radical polymerization. Polymer 1986, 27, 1823–1825. [Google Scholar] [CrossRef]
- Bevington, J.C.; Huckerby, T.N.; Vickerstaff, N. Study by 19F NMR of the end-groups in polymers of styrene prepared using bis(4-fluorobenzoyl) peroxide in the presence of stilbene and some of its derivatives. Makromol. Chem. Rapid Commun. 1988, 9, 791–796. [Google Scholar] [CrossRef]
- Bevington, J.C.; Breuer, S.W.; Huckerby, T.N. Involvement of analogues of stilbene in radical polymerization: The reactions of arylstyrenes with benzoyloxy radical. Macromolecules 1989, 22, 55–61. [Google Scholar] [CrossRef]
- Barson, C.A.; Bevington, J.C.; Dillingham, K.A.; Huckerby, T.N. Polymers with furyl groups at chain-ends: The use of 2-styrylfuran as an additive in radical polymerizations. React. Funct. Polym. 1999, 42, 273–278. [Google Scholar] [CrossRef]
- Barson, C.A.; Bevington, J.C.; Breuer, S.W. Comparison of some 4- and 4,4′-substituted derivatives of stilbene for their reactivites towards the benzoyloxy radical. Polym. Bull. 1996, 36, 423–426. [Google Scholar] [CrossRef]
- Barson, C.A.; Bevington, J.C.; Breuer, S.W. The reactivities towards the benzoyloxy radical of various isomers of fluorostilbenes. Polym. Bull. 1994, 32, 625–628. [Google Scholar] [CrossRef]
- Chiefari, J.; Chong, Y.K.; Ercole, F.; Krstina, J.; Jeffery, J.; Tam., P.T.; Le, R.T.A.; Maydunne, G.F.; Meijs, C.I.; Moad, C.L. Living Free-Radical Polymerization by Reversible Addition-Fragmentation Chain Transfer: The RAFT Process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.A. 20th anniversary perspective on the life of RAFT: RAFT coming of age. Polym. Int. 2019, 69, 658–661. [Google Scholar] [CrossRef]
- Perrier, S. 50Th Anniversary perspective: RAFT polymerization—A user guide. Macromolecules 2017, 50, 7433–7447. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Discovery of the RAFT process and its impact on radical polymerization. Macromolecules 2020, 53, 495–497. [Google Scholar] [CrossRef]
- Shim, S.E.; Lee, H.; Choe, S. Synthesis of functionalized monodisperse poly(methyl methacrylate nanoparticles by a RAFT agent carrying carboxyl end group. Macromolecules 2004, 37, 5565–5571. [Google Scholar] [CrossRef]
- Moad, G.; Chong, Y.K.; Postma, A.; Rizzardo, E.; Thang, S.H. Advances in RAFT polymerization: The synthesis of polymers with defined end-groups. Polymer 2005, 46, 8458–8468. [Google Scholar] [CrossRef]
- Patton, D.L.; Mullins, M.; Fulghum, T.; Advincula, R.C. A facile synthesis route to α,ω-telechelic polymers via reversible addition fragmentation chain transfer polymerization. Macromolecules 2005, 38, 8597–8602. [Google Scholar] [CrossRef]
- Qiu, X.-P.; Winnick, F.M. Synthesis of α,ω-dimercapto poly(N-isopropylacrylamides) by RAFT polymerization with a hydrophilic difunctional chain transfer agent. Macromolecules 2007, 40, 872–878. [Google Scholar] [CrossRef]
- Iha, R.K.; Wooley, K.; Nystrom, A.N.; Burke, D.J.; Kade, M.J.; Hawker, C.J. Applications of orthogonal “click” chemistries in the synthesis of functional soft materials. Chem. Rev. 2009, 109, 5620–5686. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Q.; Guo, F.; Gu, J.; Yin, C. Preparation of diblock copolymer PBA-b-PSt by DPE method in emulsion. J. Poly. Res. 2011, 18, 1229–1235. [Google Scholar] [CrossRef]
- Novotny, J.; Collins, C.H.; Starks, F.W. Synthesis and antimalarial activity of benzotrifluoride derivatives. J. Pharm. Sci. 1973, 62, 910–913. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.C.; Giansiracusa, J.J.; Kelly, D.P.; Periasamy, M.; Perumal, P.T. Structural effects in solvolytic reactions. 45. Carbon-13 NMR studies of carbocations. 9. Variation of the cationic carbon chemical shifts with increasing electron demand in 1,1-diaryl-1-ethyl carbocations. Importance of the inductive localized π-polarization effect in causing deviations from linearity. J. Am. Chem. Soc. 1983, 105, 6300–6305. [Google Scholar]
- Baldwin, J.E.; Kapecki, J.A. Kinetics of the cycloadditions of diphenylketene with 1,1-diarylethylenes and styrenes. J. Am. Chem. Soc. 1970, 92, 4868–4873. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Monomer | Initiator | Percentage of Initiator(s) b | Polymer Yield | Polymer Designation | Mn c | Mw c | D c | Percentage “Living” d |
|---|---|---|---|---|---|---|---|---|---|
| 1 | MMA | (4) | 0.5 | 65 | ---- | 75.0 | 159.2 | 2.12 | -- |
| 2 | S | (4) | 0.5 | 59 | ---- | 26.2 | 50.5 | 1.92 | -- |
| 3 | MMA | (4) + (5) | 0.5 + 1.0 e | 14 | (13) | 3.95 | 4.45 | 1.13 | 92 |
| 4 | S | (4) + (5) | 0.5 + 1.0 e | 25 | (14) | 3.30 | 3.65 | 1.11 | 93 |
| 5 | MMA | (4) + (5) | 0.033 + 0.0667 e | 27 | (15) | 49.6 | 60.2 | 1.21 | 75 |
| Entry | Capping Agent | PMMA (13) | PS (14) | |||
|---|---|---|---|---|---|---|
| Capped b | Living b,c | Designation | Capped b | Living b,c | ||
| 1 | (6) | 63% | 100% | (16) | 23% | n.d. |
| 2 | (7) | 29% | n.d. | --- | 6% | n.d. |
| 3 | (8) | 39% | n.d. | --- | 6% | n.d. |
| 4 | (9) | 50% | n.d. | --- | 15% | n.d. |
| 5 | (10) | 78% | n.d. | --- | 12% | n.d. |
| 6 | (11) | 102% d | 94% | (17) | 15% | n.d. |
| 7 | (12) | 100% e | 85% | (18) | 8% | n.d. |
| 8 | (11) f | 100% | 0 g | --- | --- | --- |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobinson, M.; Hodge, P.; Wear, T. Introduction of Stilbene Derivatives and Cinnamate Ester Derivatives at the ω-End Groups of Poly(Methyl Methacrylate) Prepared via RAFT Polymerization. Polymers 2020, 12, 2449. https://doi.org/10.3390/polym12112449
Dobinson M, Hodge P, Wear T. Introduction of Stilbene Derivatives and Cinnamate Ester Derivatives at the ω-End Groups of Poly(Methyl Methacrylate) Prepared via RAFT Polymerization. Polymers. 2020; 12(11):2449. https://doi.org/10.3390/polym12112449
Chicago/Turabian StyleDobinson, Martyn, Philip Hodge, and Trevor Wear. 2020. "Introduction of Stilbene Derivatives and Cinnamate Ester Derivatives at the ω-End Groups of Poly(Methyl Methacrylate) Prepared via RAFT Polymerization" Polymers 12, no. 11: 2449. https://doi.org/10.3390/polym12112449
APA StyleDobinson, M., Hodge, P., & Wear, T. (2020). Introduction of Stilbene Derivatives and Cinnamate Ester Derivatives at the ω-End Groups of Poly(Methyl Methacrylate) Prepared via RAFT Polymerization. Polymers, 12(11), 2449. https://doi.org/10.3390/polym12112449