Mechanistic Studies for Palladium Catalyzed Copolymerization of Ethylene with Vinyl Ethers




Abstract
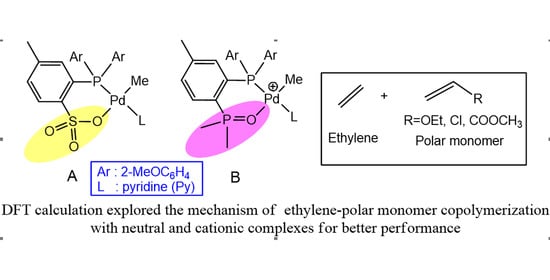
1. Introduction
2. Computational Methods Used to Elucidate the Copolymerization
3. Results and Discussion
3.1. Chain Initiation
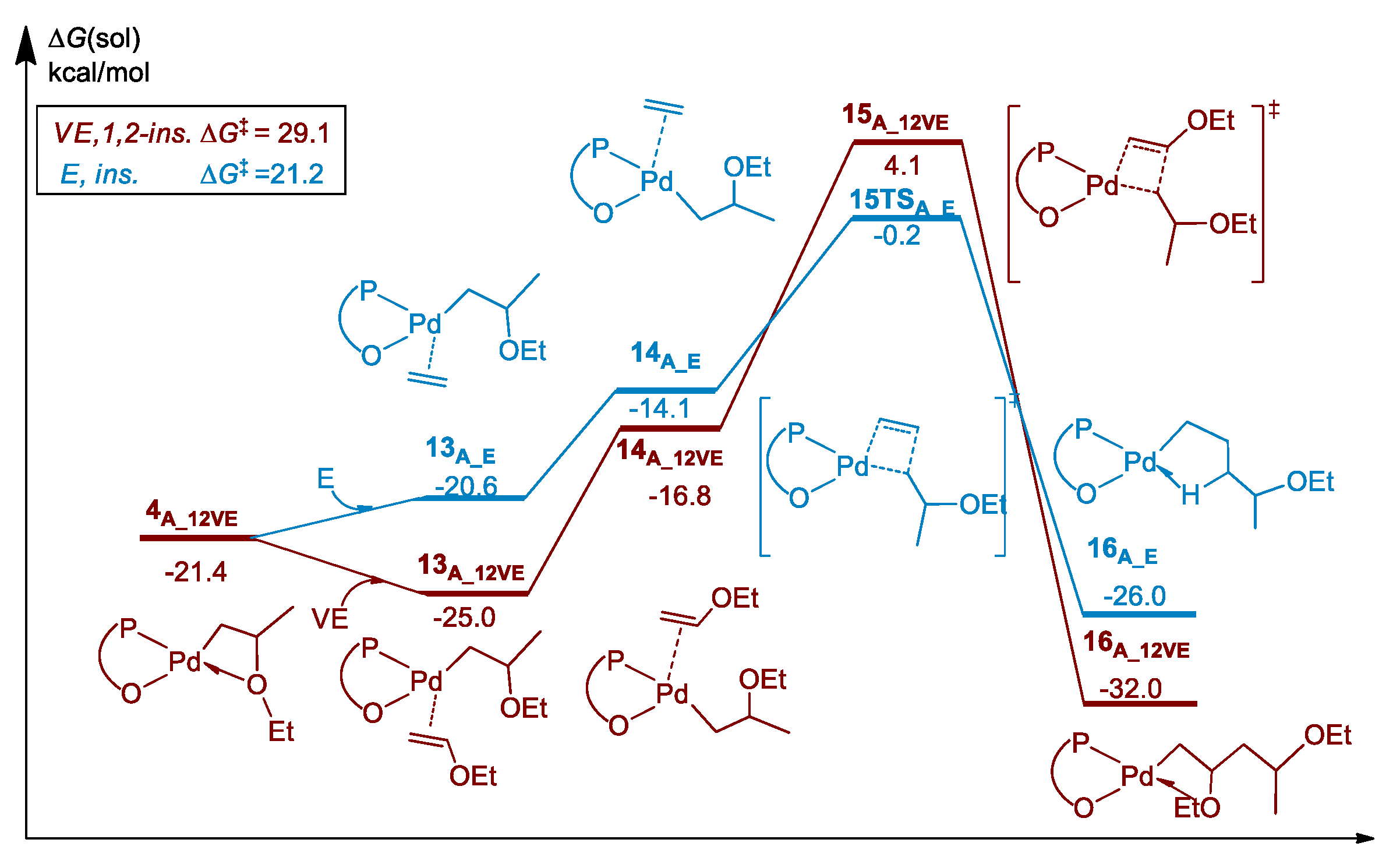
3.2. Chain Propagation
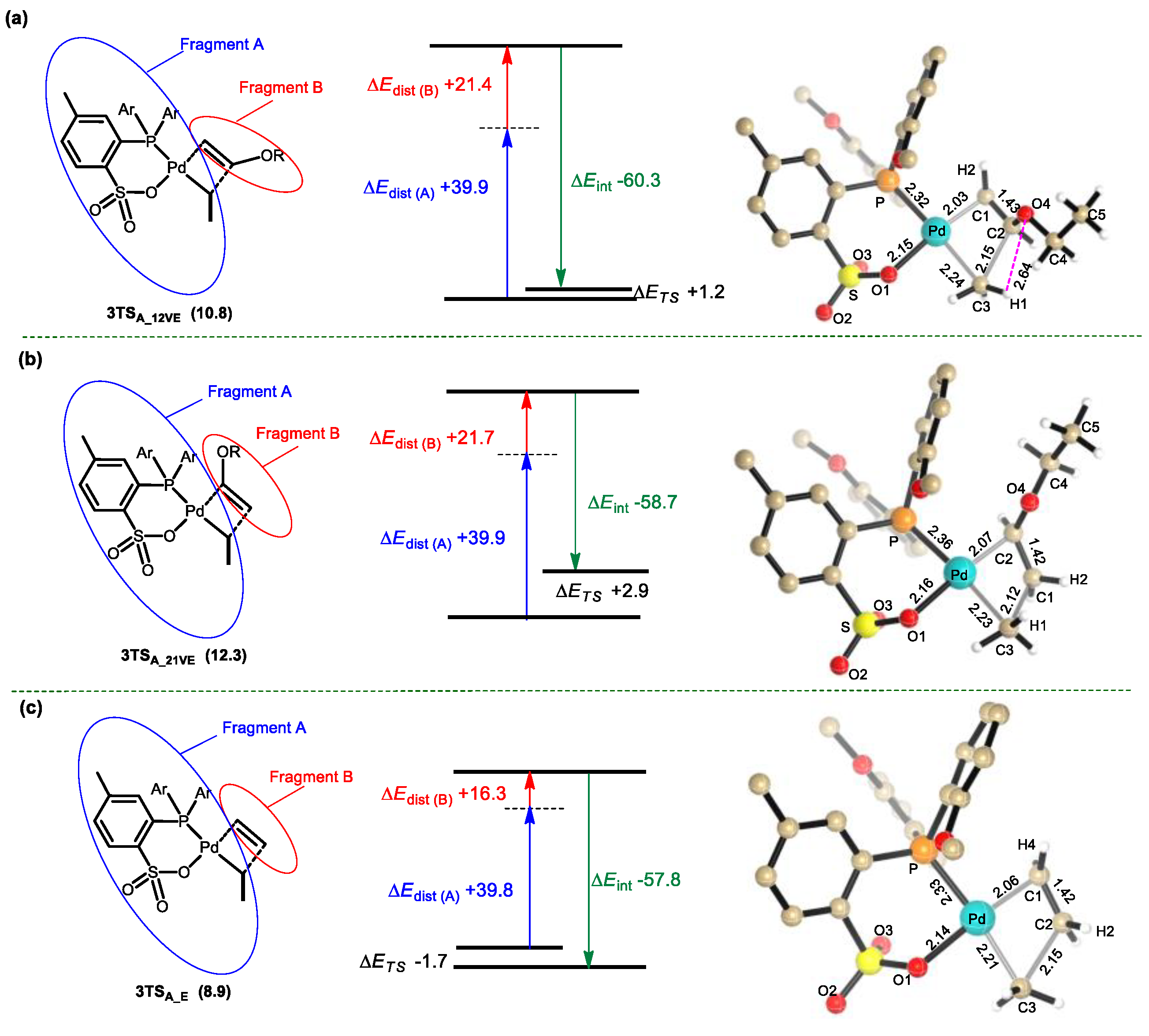
3.3. β-H and β-OEt Elimination
3.4. Catalyst Tuning
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nakamura, A.; Ito, S.; Nozaki, K. Coordination−Insertion Copolymerization of Fundamental Polar Monomers. Chem. Rev. 2009, 109, 5215–5244. [Google Scholar] [CrossRef]
- Fu, X.; Zhang, L.; Tanaka, R.; Shiono, T.; Cai, Z. Highly Robust Nickel Catalysts Containing Anilinonaphthoquinone Ligand for Copolymerization of Ethylene and Polar Monomers. Macromolecules 2017, 50, 9216–9221. [Google Scholar] [CrossRef]
- Hyatt, M.G.; Guironnet, D. Silane as Chain Transfer Agent for the Polymerization of Ethylene Catalyzed by a Palladium (II) Diimine Catalyst. ACS Catal. 2017, 7, 5717–5720. [Google Scholar] [CrossRef]
- Takeuchi, D.; Iwasawa, T.; Osakada, K. Double-Decker-Type Dipalladium Catalysts for Copolymerization of Ethylene with Acrylic Anhydride. Macromolecules 2018, 51, 5048–5054. [Google Scholar] [CrossRef]
- Wang, X.; Nozaki, K. Selective Chain-End Functionalization of Polar Polyethylenes: Orthogonal Reactivity of Carbene and Polar Vinyl Monomers in Their Copolymerization with Ethylene. J. Am. Chem. Soc. 2018, 140, 15635–15640. [Google Scholar] [CrossRef]
- Rix, F.C.; Brookhart, M.; White, P.S. Mechanistic Studies of the Palladium (II)-Catalyzed Copolymerization of Ethylene with Carbon Monoxide. J. Am. Chem. Soc. 1996, 118, 4746–4764. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, B.; Zhang, J.; Yu, W.; Liu, Z.; Zhang, Y. Transition metal-catalyzed C–H bond functionalizations by the use of diverse directing groups. Org. Chem. Front. 2015, 2, 1107–1295. [Google Scholar] [CrossRef]
- Nakamura, A.; Anselment, T.M.J.; Claverie, J.; Goodall, B.; Jordan, R.F.; Mecking, S.; Rieger, B.; Sen, A.; Van Leeuwen, P.W.N.M.; Nozaki, K. Ortho-Phosphinobenzenesulfonate: A Superb Ligand for Palladium-Catalyzed Coordination–Insertion Copolymerization of Polar Vinyl Monomers. Acc. Chem. Res. 2013, 46, 1438–1449. [Google Scholar] [CrossRef]
- Tan, C.; Chen, C. Emerging Palladium and Nickel Catalysts for Copolymerization of Olefins with Polar Monomers. Angew. Chem. Int. Ed. 2019, 58, 7192–7200. [Google Scholar] [CrossRef]
- Luo, S.; Jordan, R.F. Copolymerization of Silyl Vinyl Ethers with Olefins by (α-diimine)PdR+. J. Am. Chem. Soc. 2006, 128, 12072–12073. [Google Scholar] [CrossRef]
- Chen, C.; Luo, S.; Jordan, R.F. Cationic Polymerization and Insertion Chemistry in the Reactions of Vinyl Ethers with (α-Diimine)PdMe+Species. J. Am. Chem. Soc. 2010, 132, 5273–5284. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, W.; Daugulis, O.; Brookhart, M. Mechanistic Studies of Pd (II)-Catalyzed Copolymerization of Ethylene and Vinylalkoxysilanes: Evidence for a β-Silyl Elimination Chain Transfer Mechanism. J. Am. Chem. Soc. 2016, 138, 16120–16129. [Google Scholar] [CrossRef]
- Jian, Z.; Mecking, S. Insertion Polymerization of Divinyl Formal. Macromolecules 2016, 49, 4395–4403. [Google Scholar] [CrossRef]
- Jian, Z.; Mecking, S. Insertion Homo- and Copolymerization of Diallyl Ether. Angew. Chem. Int. Ed. 2015, 54, 15845–15849. [Google Scholar] [CrossRef]
- Luo, S.; Vela, J.; Lief, G.R.; Jordan, R.F. Copolymerization of Ethylene and Alkyl Vinyl Ethers by a (Phosphine-sulfonate)PdMe Catalyst. J. Am. Chem. Soc. 2007, 129, 8946–8947. [Google Scholar] [CrossRef]
- Nakano, R.; Nozaki, K. Copolymerization of Propylene and Polar Monomers Using Pd/IzQO Catalysts. J. Am. Chem. Soc. 2015, 137, 10934–10937. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, C. Influence of Polyethylene Glycol Unit on Palladium- and Nickel-Catalyzed Ethylene Polymerization and Copolymerization. Angew. Chem. Int. Ed. 2017, 56, 14672–14676. [Google Scholar] [CrossRef] [PubMed]
- Carrow, B.P.; Nozaki, K. Synthesis of Functional Polyolefins Using Cationic Bisphosphine Monoxide–Palladium Complexes. J. Am. Chem. Soc. 2012, 134, 8802–8805. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, C. A Versatile Ligand Platform for Palladium- and Nickel-Catalyzed Ethylene Copolymerization with Polar Monomers. Angew. Chem. Int. Ed. 2018, 57, 3094–3098. [Google Scholar] [CrossRef]
- Mitsushige, Y.; Yasuda, H.; Carrow, B.P.; Ito, S.; Kobayashi, M.; Tayano, T.; Watanabe, Y.; Okuno, Y.; Hayashi, S.; Kuroda, J.; et al. Methylene-Bridged Bisphosphine Monoxide Ligands for Palladium-Catalyzed Copolymerization of Ethylene and Polar Monomers. ACS Macro Lett. 2018, 7, 305–311. [Google Scholar] [CrossRef]
- Zhang, W.; Waddell, P.M.; Tiedemann, M.A.; Padilla, C.E.; Mei, J.; Chen, L.; Carrow, B.P. Electron-Rich Metal Cations Enable Synthesis of High Molecular Weight, Linear Functional Polyethylenes. J. Am. Chem. Soc. 2018, 140, 8841–8850. [Google Scholar] [CrossRef]
- Sui, X.; Dai, S.; Chen, C. Ethylene Polymerization and Copolymerization with Polar Monomers by Cationic Phosphine Phosphonic Amide Palladium Complexes. ACS Catal. 2015, 5, 5932–5937. [Google Scholar] [CrossRef]
- Mitsushige, Y.; Carrow, B.P.; Ito, S.; Nozaki, K. Ligand-controlled insertion regioselectivity accelerates copolymerisation of ethylene with methyl acrylate by cationic bisphosphine monoxide–palladium catalysts. Chem. Sci. 2016, 7, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Avcı, D.; Bahçeli, S.; Tamer, Ö.; Atalay, Y. Comparative study of DFT/B3LYP, B3PW91, and HSEH1PBE methods applied to molecular structures and spectroscopic and electronic properties of flufenpyr and amipizone. Can. J. Chem. 2015, 93, 1147–1156. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Sundermann, A. Correlation consistent valence basis sets for use with the Stuttgart–Dresden–Bonn relativistic effective core potentials: The atoms Ga–Kr and In–Xe. J. Chem. Phys. 2001, 114, 3408–3420. [Google Scholar] [CrossRef]
- Dolg, M.; Stoll, H.; Preuss, H. Energy-adjustedabinitiopseudopotentials for the rare earth elements. J. Chem. Phys. 1989, 90, 1730–1734. [Google Scholar] [CrossRef]
- Steinbrenner, U.; Bergner, A.; Dolg, M.; Stoll, H. On the transferability of energy adjusted pseudopotentiais: A calibration study for XH4(X = C, Si, Ge, Sn, Pb). Mol. Phys. 1994, 82, 3–11. [Google Scholar] [CrossRef]
- Kaupp, M.; Schleyer, P.V.R.; Stoll, H.; Preuss, H. ChemInform Abstract: Pseudopotential Approaches to Ca, Sr, and Ba Hydrides. Why are Some Alkaline Earth MX2 Compounds Bent? J. Chem. Phys. 2010, 22, 1360–1366. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Kozuch, S.; Shaik, S. How to Conceptualize Catalytic Cycles? The Energetic Span Model. Acc. Chem. Res. 2011, 44, 101–110. [Google Scholar] [CrossRef]
- Legault, C. CYLVIEW, 1.0 b; Université de Sherbrooke: Shebrooke, QC, Canada, 2009. [Google Scholar]
- Lu, T.; Fei-Wu, C. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2011, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Falivene, L.; Credendino, R.; Poater, A.; Petta, A.; Serra, L.; Oliva, R.; Scarano, V.; Cavallo, L. SambVca 2. A Web Tool for Analyzing Catalytic Pockets with Topographic Steric Maps. Organometallics 2016, 35, 2286–2293. [Google Scholar] [CrossRef]
- Nakano, R.; Chung, L.W.; Watanabe, Y.; Okuno, Y.; Okumura, Y.; Ito, S.; Morokuma, K.; Nozaki, K. Elucidating the Key Role of Phosphine−Sulfonate Ligands in Palladium-Catalyzed Ethylene Polymerization: Effect of Ligand Structure on the Molecular Weight and Linearity of Polyethylene. ACS Catal. 2016, 6, 6101–6113. [Google Scholar] [CrossRef]
- Noda, S.; Nakamura, A.; Kochi, T.; Chung, L.W.; Morokuma, K.; Nozaki, K. Mechanistic Studies on the Formation of Linear Polyethylene Chain Catalyzed by Palladium Phosphine−Sulfonate Complexes: Experiment and Theoretical Studies. J. Am. Chem. Soc. 2009, 131, 14088–14100. [Google Scholar] [CrossRef] [PubMed]
- Rezabal, E.; Ugalde, J.; Frenking, G. The Trans Effect in Palladium Phosphine Sulfonate Complexes. J. Phys. Chem. A 2017, 121, 7709–7716. [Google Scholar] [CrossRef]
- Runzi, T.; Guironnet, D.; Gottker-Schnetmann, I.; Mecking, S. Reactivity of Methacrylates in Insertion Polymerization. J. Am. Chem. Soc. 2010, 132, 16623–16630. [Google Scholar] [CrossRef]
- Michalak, A.; Ziegler, T. Palladium-Catalyzed Polymerization of Propene: DFT Model Studies. Organometallics 1999, 18, 3998–4004. [Google Scholar] [CrossRef]
- Kitaura, K.; Morokuma, K. A new energy decomposition scheme for molecular interactions within the Hartree-Fock approximation. Int. J. Quantum Chem. 1976, 10, 325–340. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Houk, K.N. Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem. Int. Ed. 2017, 56, 10070–10086. [Google Scholar] [CrossRef]
- Liu, F.; Liang, Y.; Houk, K.N. Bioorthogonal Cycloadditions: Computational Analysis with the Distortion/Interaction Model and Predictions of Reactivities. Acc. Chem. Res. 2017, 50, 2297–2308. [Google Scholar] [CrossRef] [PubMed]
- Fernández, I.; Bickelhaupt, F.M. The activation strain model and molecular orbital theory: Understanding and designing chemical reactions. Chem. Soc. Rev. 2014, 43, 4953–4967. [Google Scholar] [CrossRef]
- Xu, X.; He, G.; Wei, N.-N.; Hao, C.; Pan, Y. Selective Insertion in Copolymerization of Ethylene and Styrene Catalyzed by Half-Titanocene System Bearing Ketimide Ligand: A Theoretical Study. Chin. J. Chem. 2017, 35, 1731–1738. [Google Scholar] [CrossRef]
- Luo, Y.; Luo, Y.; Qu, J.; Hou, Z. QM/MM Studies on Scandium-Catalyzed Syndiospecific Copolymerization of Styrene and Ethylene. Organometallics 2011, 30, 2908–2919. [Google Scholar] [CrossRef]
- Hong, C.; Wang, X.-B.; Chen, C. Palladium-Catalyzed Dimerization of Vinyl Ethers: Mechanism, Catalyst Optimization, and Polymerization Applications. Macromolecules 2019, 52, 7123–7129. [Google Scholar] [CrossRef]
- Zou, C.; Pang, W.; Chen, C. Influence of chelate ring size on the properties of phosphine-sulfonate palladium catalysts. Sci. China Ser. B Chem. 2018, 61, 1175–1178. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | EC1 | ETS1 | EC2 | ETS2 | ∆G‡E2 | E-VEC3 | E-VETS3 | ∆G‡E-VE |
|---|---|---|---|---|---|---|---|---|
| a | −9.9 | 7.6 | −23.0 | −5.0 | 18.0 | −22.5 | −7.5 | 15.0 |
| b | −10.3 | 7.6 | −26.1 | −5.7 | 20.4 | −23.5 | −7.9 | 15.6 |
| c | −10.2 | 7.2 | −24.9 | −8.1 | 19.0 | −23.4 | −8.1 | 15.3 |
| d | −12.1 | 6.0 | −25.8 | −7.3 | 18.4 | −26.2 | −10.0 | 16.2 |
| e | −12.4 | 5.5 | −26.1 | −7.4 | 18.7 | 26.7 | −6.7 | 20.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehmood, A.; Xu, X.; Raza, W.; Kim, K.-H.; Luo, Y. Mechanistic Studies for Palladium Catalyzed Copolymerization of Ethylene with Vinyl Ethers. Polymers 2020, 12, 2401. https://doi.org/10.3390/polym12102401
Mehmood A, Xu X, Raza W, Kim K-H, Luo Y. Mechanistic Studies for Palladium Catalyzed Copolymerization of Ethylene with Vinyl Ethers. Polymers. 2020; 12(10):2401. https://doi.org/10.3390/polym12102401
Chicago/Turabian StyleMehmood, Andleeb, Xiaowei Xu, Waseem Raza, Ki-Hyun Kim, and Yi Luo. 2020. "Mechanistic Studies for Palladium Catalyzed Copolymerization of Ethylene with Vinyl Ethers" Polymers 12, no. 10: 2401. https://doi.org/10.3390/polym12102401
APA StyleMehmood, A., Xu, X., Raza, W., Kim, K.-H., & Luo, Y. (2020). Mechanistic Studies for Palladium Catalyzed Copolymerization of Ethylene with Vinyl Ethers. Polymers, 12(10), 2401. https://doi.org/10.3390/polym12102401