2.1. Synthesis and Post-Processing
1,2,6-hexanetriol (HT, 96%, Alfa Aesar, Haverhill, MA, USA), glycerol (G, ≥99.5%, Sigma-Aldrich, St. Louis, MO, USA), 2-butyl-2-ethyl-1,3-propanediol (BEP, >98% TCI America Inc., Portland, OR, USA), hexamethylene diisocyanate (HDI, TCI America Inc., Portland, OR, USA), Vorasurf DC 198 (Dow Corning, Midland, MI, USA), Vorasurf DC 5943 (Dow Corning, Midland, MI, USA), DABCO T-131 (Evonik Industries AG, Essen, Germany), DABCO BL-22 (Evonik Industries AG, Essen, Germany), Enovate 245fa Blowing Agent (Honeywell International, Inc., Charlotte, NC, USA) deionized (DI) water (ASTM Type II; LabChem, <1 µS/cm), HPLC Grade Water (PHARMCO, 0.5 µS/cm, Greenfield Global USA Inc., Brookfield, CT, USA), reverse osmosis water (RO Water, Texas A&M University, College Station, TX, USA) and isopropyl alcohol (IPA, ≥99.5%; Fisher Scientific, Waltham, MA, USA) were used as received.
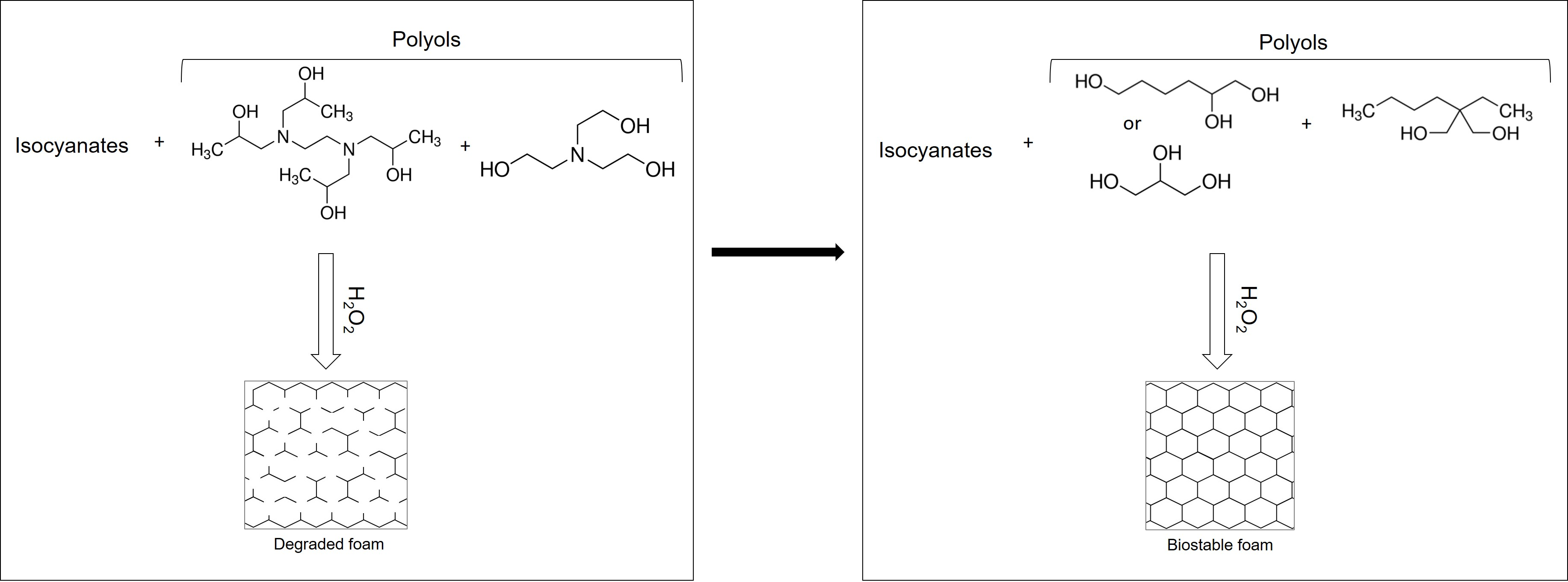
SMP foams were synthesized using a gas-blowing process described by Hasan et al. [
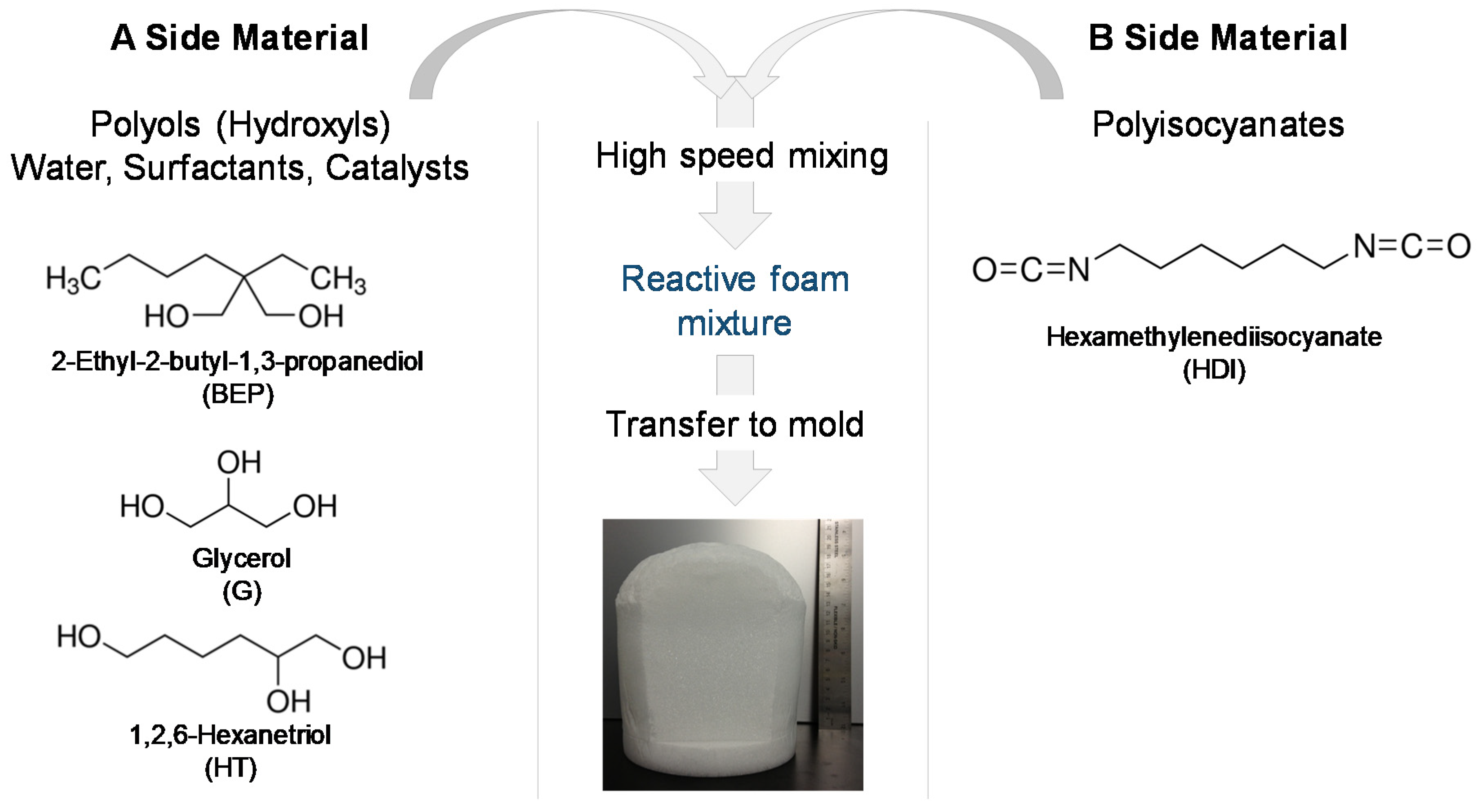
12]. To synthesize glycerol foams, first, an isocyanate (NCO) prepolymer was synthesized with appropriate molar ratios of G, BEP, and HDI. Next, a hydroxyl (OH) mixture was blended with the remaining molar equivalents of G and BEP, along with foaming additives such as DI water (<1 µS/cm), and catalysts. The NCO prepolymer and the OH mixture were combined in the foam cup along with surfactants, DC 198 and DC 5943, and Enovate. This mixture was mixed in a FlackTek speedmixer (FlackTek, Inc., Landrum, SC, USA) and poured in the foam bucket. The foam was allowed to cure fully before further processing. To synthesize hexanetriol foams, the abovementioned process was utilized with glycerol (G) being replaced by hexanetriol (HT).
Table 1 shows the weight percent of each component used for foam synthesis and
Figure 1 shows the corresponding molecular structure of each monomer and a schematic of the foaming process.
Post-synthesis, the bulk foam was cut into 2 cm long cylinders with 6 mm diameter using a biopsy punch. The samples were annealed at 90 °C for 30 min and allowed to cool down completely before further processing. Foam cylinders were cleaned in 32 oz glass jars using one 30 min sonication wash in DI water and four 30 min sonication washes in IPA. After each wash, the solvent was discarded and the jars were replenished with fresh solvent. Prior to testing, foam cylinders were dried at 100 °C under vacuum for at least 12 h after which they were stored in a plastic storage container with dessicant.
2.2. Chemical and Physical Characterization
2.2.1. Cell Structure and Pore Size
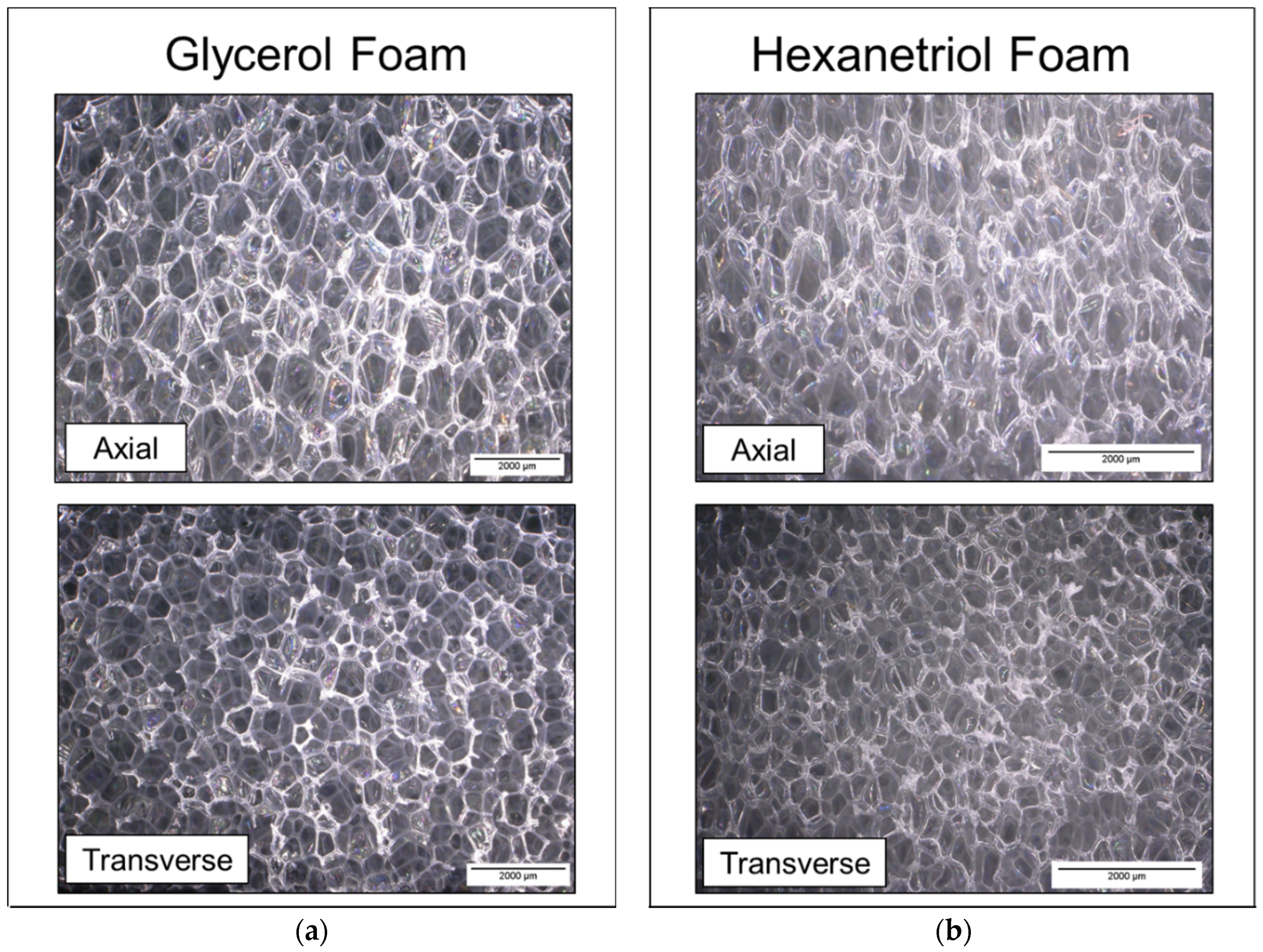
Foam cubes were cut from the top, middle, and bottom sections of the foams using a resistive hot-wire cutter for pore size characterization. Sample preparation and instrumentation for pore size analysis were determined using ASTM D3576 [
20]. Pore size was measured using the Keyence Inspection System (VHX-5000) with the variable angle adapter lens via 2-point linear measurements. Pore images were collected from samples that were axial (parallel) and transverse (perpendicular) to the foam rise, and average pore size values were calculated for each foam (10 points measured per image).
2.2.2. Fourier-Transform Infrared (FTIR) Spectroscopy
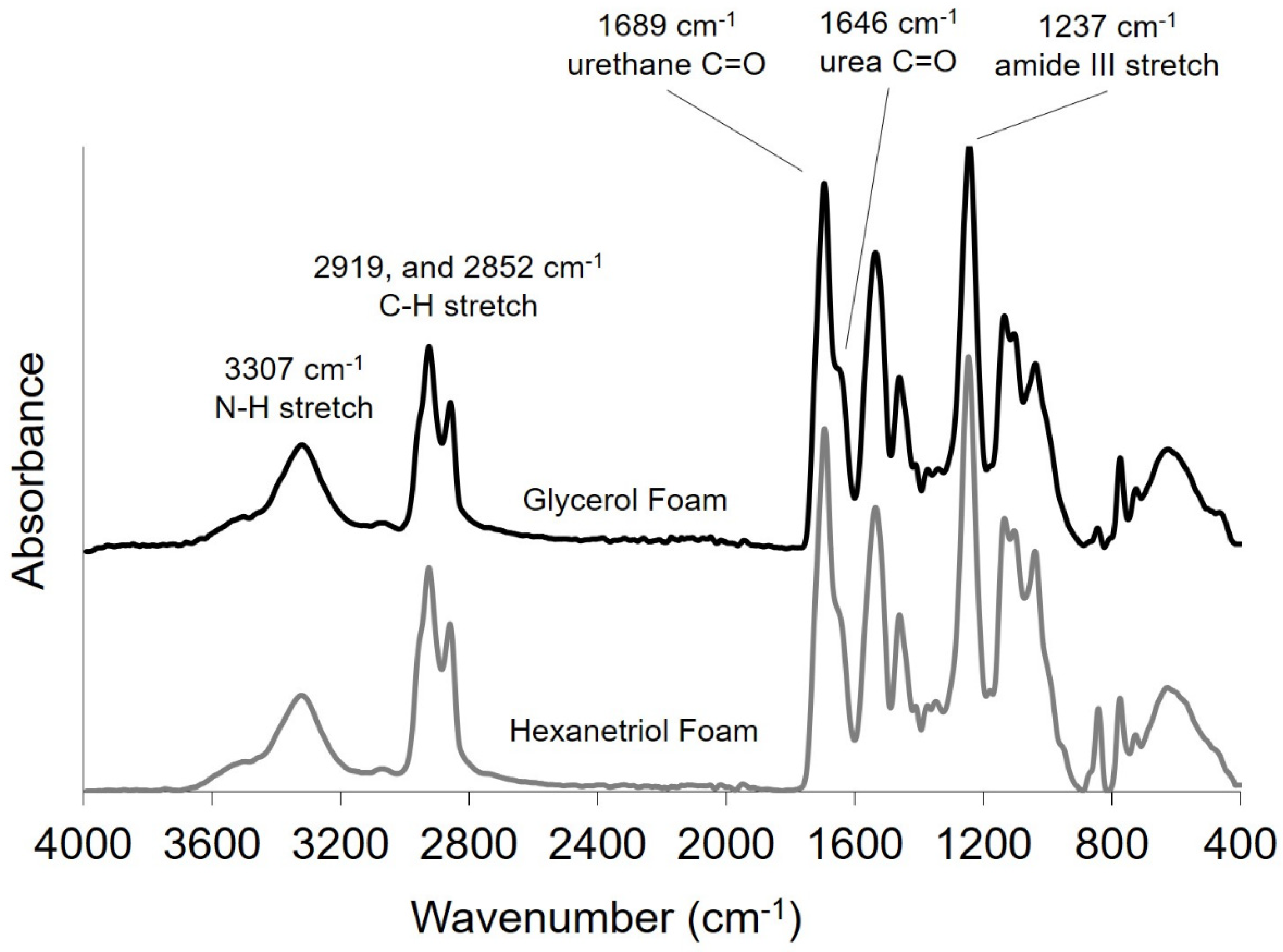
Thin foam samples were cut (2–3 mm) from bulk foams, and FTIR spectra was collected using a Bruker ALPHA Infrared Spectrometer (Bruker, Billerica, MA, USA) via the Platinum ATR Sampling Module. Sixty-four background scans of the empty chamber were taken followed by 32 sample scans of the various foam compositions. FTIR spectra was collected in absorption mode at a resolution of 4 cm−1 within the wavenumber range of 400 to 4000 cm−1 with atmospheric compensation. OPUS software (Bruker, Billerica, MA, USA) was utilized to subtract the background scans from the spectra and to conduct a baseline correction for IR beam scattering and an atmospheric compensation to remove any peaks acquired due to carbon dioxide or water in the air.
2.2.3. Differential Scanning Calorimetry (DSC)
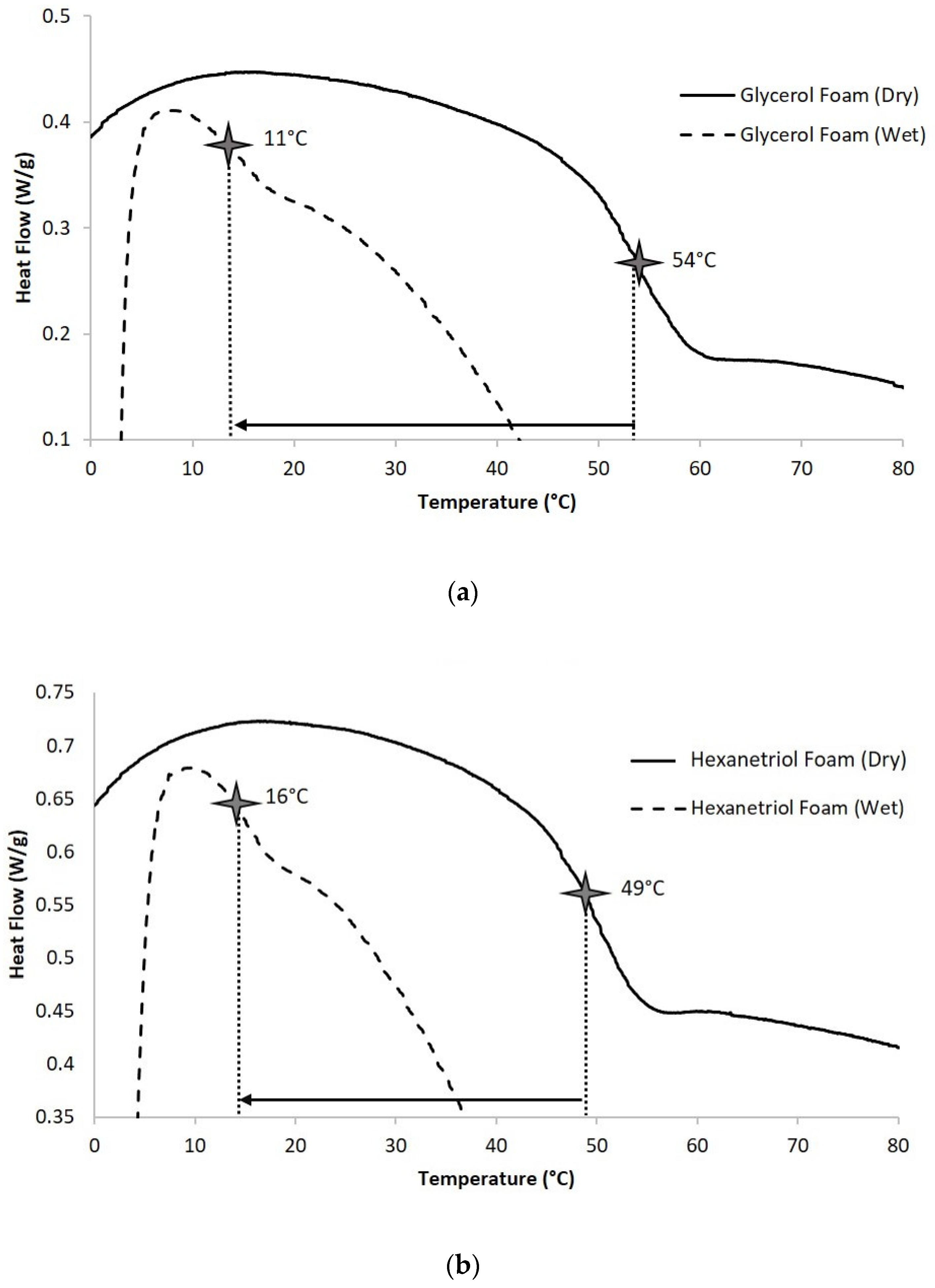
Foam samples (4–5 mg) were cut from bulk foam and thermally characterized using the Q-2000 DSC (TA Instruments, Inc., New Castle, DE, USA). The first cycle consisted of decreasing the temperature to −40 °C at 30 °C min−1 and holding it isothermally for 2 min. The temperature was then increased to 120 °C at 30 °C min−1 and held isothermally for 2 min. In the second cycle, the temperature was reduced to −40 °C at 30 °C min−1, held isothermally for 2 min, and raised to 120 °C at 10 °C min−1. Tg (dry) was recorded from the second cycle based on the inflection point of the thermal transition curve using TA instruments software. The aluminum pan was vented during this test to remove moisture from the sample during the first cycle. N = 3 was utilized per foam composition.
2.2.4. Water Moisture Plasticized Glass Transition Temperature
Foam samples (4–5 mg) were cut from bulk foam and submerged in RO water at 50 °C for 5 min to allow full plasticization. After the samples were removed from water, they were pressed dry with Kim Wipes (Kimberly-Clark Professionals, Roswell, GA, USA), weighed, and placed in an aluminum pan sealed with a vented aluminum lid. A Q-2000 DSC was used to cool the samples to −40 °C, hold them isothermally for 2 min, and heat them to 80 °C at 10 °C·min−1. TA instruments software was used to generate the thermogram and acquire the Tg (wet), after water plasticization, using the inflection point of the thermal transition. N = 3 was utilized per foam composition.
2.2.5. Thermogravimetric Analysis
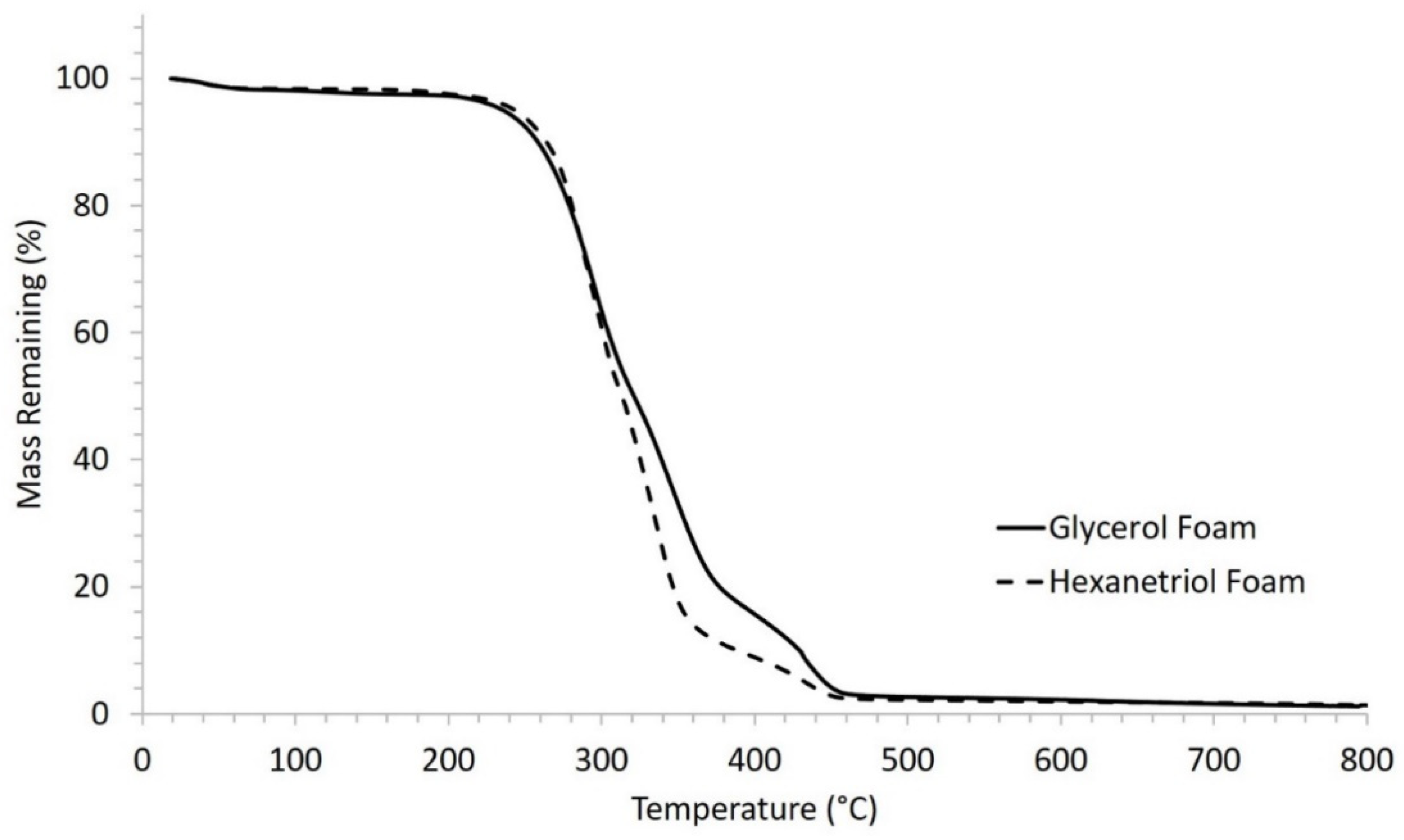
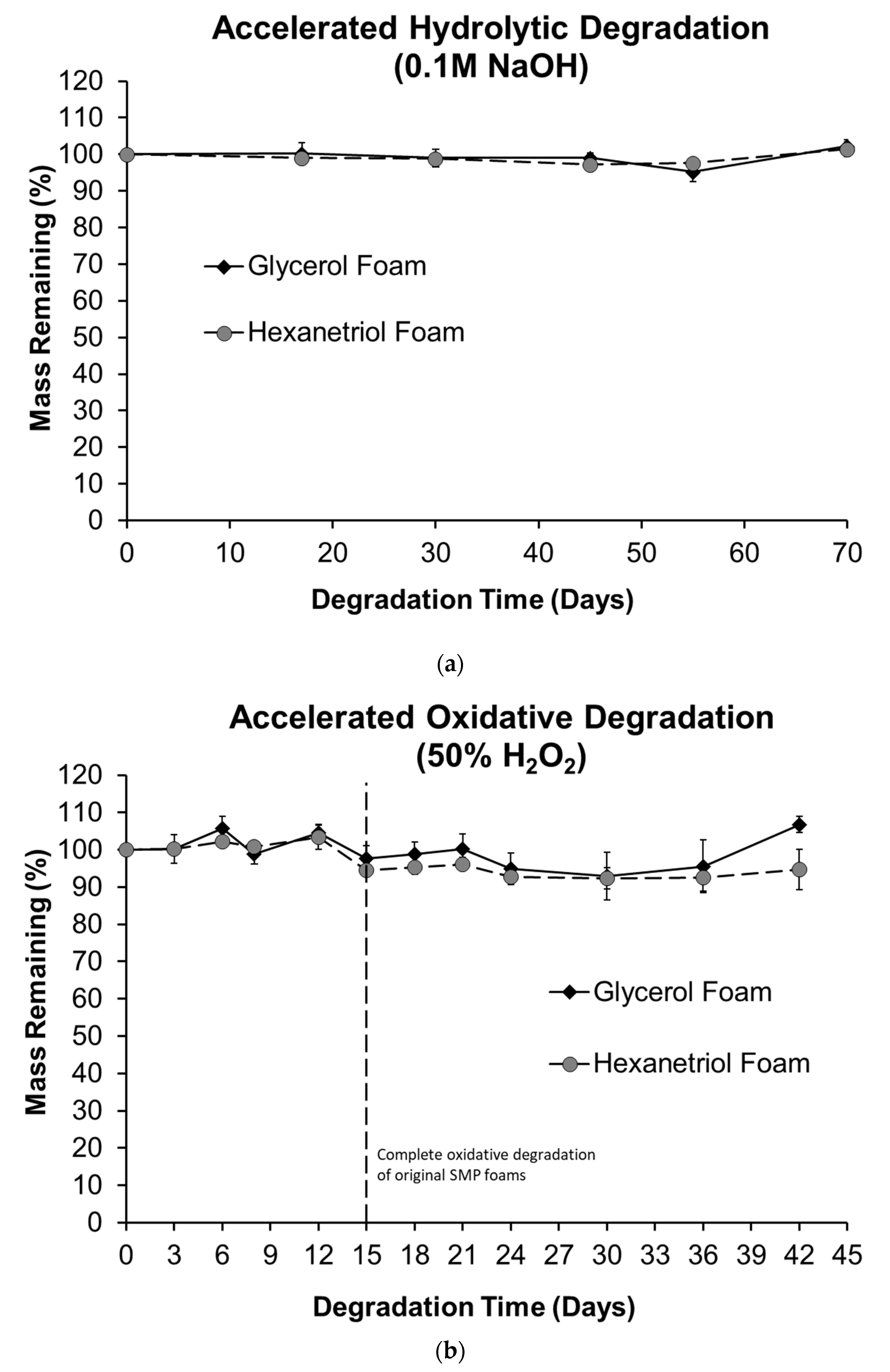
Thermal stability of the SMP foams was determined using thermogravimetric analysis (TGA). Samples (10–15 mg, n = 1) were prepared from the bulk foam. A platinum pan was used to hold each sample and tared before each run. The samples were heated from 25 to 800 °C at 10 °C·min−1 under nitrogen flow of 60 mL·min−1 using a TGA Q 50 (TA Instruments, New Castle, DE, USA). At 800 °C, the gas was switched to air flow at 60 mL·min−1, and the samples were heated to 1000 °C. The thermograms were evaluated using TA Universal Analysis software, and percent mass remaining (%) versus temperature (°C) curves were graphed for each foam composition.
2.2.6. Mechanical Testing
Mechanical properties of the SMP foams were determined using MTS Criterion Model 42 (MTS Systems Corporation, Inc., Eden Prairie, MN, USA), A 10 N load cell was used for conducting tensile (extension) testing with a test speed of 5 mm/min. N = 10 samples were prepared for each SMP foam formulation. Then, 2 mm thick foam slices were cut from the bulk foam using the Proxxon 37080 Hot Wire Cutter (PROXXON Inc., Hickory, NC, USA). The foam slices were further cut into dog bones using a laser engraving system (Orion Motor Tech, Lake Forest, CA, USA). The foam dog bones were endcapped with wooden blocks to prevent material damage during clamping and annealed for 30 min at 90 °C. The samples were allowed to cool down to room temperature before testing. Five samples, for each formulation, were tensile tested under dry, ambient conditions and the remaining 5 samples were soaked in room temperature DI water for 48 h, to ensure complete water-plasticization, prior to tensile testing. Strain at break (%) and Young’s Modulus (kPa) was recorded for both the glycerol and the hexanetriol SMP foams under dry and water-plasticized conditions.
2.2.7. Shape Fixity and Shape Recovery
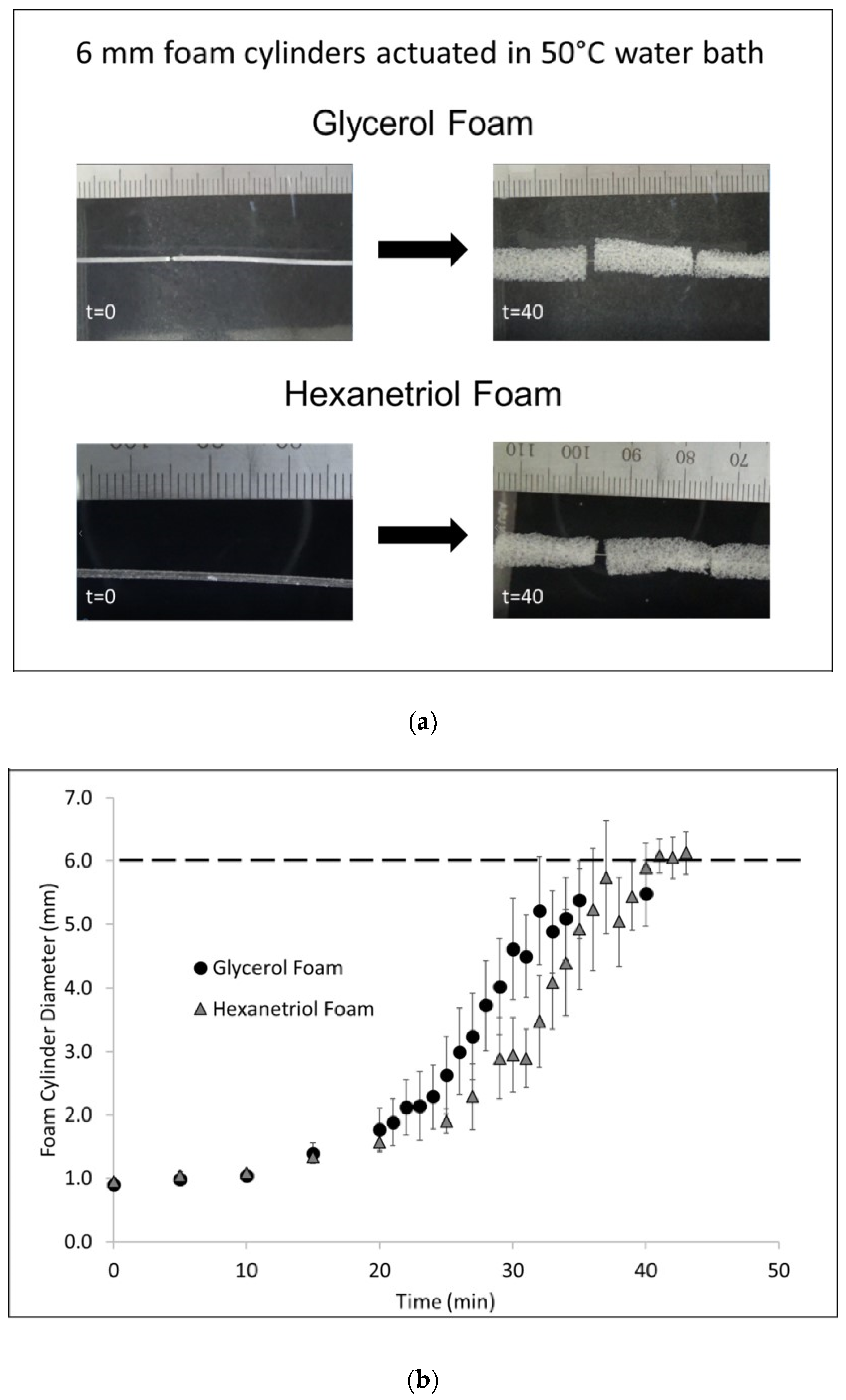
Foam cylinders (n = 3) with a diameter of 10 mm and height of 10 mm were cut out of the bulk foam for each SMP formulation. The samples were annealed for 30 min at 90 °C and allowed to cool to room temperature, prior to testing. Each sample, from the glycerol and hexanetriol formulation, was heated to 100 °C, using a heat gun, and programmed to a compressed height of 3 mm. Shape setting occurred by cooling the sample to room temperature while the sample remained under load. Once the sample was completely cool, the load was removed and the programmed height (
h1) was measured using a microcaliper. The sample was allowed to remain in the compressed shape, without load, at room temperature, for 15 min after which the programmed height was remeasured (
h2). After this step, the sample was reheated to 100 °C, using a heat gun, allowed to recover to its original shape without any load. The recovered height was measured using micro-calipers. Shape fixity (R
f) and shape recovery (R
r) was calculated for each sample using Equations (1) and (2), respectively.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






