The Mechanical Properties of Poly (Urea-Formaldehyde) Incorporated with Nano-SiO2 by Molecular Dynamics Simulation


Abstract
1. Introduction
2. Simulation and Experimental Methods
2.1. Materials Models
2.1.1. PUF Model
2.1.2. Nano-SiO2 Model
2.1.3. PUF-SiO2 Models
2.1.4. PUF–SiO2 Interfacial Interaction Model
2.2. Molecular Dynamics (MD) Simulation
2.3. Preparation Method of DCPD/PUF Microcapsules

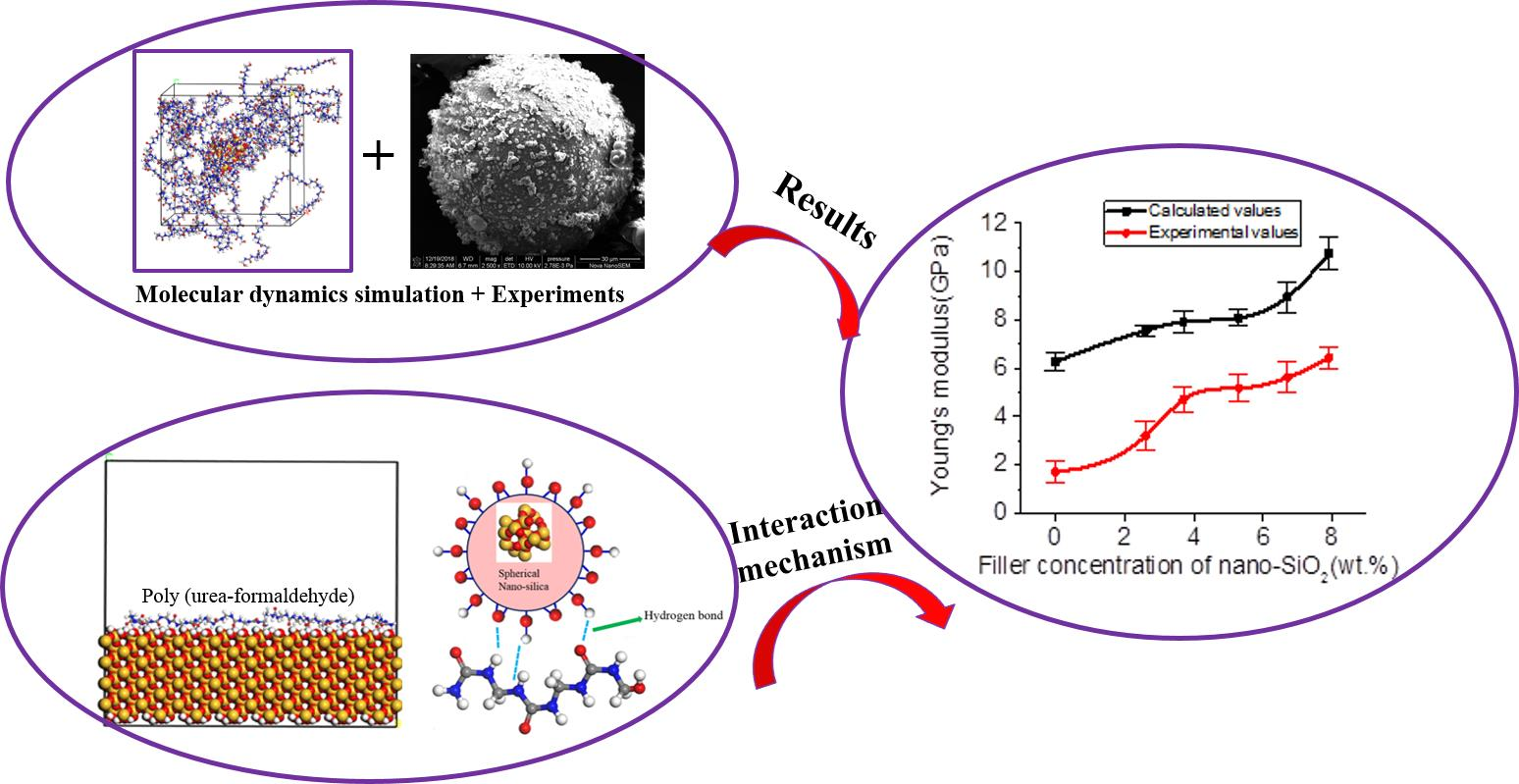
3. Results
3.1. Density
3.2. Fractional Free Volume
3.3. Mechanical Properties
3.3.1. Micromechanical Properties
3.3.2. Mechanical Properties of DCPD/PUF Microcapsules
4. Mechanism Analysis
4.1. Interfacial Binding Energy
4.2. The Interfacial Hydrogen Bond Number
4.3. Interfacial Interaction Mechanism
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tong, X.M.; Zhang, T.; Yang, M.Z.; Zhang, Q. Preparation and characterization of novel melamine modified poly (urea–formaldehyde) self-repairing microcapsules. Colloids Surf. B Eng. 2010, 371, 91–97. [Google Scholar] [CrossRef]
- Jin, H.; Miller, G.M.; Sottos, N.R.; White, S.R. Fracture and fatigue response of a self-healing epoxy adhesive. Polymer 2011, 52, 1628–1634. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, M.; Tong, X.M.; Chen, F.; Qiu, J.H. Optimal preparation and characterization of poly (urea–formaldehyde) microcapsules. J. Appl. Polym. Sci. 2010, 115, 2162–2169. [Google Scholar]
- Wang, R.; Li, H.; Hu, H.; He, X.; Liu, W. Preparation and characterization of self-healing microcapsules with poly(urea–formaldehyde) grafted epoxy functional group shell. J. Appl. Polym. Sci. 2009, 113, 1501–1506. [Google Scholar] [CrossRef]
- Blaiszik, B.J.; Caruso, M.M.; McIlroy, D.A.; Moore, J.S.; White, S.R.; Sottos, N.R. Microcapsules filled with reactive solutions for self-healing materials. Polymer 2009, 50, 990–997. [Google Scholar] [CrossRef]
- Ghorbanzadeh Ahangari, M.; Fereidoon, A.; Jahanshahi, M.; Sharifi, N. Effect of nanoparticles on the micromechanical and surface properties of poly(urea–formaldehyde) composite microcapsules. Compos. Part B-Eng. 2014, 56, 450–455. [Google Scholar] [CrossRef]
- Corona-Gomez, J.; Chen, X.; Yang, Q. Effect of nanoparticle incorporation and surface coating on mechanical properties of bone scaffolds: A brief review. J. Funct. Biomater. 2016, 7, 18–28. [Google Scholar] [CrossRef]
- Qiao, B.; Liang, Y.; Wang, T.J.; Jiang, Y. Surface modification to produce hydrophobic nano-silica particles using sodium dodecyl sulfate as a modifier. Appl. Surf. Sci. 2016, 364, 103–109. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, C.A. Nano-network MnO2/polyaniline composites with enhanced electrochemical properties for supercapacitors. Mater. Des. 2016, 97, 512–518. [Google Scholar] [CrossRef]
- Fan, C.; Zhou, X. Preparation and barrier properties of the microcapsules added nanoclays in the wall. Polym. Adv. Technol. 2009, 20, 934–939. [Google Scholar]
- Fereidoon, A.; Ghorbanzadeh Ahangari, M.; Jahanshahi, M. Effect of nanoparticles on the morphology and thermal properties of self-healing poly(urea-formaldehyde) microcapsules. J. Polym. Res. 2013, 20, 151–158. [Google Scholar] [CrossRef]
- Jia, J.; Yang, J.; Zhao, Y.; Liang, H.; Chen, M. The crystallization behaviors and mechanical properties of poly(L-lactic acid)/magnesium oxide nanoparticle composites. RSC Adv. 2016, 6, 43855–43863. [Google Scholar] [CrossRef]
- Griffin, M.; Nayyer, L.; Butler, P.E.; Palgrave, R.G.; Seifalian, A.M.; Kalaskar, D.M. Development of mechano-responsive polymeric scaffolds using functionalised silica nano fillers for the control of cellular functions. Nanomed-Nanotechnol 2016, 12, 1725–1733. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sowjanya, J.A.; Singh, J.; Mohita, T.; Sarvanan, S.; Moorthi, A.; Srinivasan, N.; Selvamurugan, N. Biocomposite scaffolds containing chitosan/alginate/nano-silica for bone tissue engineering. Colloids Surf. B Biointerfaces 2013, 109, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Taheri-Behrooz, F.; Memar Maher, B.; Shokrieh, M.M. Mechanical properties modification of a thin film phenolic resin filled with nano silica particles. Comput. Mater. Sci. 2015, 96, 411–415. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, H.Y.; Zhou, H.; Zhang, Y.; Gao, X.; Mai, Y.W. On adhesive properties of nanosilica/epoxy bonded single-lap joints. Mater. Des. 2016, 95, 212–218. [Google Scholar] [CrossRef]
- Malaki, M.; Hashemzadeh, Y.; Karevan, M. Effect of nano-silica on the mechanical properties of acrylic polyurethane coatings. Prog. Org. Coat. 2016, 101, 477–485. [Google Scholar] [CrossRef]
- Fallah, S.; Nematzadeh, M. Mechanical properties and durability of high-strength concrete containing macro-polymeric and polypropylene fibers with nano-silica and silica fume. Constr. Build. Mater. 2017, 132, 170–187. [Google Scholar] [CrossRef]
- Yang, M.; Xia, Y.; Wang, Y.; Zhao, X.; Xue, Z.; Quan, F.; Geng, C.; Zhao, Z. Preparation and property investigation of crosslinked alginate/silicon dioxide nanocomposite films. J. Appl. Polym. Sci. 2016, 133, 43489–43498. [Google Scholar] [CrossRef]
- Dil, E.J.; Virgilio, N.; Favis, B.D. The effect of the interfacial assembly of nano-silica in poly(lactic acid)/poly(butylene adipate-co-terephthalate) blends on the morphology, rheology and mechanical properties. Eur. Polym. J. 2016, 85, 635–646. [Google Scholar]
- Wei, Q.; Wang, Y.; Chai, W.; Wang, T.; Zhang, Y. Effects of composition ratio on the properties of poly(vinyl alcohol)/poly(acrylic acid) blend membrane: A molecular dynamics simulation study. Mater. Des. 2016, 89, 848–855. [Google Scholar] [CrossRef]
- Roussou, R.E.; Karatasos, K. Graphene/poly (ethylene glycol) nanocomposites as studied by molecular dynamics simulations. Mater. Des. 2016, 97, 163–174. [Google Scholar] [CrossRef]
- Moyassari, A.; Gkourmpis, T.; Hedenqvist, M.S.; Gedde, U.W. Molecular dynamics simulations of short-chain branched bimodal polyethylene: Topological characteristics and mechanical behavior. Macromolecules 2019, 52, 807–818. [Google Scholar] [CrossRef]
- Moyassaria, A.; Gkourmpisb, T.; Hedenqvista, M.S.; Gedde, U.W. Molecular dynamics simulation of linear polyethylene blends: Effect of molar mass bimodality on topological characteristics and mechanical behavior. Polymer 2019, 161, 139–150. [Google Scholar] [CrossRef]
- Wei, Q.; Zhang, Y.; Wang, Y.; Chai, W.; Yang, M. Measurement and modeling of the effect of composition ratios on the properties of poly (vinyl alcohol)/poly (vinyl pyrrolidone) membranes. Mater. Des. 2016, 103, 249–258. [Google Scholar] [CrossRef]
- Wei, Q.; Wang, Y.; Che, Y.; Yang, M.; Li, X.; Zhang, Y. Molecular mechanisms in compatibility and mechanical properties of Polyacrylamide/Polyvinyl alcohol blends. J. Mech. Behav. Biomed. 2017, 65, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Shariatinia, Z.; Jalali, A.M.; Taromi, F.A. Molecular dynamics simulations on desulfurization of n-octane/thiophene mixture using silica filled polydimethylsiloxane nanocomposite membranes. Model. Simul. Mater. Sci. Eng. 2016, 24, 035002–035022. [Google Scholar] [CrossRef]
- Wei, Q.; Wang, Y.; Rao, Y.; Jiang, A.; Zhang, K.; Lu, T.; Chen, X. Evaluating the effects of nanosilica on mechanical and tribological properties of polyvinyl alcohol/polyacrylamide polymer composites for artificial cartilage from an atomic level. Polymers 2019, 11, 76–89. [Google Scholar] [CrossRef]
- Akhlaghi, S.; Pourrahimi, A.M.; Sjostedt, C.; Hedenqvist, M.S. Effects of ageing conditions on degradation of acrylonitrile butadiene rubber filled with heat-treated ZnO star-shaped particles in rapeseed biodiesel. Polym. Degrad. Stabil. 2017, 138, 27–39. [Google Scholar] [CrossRef]
- Akhlaghi, S.; Pourrahimi, A.M.; Sjostedt, C.; Bellander, M.; Hedenqvist, M.S. Degradation of fluoroelastomers in rapeseed biodiesel at different oxygen concentrations. Polym. Degrad. Stab. 2017, 136, 10–19. [Google Scholar] [CrossRef]
- Hedenqvist, M.; Gedde, U.W. Diffusion of small-molecule penetrants in semicrystalline polymers. Prog. Polym. Sci. 1996, 21, 299–333. [Google Scholar] [CrossRef]
- Lai, Z.B.; Wang, M.; Yan, C.; Oloyede, A. Molecular dynamics simulation of mechanical behavior of osteopontin-hydroxyapatite interfaces. J. Mech. Behav. Biomed. 2014, 36, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wei, Q.; Pan, F.; Yang, M.; Wei, S. Molecular dynamics simulations for the examination of mechanical properties of hydroxyapatite/poly α-n-butyl cyanoacrylate under additive manufacturing. Bio-Med. Mater. Eng. 2014, 24, 825–833. [Google Scholar]
- Wei, Q.; Wang, Y.; Wang, S.; Zhang, Y.; Chen, X. Investigating the Properties and Interaction Mechanism of Nano-Silica in Polyvinyl Alcohol/Polyacrylamide Blends at an Atomic Level. J. Mech. Behav. Biomed. Mater. 2017, 75, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Zhang, Y.; Wang, Y.; Yang, M. A molecular dynamic simulation method to elucidate the interaction mechanism of nano-SiO2 in polymer blends. J. Mater. Sci. 2017, 52, 12889–12901. [Google Scholar] [CrossRef]
- Kubyshkina1, E.; Unge, M. Impact of interfacial structure on the charge dynamics in nanocomposite dielectrics. J. Appl. Phys. 2019, 125, 045109–045117. [Google Scholar] [CrossRef]
- Pourrahimi, A.M.; Olsson, R.T.; Hedenqvist, M.S. The role of interfaces in polyethylene/metal-oxide nanocomposites for ultrahigh-voltage insulating materials. Adv. Mater. 2018, 30, 1703624–1703648. [Google Scholar] [CrossRef] [PubMed]
- Arab, B.; Shokuhfar, A. Molecular dynamics simulation of cross-linked urea-formaldehyde polymers for self-healing nanocomposites: Prediction of mechanical properties and glass transition temperature. J. Mol. Model. 2013, 19, 5053–5062. [Google Scholar] [CrossRef] [PubMed]
- Sun, H. COMPASS: An ab initio Force-Field Optimized for Condensed-Phase Applications-Overview with Details on Alkane and Benzene Compounds. J. Phys. Chem. B 1988, 102, 7338–7364. [Google Scholar] [CrossRef]
- Wei, Q.; Wang, Y.; Li, X.; Yang, M.; Chai, W.; Wang, K.; Zhang, Y. Study the bonding mechanism of binders on hydroxyapatite surface and mechanical properties for 3DP fabrication bone scaffolds by a molecular dynamics simulation. J. Mech. Behav. Biomed. Mater. 2016, 57, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Andersen, H.C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 1980, 72, 2384–2393. [Google Scholar] [CrossRef]
- Peng, F.; Jiang, Z.; Hoek, E.M.V. Tuning the molecular structure, separation performance and interfacial properties of poly(vinyl alcohol)-polysulfone interfacial composite membranes. J. Membr. Sci. 2011, 368, 26–33. [Google Scholar] [CrossRef]
- Chen, G.; Li, A.; Liu, H.; Huang, S.; Zhang, Z.; Liu, W.; Zha, C.; Li, B.; Wang, Z. Mechanical and dynamic properties of resin blend and composite systems: A molecular dynamics study. Compos. Struct. 2018, 190, 160–168. [Google Scholar] [CrossRef]
- Watt, J.P.; Davies, G.F.; O’Connell, R.J. The elastic properties of composite materials. Rev. Geophys. 1976, 14, 541–563. [Google Scholar] [CrossRef]
- Nazarychev, V.M.; Lyulin, A.V.; Larin, S.V.; Gurtovenko, A.A.; Kenny, J.M.; Lyulin, S.V. Molecular dynamics simulations of uniaxial deformation of thermoplastic polyimides. Soft Matter 2016, 12, 3972–3981. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Li, X.; Li, Z.; Hao, J. Interfacial hydrogen bonds and their influence mechanism on increasing the thermal stability of nano-SiO2-modified meta-aramid fibres. Polymers 2017, 9, 504–521. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Before NPT-MD | After NPT-MD | ||
|---|---|---|---|---|
| Cell Length (Å) | Density (g/cm³) | Cell Length (Å) | Density (g/cm³) | |
| 37PUF-0SiO2-0% | 56.4 | 0.5 | 42.30 ± 0.02 | 1.184 ± 0.002 |
| 37PUF-1SiO2-2.6% | 56.9 | 0.5 | 41.28 ± 0.03 | 1.307 ± 0.003 |
| 25PUF-1SiO2-3.7% | 50.1 | 0.5 | 35.81 ± 0.04 | 1.369 ± 0.004 |
| 18PUF-1SiO2-5.3% | 45.1 | 0.5 | 31.87 ± 0.03 | 1.418 ± 0.003 |
| 14PUF-1SiO2-6.7% | 41.7 | 0.5 | 29.89 ± 0.07 | 1.424 ± 0.008 |
| 12PUF-1SiO2-7.9% | 39.7 | 0.5 | 27.59 ± 0.01 | 1.491 ± 0.001 |
| Model | Free Volume (Å3) | Occupied Volume (Å3) | FFV (%) |
|---|---|---|---|
| 37PUF-0SiO2 -0% | 21,173.85 ± 50.05 | 54,427.06 ± 50.05 | 28.01 ± 0.07 |
| 37PUF-1SiO2 -2.6% | 14,257.58 ± 19.32 | 55,946.39 ± 19.32 | 20.31 ± 0.03 |
| 25PUF-1SiO2-3.7% | 7626.71 ± 35.83 | 38,145.95 ± 35.83 | 16.66 ± 0.08 |
| 18PUF-1SiO2 -5.3% | 4422.97 ± 38.81 | 27,870.26 ± 38.81 | 13.70 ± 0.12 |
| 14PUF-1SiO2-6.7% | 3253.46 ± 30.22 | 23,026.51 ± 30.22 | 12.38 ± 0.09 |
| 12PUF-1SiO2 -7.9% | 2006.52 ± 14.26 | 18,961.32 ± 14.26 | 9.56 ± 0.07 |
| Model | 37PUF-0SiO2-0% | 37PUF-1SiO2-2.6% | 25PUF-1SiO2-3.7% | 18PUF-1SiO2-5.3% | 14PUF-1SiO2-6.7% | 12PUF-1SiO2-7.9% |
|---|---|---|---|---|---|---|
| 2.7527 ± 0.3781 | 3.2839 ± 0.3051 | 3.8483 ± 0.1840 | 4.7356 ± 0.4571 | 4.9852 ± 0.5445 | 5.7648 ± 0.2067 | |
| 7.6535 ± 0.5467 | 9.4918 ± 0.3322 | 12.1510 ± 0.2894 | 10.7983 ± 1.3888 | 12.3678 ± 1.5087 | 15.0725 ± 0.2480 | |
| 8.5477 ± 0.2839 | 10.3343 ± 0.3099 | 10.5001 ± 0.1673 | 12.5241 ± 0.8194 | 12.8983 ± 1.4587 | 13.3956 ± 0.2276 | |
| 9.0590 ± 0.6414 | 9.8329 ± 0.1831 | 10.6432 ± 0.1875 | 11.5590 ± 0.4832 | 11.3454 ± 1.2693 | 12.5315 ± 0.2640 | |
| 2.5086 ± 0.1973 | 2.9983 ± 0.1460 | 3.1127 ± 0.1185 | 3.8134 ± 0.2170 | 3.8561 ± 0.2649 | 4.0825 ± 0.2050 | |
| 2.5624 ± 0.0687 | 2.9176 ± 0.1447 | 3.1466 ± 0.0813 | 3.3067 ± 0.6514 | 3.5843 ± 1.2084 | 4.5313 ± 0.1048 | |
| 2.1790 ± 0.0503 | 2.8749 ± 0.1844 | 2.7866 ± 0.0943 | 2.0627 ± 1.1979 | 2.8310 ± 1.1607 | 4.0137 ± 0.0595 | |
| 6.28 ± 0.38 | 7.56 ± 0.21 | 7.92 ± 0.47 | 8.09 ± 0.33 | 8.94 ± 0.62 | 10.75 ± 0.67 | |
| 5.20 ± 0.04 | 5.98 ± 0.02 | 7.08 ± 0.02 | 7.55 ± 0.04 | 7.64 ± 0.24 | 8.05 ± 0.06 | |
| 2.42 ± 0.02 | 2.93 ± 0.02 | 3.02 ± 0.01 | 3.06 ± 0.07 | 3.42 ± 0.18 | 4.21 ± 0.03 | |
| 0.30 ± 0.01 | 0.29 ± 0.01 | 0.31 ± 0.02 | 0.32 ± 0.02 | 0.31 ± 0.01 | 0.28 ± 0.03 | |
| 0.24 ± 0.18 | 0.29 ± 0.16 | 0.74 ± 0.06 | 0.92 ± 0.24 | 1.13 ± 0.28 | 1.68 ± 0.00 |
| System | ||||||
|---|---|---|---|---|---|---|
| PUF–SiO2 | 36,578,757.7 ± 6.9 | 36,579,050.3 ± 0.2 | –225.7 ± 7.3 | –66.8 ± 0.2 | 66.8 ± 0.2 | –45.1 ± 0.8 |
| Model | |||||
|---|---|---|---|---|---|
| 37PUF-0SiO2-0% | 4369 ± 18 | 4369 ± 18 | 0 ± 0 | 0 ± 0 | 0.00 ± 0.00 |
| 37PUF-1SiO2-2.6% | 4557 ± 10 | 4369 ± 18 | 27 ± 0 | 161 ± 8 | 3.69 ± 0.17 |
| 25PUF-1SiO2-3.7% | 3046 ± 9 | 2880 ± 11 | 27 ± 0 | 139 ± 2 | 4.83 ± 0.05 |
| 18PUF-1SiO2-5.3% | 2272 ± 4 | 2120 ± 6 | 27 ± 0 | 125 ± 2 | 5.90 ± 0.08 |
| 14PUF-1SiO2-6.7% | 1710 ± 8 | 1568 ± 10 | 27 ± 0 | 113 ± 2 | 7.20 ± 0.08 |
| 12PUF-1SiO2-7.9% | 1593 ± 9 | 1436 ± 12 | 27 ± 0 | 126 ± 3 | 8.75 ± 0.14 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wang, Y.; Li, Y.; Zhang, Z. The Mechanical Properties of Poly (Urea-Formaldehyde) Incorporated with Nano-SiO2 by Molecular Dynamics Simulation. Polymers 2019, 11, 1447. https://doi.org/10.3390/polym11091447
Zhang Y, Wang Y, Li Y, Zhang Z. The Mechanical Properties of Poly (Urea-Formaldehyde) Incorporated with Nano-SiO2 by Molecular Dynamics Simulation. Polymers. 2019; 11(9):1447. https://doi.org/10.3390/polym11091447
Chicago/Turabian StyleZhang, Yanfang, Youyuan Wang, Yudong Li, and Zhanxi Zhang. 2019. "The Mechanical Properties of Poly (Urea-Formaldehyde) Incorporated with Nano-SiO2 by Molecular Dynamics Simulation" Polymers 11, no. 9: 1447. https://doi.org/10.3390/polym11091447
APA StyleZhang, Y., Wang, Y., Li, Y., & Zhang, Z. (2019). The Mechanical Properties of Poly (Urea-Formaldehyde) Incorporated with Nano-SiO2 by Molecular Dynamics Simulation. Polymers, 11(9), 1447. https://doi.org/10.3390/polym11091447