Hydrogen Bonding-Induced Assembled Structures and Photoresponsive Behavior of Azobenzene Molecule/Polyethylene Glycol Complexes



Abstract
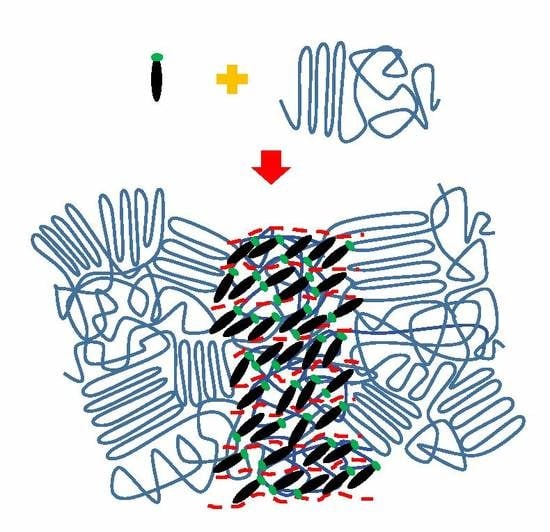
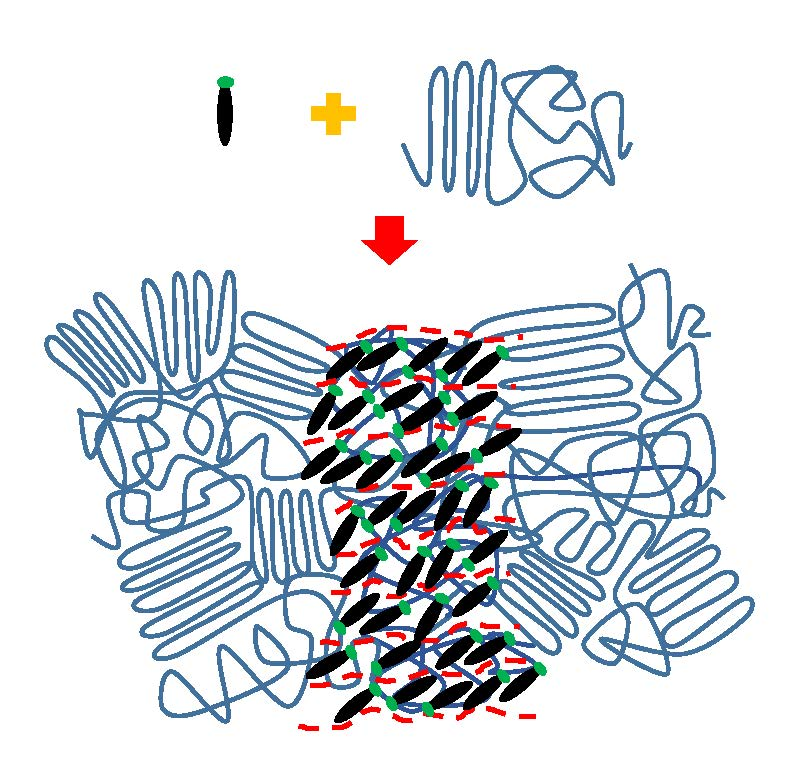
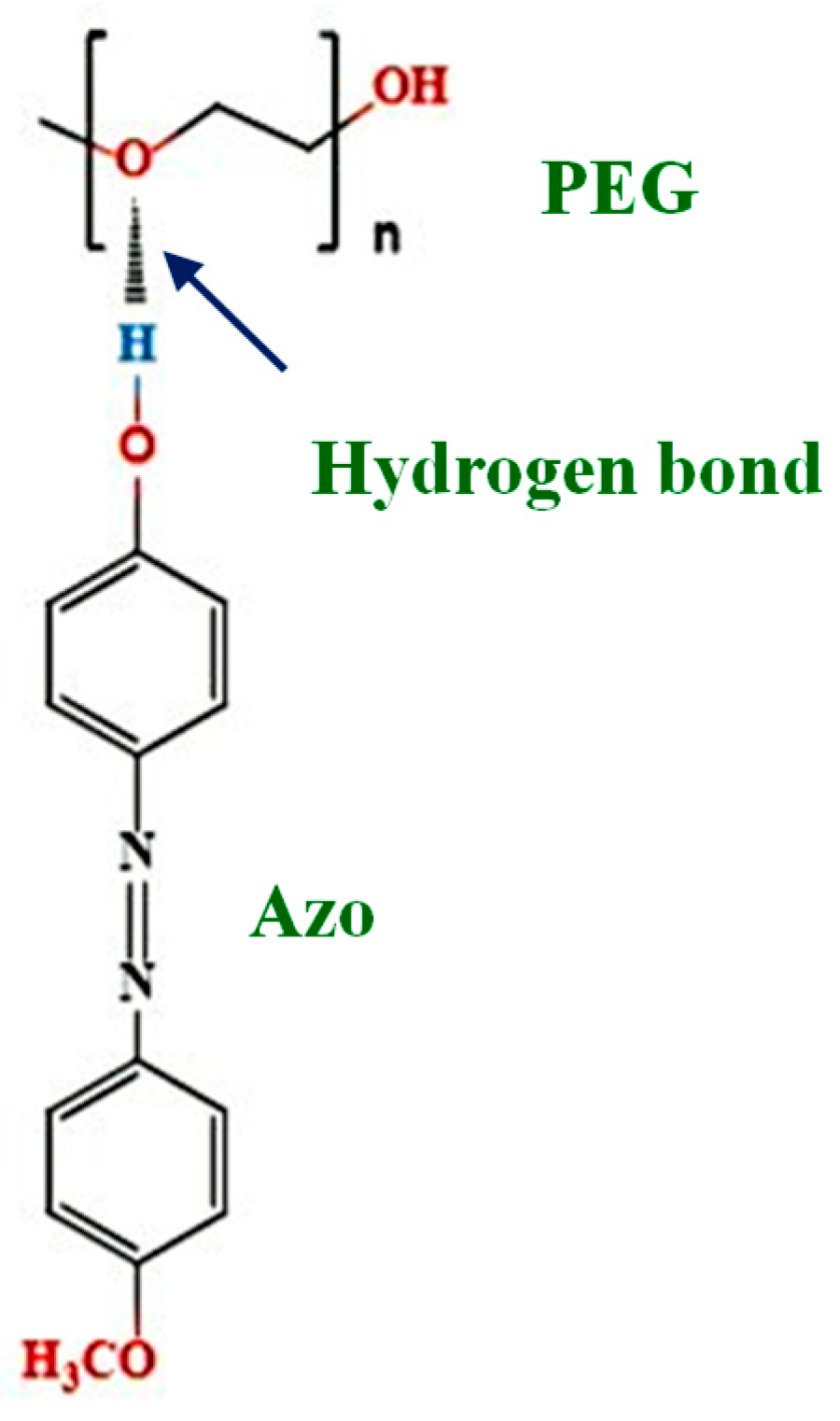
1. Introduction
2. Materials and Methods
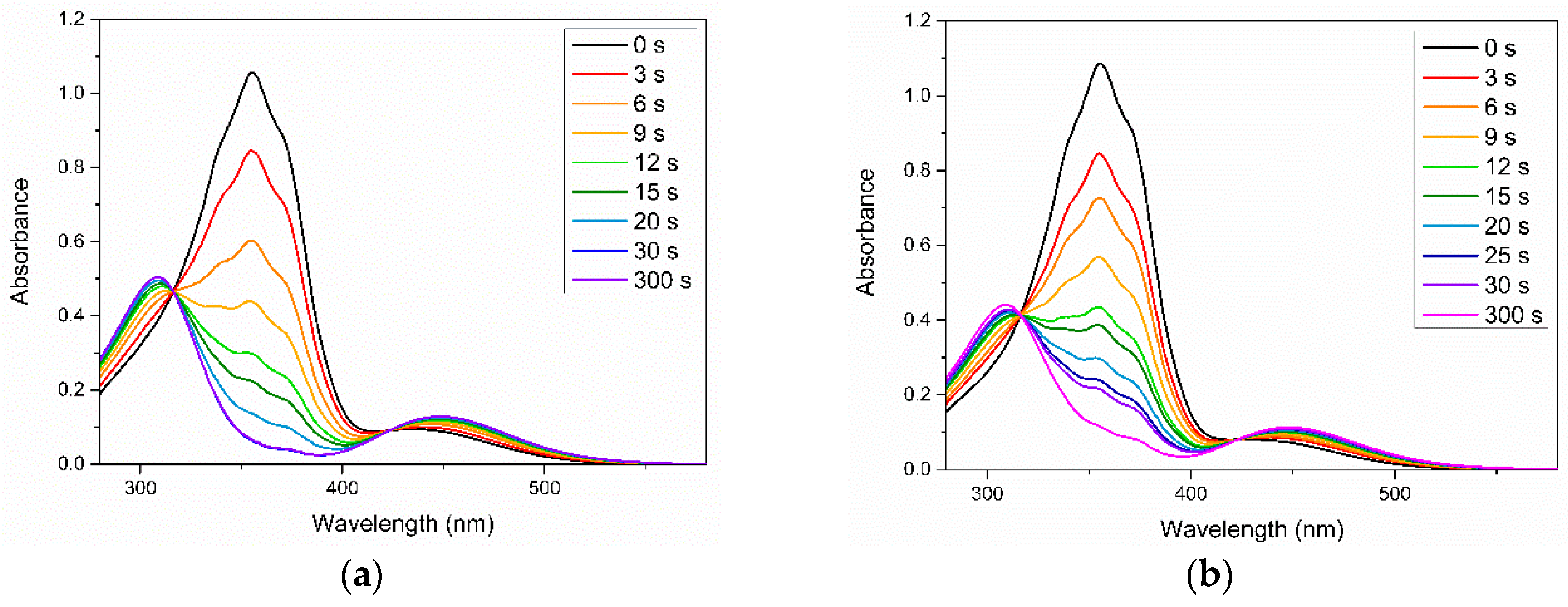
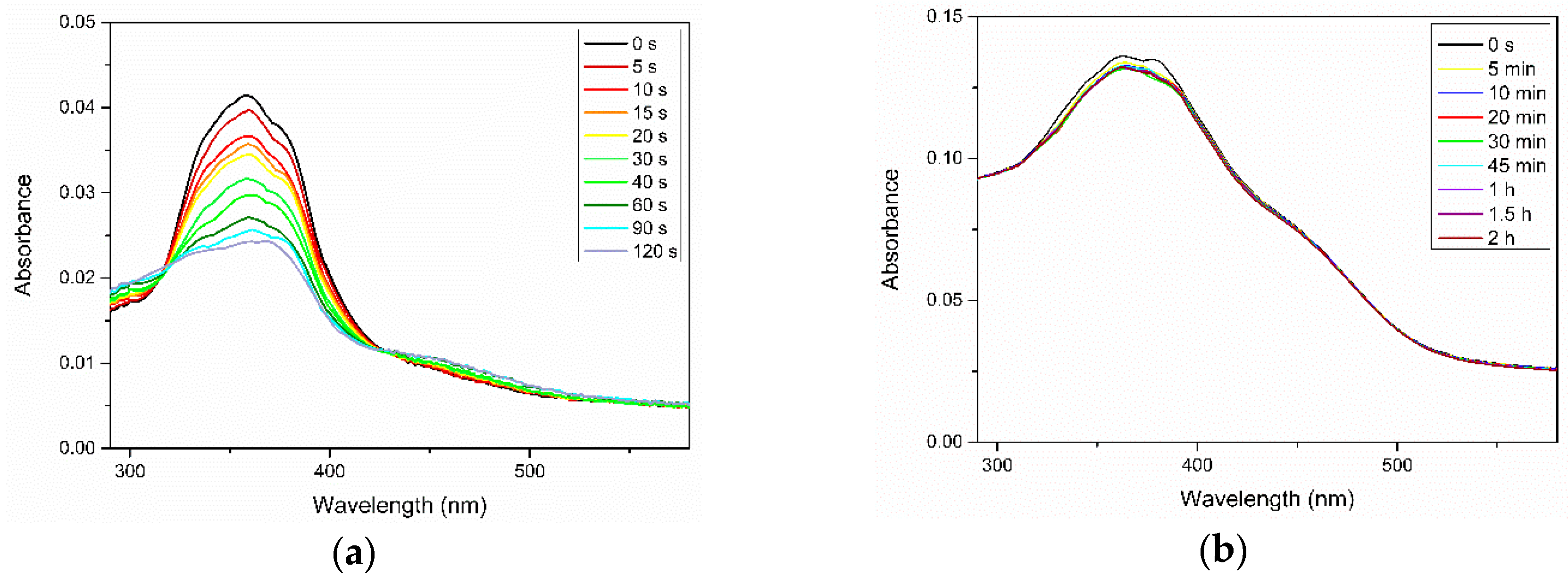
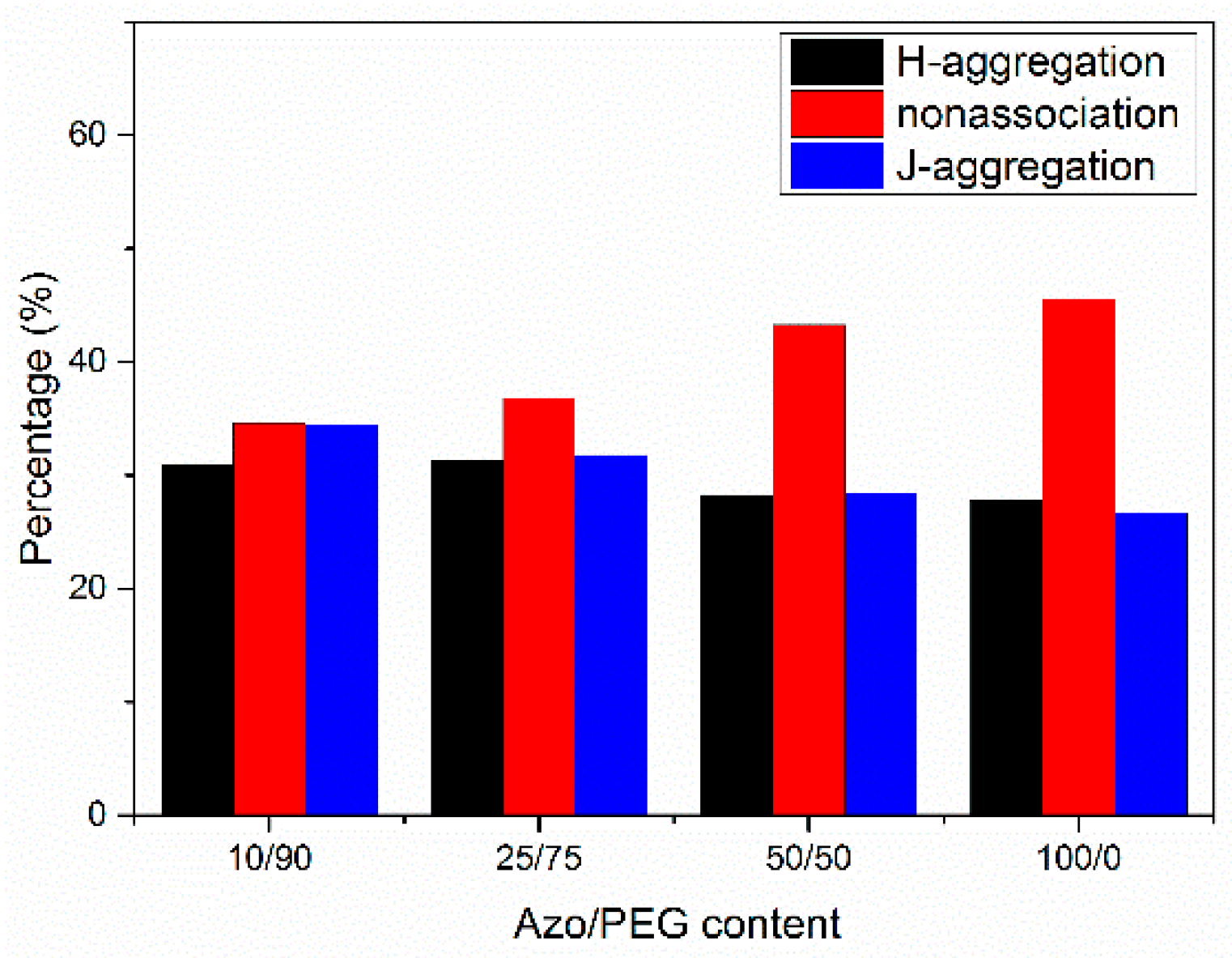
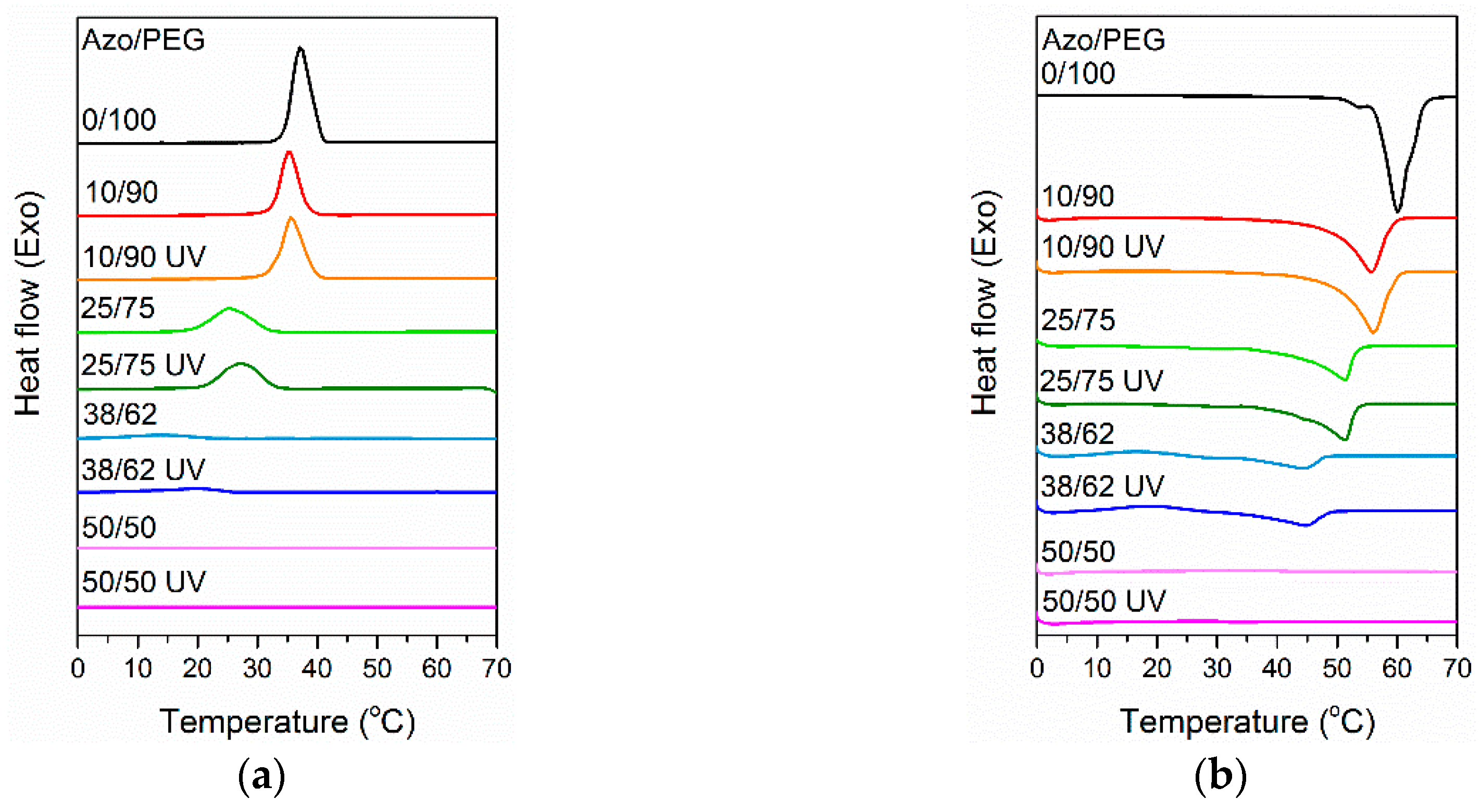
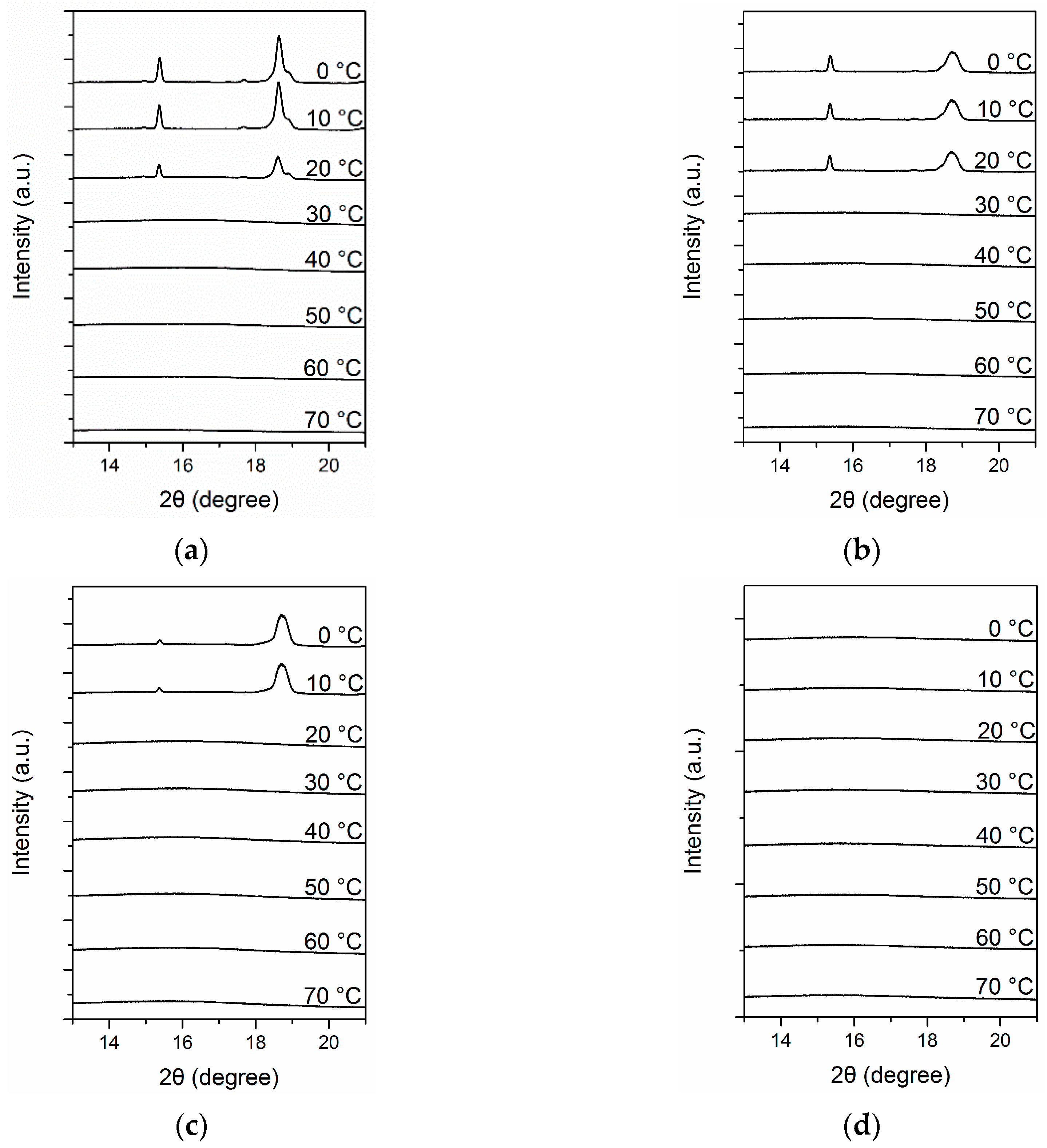
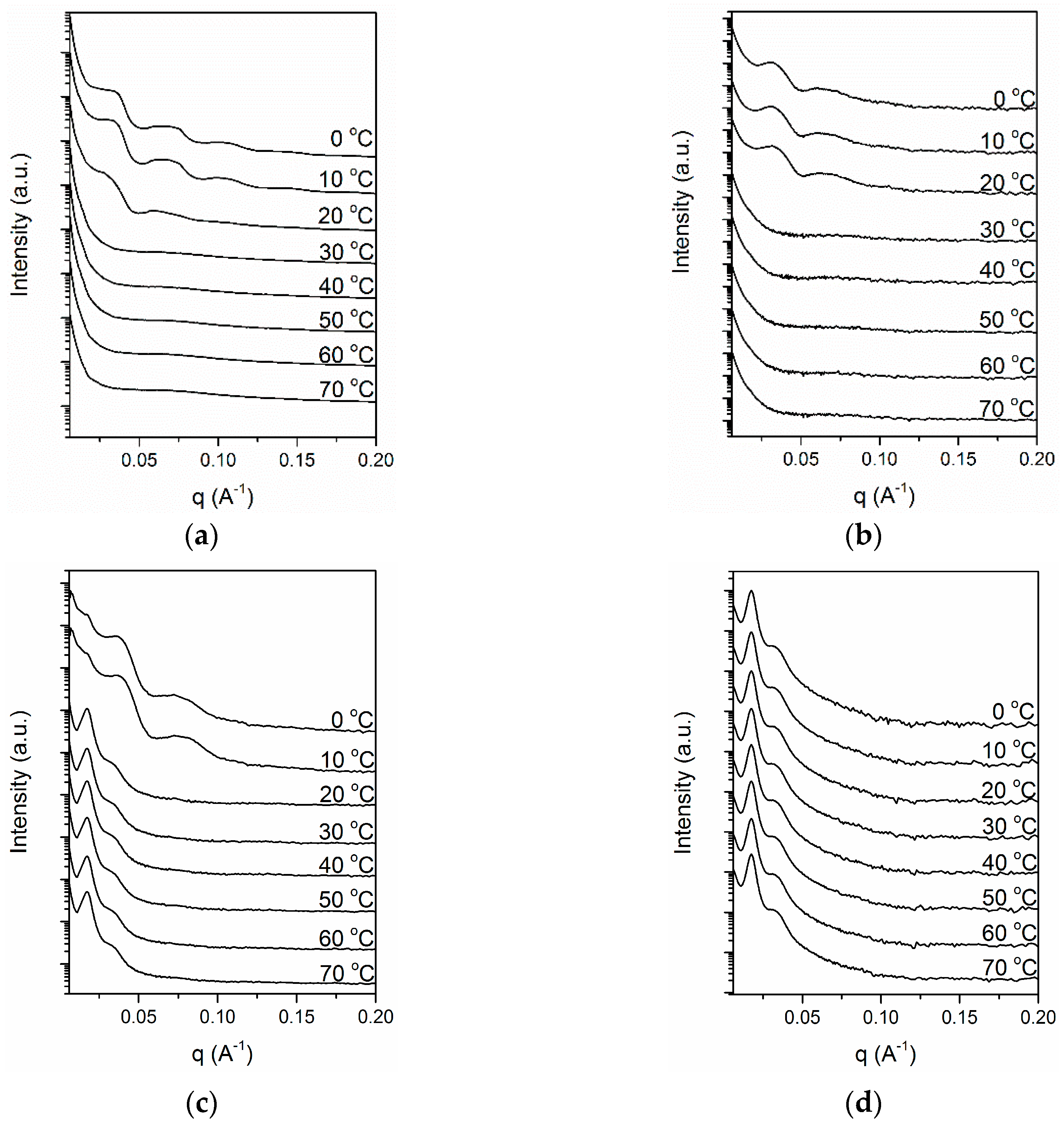
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yu, Z.P.; Liu, N.; Yang, L.; Jiang, Z.Q.; Wu, Z.Q. One-Pot Synthesis, Stimuli Responsiveness, and White-Light Emissions of Sequence-Defined ABC Triblock Copolymers Containing Polythiophene, Polyallene, and Poly(phenyl isocyanide) Blocks. Macromolecules 2017, 50, 3204–3214. [Google Scholar] [CrossRef]
- Zhou, L.; Jiang, Z.Q.; Xu, L.; Liu, N.; Wu, Z.Q. Polythiophene-block-poly(phenyl isocyanide) Copolymers: One-pot Synthesis, Properties and Applications. Chin. J. Polym. Sci. 2017, 35, 1447–1456. [Google Scholar] [CrossRef]
- Zhou, L.; Shen, L.; Huang, J.; Liu, N.; Zhu, Y.Y.; Wu, Z.Q. Optically Active Helical Polyisocyanides Bearing Chiral Phosphine Pendants: Facile Synthesis and Application in Enantioselective Rauhut-Currier Reaction. Chin. J. Polym. Sci. 2018, 36, 163–170. [Google Scholar] [CrossRef]
- Jiang, Z.Q.; Zhao, S.Q.; Su, Y.X.; Liu, N.; Wu, Z.Q. Combination of RAFT and Pd(II)-Initiated Isocyanide Polymerizations: A Versatile Method for Facile Synthesis of Helical Poly(phenyl isocyanide) Block and Star Copolymers. Macromolecules 2018, 51, 737–745. [Google Scholar] [CrossRef]
- Wang, Q.; Chu, B.F.; Chu, J.H.; Liu, N.; Wu, Z.Q. Facile Synthesis of Optically Active and Thermoresponsive Star Block Copolymers Carrying Helical Polyisocyanide Arms and Their Thermo-Triggered Chiral Resolution Ability. ACS Macro Lett. 2018, 7, 127–131. [Google Scholar] [CrossRef]
- Zhao, S.Q.; Hu, G.; Xu, X.H.; Kang, S.M.; Liu, N.; Wu, Z.Q. Synthesis of Redox-Responsive Core Cross-Linked Micelles Carrying Optically Active Helical Poly(phenyl isocyanide) Arms and Their Applications in Drug Delivery. ACS Macro Lett. 2018, 7, 1073–1079. [Google Scholar] [CrossRef]
- Natansohn, A.; Rochon, P. Photoinduced Motions in Azobenzene-Based Amorphous Polymers: Possible Photonic Devices. Adv. Mater. 1999, 11, 1387–1391. [Google Scholar] [CrossRef]
- Hagen, R.; Bieringer, T. Photoaddressable Polymers for Optical Data Storage. Adv. Mater. 2001, 13, 1805–1810. [Google Scholar] [CrossRef]
- Yu, H.; Iyoda, T.; Ikeda, T. Photoinduced Alignment of Nanocylinders by Supramolecular Cooperative Motions. J. Am. Chem. Soc. 2006, 128, 11010–11011. [Google Scholar] [CrossRef]
- Hackel, M.; Kador, L.; Kropp, D.; Schmidt, H.W. Polymer Blends with Azobenzene-Containing Block Copolymers as Stable Rewritable Volume Holographic Media. Adv. Mater. 2007, 19, 227–231. [Google Scholar] [CrossRef]
- Ikeda, T.; Mamiya, J.I.; Yu, Y. Photomechanics of Liquid-Crystalline Elastomers and Other Polymers. Angew. Chem. Int. Ed. 2007, 46, 506–528. [Google Scholar] [CrossRef] [PubMed]
- Mita, I.; Horie, K.; Hirao, K. Photochemistry in polymer solids. 9. Photoisomerization of azobenzene in a polycarbonate film. Macromolecules 1989, 22, 558–563. [Google Scholar] [CrossRef]
- Han, M.; Ichimura, K. In-Plane and Tilt Reorientation ofp-Methoxyazobenzene Side Chains Tethered to Liquid Crystalline Polymethacrylates by Irradiation with 365 nm Light. Macromolecules 2001, 34, 90–98. [Google Scholar] [CrossRef]
- Kawatsuki, N.; Goto, K.; Kawakami, T.; Yamamoto, T. Reversion of Alignment Direction in the Thermally Enhanced Photoorientation of Photo-Cross-Linkable Polymer Liquid Crystal Films. Macromolecules 2002, 35, 706–713. [Google Scholar] [CrossRef]
- Tong, X.; Cui, L.; Zhao, Y. Confinement Effects on Photoalignment, Photochemical Phase Transition, and Thermochromic Behavior of Liquid Crystalline Azobenzene-Containing Diblock Copolymers. Macromolecules 2004, 37, 3101–3112. [Google Scholar] [CrossRef]
- Zhao, Y.; Qi, B.; Tong, X.; Zhao, Y. Synthesis of Double Side-Chain Liquid Crystalline Block Copolymers Using RAFT Polymerization and the Orientational Cooperative Effect. Macromolecules 2008, 41, 3823–3831. [Google Scholar] [CrossRef]
- Menzel, H.; Weichart, B.; Schmidt, A.; Paul, S.; Knoll, W.; Stumpe, J.; Fischer, T. Small-Angle X-ray Scattering and Ultraviolet-Visible Spectroscopy Studies on the Structure and Structural Changes in Langmuir-Blodgett Films of Polyglutamates with Azobenzene Moieties Tethered by Alkyl Spacers of Different Length. Langmuir 1994, 10, 1926–1933. [Google Scholar] [CrossRef]
- Wang, G.; Tong, X.; Zhao, Y. Preparation of Azobenzene-Containing Amphiphilic Diblock Copolymers for Light-Responsive Micellar Aggregates. Macromolecules 2004, 37, 8911–8917. [Google Scholar] [CrossRef]
- Yu, H.; Shishido, A.; Ikeda, T.; Iyoda, T. Novel Amphiphilic Diblock and Triblock Liquid-Crystalline Copolymers with Well-Defined Structures Prepared by Atom Transfer Radical Polymerization. Macromol. Rapid Commun. 2005, 26, 1594–1598. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, W.; Zhou, N.; Zhu, J.; Cheng, Z.; Xu, Y.; Zhu, X. Synthesis of azobenzene-containing polymers via RAFT polymerization and investigation on intense fluorescence from aggregates of azobenzene-containing amphiphilic diblock copolymers. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 5652–5662. [Google Scholar] [CrossRef]
- Chen, D.; Liu, H.; Kobayashi, T.; Yu, H. Multiresponsive reversible gels based on a carboxylic azo polymer. J. Mater. Chem. 2010, 20, 3610–3614. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, N.; Zhang, W.; Zhang, F.; Zhu, J.; Zhang, Z.; Cheng, Z.; Tu, Y.; Zhu, X. Light-driven and aggregation-induced emission from side-chain azoindazole polymers. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 4911–4920. [Google Scholar] [CrossRef]
- Tsao, S.C.; Lo, C.T. Photoresponsive Behavior and Self-Organization of Azobenzene-Containing Block Copolymers. RSC Adv. 2014, 4, 23585–23594. [Google Scholar] [CrossRef]
- Fu, Z.; Ren, H.; Chen, D.; Shi, Y.; Yu, H. A carboxylic azo monomer and its homopolymer: Synthesis, self-organization and fluorescence behaviour in solution. Polym. Chem. 2015, 6, 270–277. [Google Scholar]
- Lee, T.L.; Lo, C.T. Photoresponsive and Fluorescence Behaviors of Azobenzene-Containing Amphiphilic Block Copolymers. J. Polym. Sci. Polym. Phys. Ed. 2017, 55, 793–803. [Google Scholar] [CrossRef]
- Huang, P.C.; Mata, J.P.; Wu, C.M.; Lo, C.T. Morphology-Mediated Photoresponsive and Fluorescence Behaviors of Azobenzene-Containing Block Copolymers. Langmuir 2018, 34, 7416–7427. [Google Scholar] [CrossRef]
- Banach, M.J.; Alexander, M.D.; Caracci, S.; Vaia, R.A. Enhancement of Electrooptic Coefficient of Doped Films through Optimization of Chromophore Environment. Chem. Mater. 1999, 11, 2554–2561. [Google Scholar] [CrossRef]
- Priimagi, A.; Kaivola, M.; Rodriguez, F.J.; Kauranen, M. Enhanced photoinduced birefringence in polymer-dye complexes: Hydrogen bonding makes a difference. Appl. Phys. Lett. 2007, 90, 121103. [Google Scholar] [CrossRef]
- Priimagi, A.; Vapaavuori, J.; Rodriguez, F.J.; Faul, C.F.J.; Heino, M.T.; Ikkala, O.; Kauranen, M.; Kaivola, M. Hydrogen-Bonded Polymer−Azobenzene Complexes: Enhanced Photoinduced Birefringence with High Temporal Stability through Interplay of Intermolecular Interactions. Chem. Mater. 2008, 20, 6358–6363. [Google Scholar] [CrossRef]
- Vapaavuori, J.; Priimagi, A.; Kaivola, M. Photoinduced surface-relief gratings in films of supramolecular polymer–bisazobenzene complexes. J. Mater. Chem. 2010, 20, 5260–5264. [Google Scholar] [CrossRef]
- Del Barrio, J.; Blasco, E.; Oriol, L.; Alcalá, R.; Sánchez-Somolinos, C. Diblock copolymer-azobenzene complexes through hydrogen bonding: Self-assembly and stable photoinduced optical anisotropy. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 1716–1725. [Google Scholar] [CrossRef]
- Vapaavuori, J.; Heikkinen, I.T.S.; Dichiarante, V.; Resnati, G.; Metrangolo, P.; Sabat, R.G.; Bazuin, C.G.; Priimagi, A.; Pellerin, C. Photomechanical Energy Transfer to Photopassive Polymers through Hydrogen and Halogen Bonds. Macromolecules 2015, 48, 7535–7542. [Google Scholar] [CrossRef]
- Poutanen, M.; Ikkala, O.; Priimägi, A. Structurally Controlled Dynamics in Azobenzene-Based Supramolecular Self-Assemblies in Solid State. Macromolecules 2016, 49, 4095–4101. [Google Scholar] [CrossRef]
- Tian, Y.; Watanabe, K.; Kong, X.; Abe, J.; Iyoda, T. Synthesis, Nanostructures, and Functionality of Amphiphilic Liquid Crystalline Block Copolymers with Azobenzene Moieties. Macromolecules 2002, 35, 3739–3747. [Google Scholar] [CrossRef]
- Lin, S.; Lin, J.; Nose, T.; Iyoda, T. Micellar structures of block-copolymers with ordered cores in dilute solution as studied by polarized and depolarized light scattering. J. Polym. Sci. Part B Polym. Phys. 2007, 45, 1333–1343. [Google Scholar] [CrossRef]
- Li, J.; Kamata, K.; Watanabe, S.; Iyoda, T. Template- and Vacuum-Ultraviolet- Assisted Fabrication of a Ag-Nanoparticle Array on Flexible and Rigid Substrates. Adv. Mater. 2007, 19, 1267–1271. [Google Scholar] [CrossRef]
- Li, J.; Kamata, K.; Komura, M.; Yamada, T.; Yoshida, H.; Iyoda, T. Anisotropic Ion Conductivity in Liquid Crystalline Diblock Copolymer Membranes with Perpendicularly Oriented PEO Cylindrical Domains. Macromolecules 2007, 40, 8125–8128. [Google Scholar] [CrossRef]
- Alam, M.Z.; Shibahara, A.; Ogata, T.; Kurihara, S. Synthesis of azobenzene-functionalized star polymers via RAFT and their photoresponsive properties. Polymer 2011, 52, 3696–3703. [Google Scholar] [CrossRef]
- Lim, J.C.; Park, J.K.; Song, H.Y. FTIR Investigation of ion-dipole interaction in styrene ionomer/poly(ethylene oxide) blends. J. Polym. Sci. Part B Polym. Phys. 1994, 32, 29–35. [Google Scholar] [CrossRef]
- Stockton, W.B.; Rubner, M.F. Molecular-Level Processing of Conjugated Polymers. 4. Layer-by-Layer Manipulation of Polyaniline via Hydrogen-Bonding Interactions. Macromolecules 1997, 30, 2717–2725. [Google Scholar] [CrossRef]
- Yang, X.; Painter, P.C.; Coleman, M.M.; Pearce, E.M.; Kwei, T.K. Equilibrium constants and predictions of miscibility windows and maps for polymer blends involving p-(hexafluoro-2-hydroxy-2-propyl)styrene with methacrylate and acetoxy groups. Macromolecules 1992, 25, 2156–2165. [Google Scholar] [CrossRef]
- Wang, S.; Ji, Q.; Tchatchoua, C.N.; Shultz, A.R.; McGrath, J.E. Phosphonyl/hydroxyl hydrogen bonding-induced miscibility of poly(arylene ether phosphine oxide/sulfone) statistical copolymers with poly(hydroxy ether) (phenoxy resin): Synthesis and characterization. J. Polym. Sci. Part B Polym. Phys. 1999, 37, 1849–1862. [Google Scholar] [CrossRef]
- Sim, L.; Gan, S.N.; Chan, C.H.; Yahya, R. ATR-FTIR studies on ion interaction of lithium perchlorate in polyacrylate/poly(ethylene oxide) blends. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2010, 76, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, L.; Hou, L.; Zhu, L.; Zhang, Y.; Zhang, B.; Zhang, H. Photoresponsive side-chain liquid crystalline polymers with amide group-substituted azobenzene mesogens: Effects of hydrogen bonding, flexible spacers, and terminal tails. Soft Matter 2012, 8, 5532. [Google Scholar] [CrossRef]
- Liu, Z.F.; Morigaki, K.; Enomoto, T.; Hashimoto, K.; Fujishima, A. Kinetic-Studies on the Thermal Cis Trans Isomerization of an Azo Compound in the Assembled Monolayer Film. J. Phys. Chem. 1992, 96, 1875–1880. [Google Scholar] [CrossRef]
- Frenz, C.; Fuchs, A.; Schmidt, H.W.; Theissen, U.; Haarer, D. Diblock Copolymers with Azobenzene Side-Groups and Polystyrene Matrix: Synthesis, Characterization and Photoaddressing. Macromol. Chem. Phys. 2004, 25, 1246–1258. [Google Scholar] [CrossRef]
- Coleman, M.; Moskala, E. FTi.r. studies of polymer blends containing the poly(hydroxy ether of bisphenol A) and poly(ε-caprolactone). Polymer 1983, 24, 251–257. [Google Scholar] [CrossRef]
- Zhu, L.; Cheng, S.Z.D.; Calhoun, B.H.; Ge, Q.; Quirk, R.P.; Thomas, E.L.; Hsiao, B.S.; Yeh, F.; Lotz, B. Crystallization Temperature-Dependent Crystal Orientations within Nanoscale Confined Lamellae of a Self-Assembled Crystalline−Amorphous Diblock Copolymer. J. Am. Chem. Soc. 2000, 122, 5957–5967. [Google Scholar] [CrossRef]
- Strobl, G.R.; Schneider, M. Direct evaluation of the electron density correlation function of partially crystalline polymers. J. Polym. Sci. Polym. Phys. Ed. 1980, 18, 1343–1359. [Google Scholar] [CrossRef]
- Cottrell, T.L. The Strengths of Chemical Bonds; Butterworths Scientific Publications: London, UK, 1958. [Google Scholar]
- Hofman, A.H.; Chen, Y.; Brinke, G.T.; Loos, K. Interaction Strength in Poly(4-vinylpyridine)–n-Alkylphenol Supramolecular Comb-Shaped Copolymers. Macromolecules 2015, 48, 1554–1562. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Azo/PEG (wt %/wt %) | Rate Constant (s−1) | |||
|---|---|---|---|---|
| Benzene | Toluene | THF | CH2Cl2 | |
| 100/0 | 0.140 | 0.139 | 0.133 | 0.118 |
| 50/50 | 0.083 | 0.099 | 0.132 | 0.089 |
| Azo/PEG | Tc (°C) | Tm (°C) | Degree of Crystallinity (%) |
|---|---|---|---|
| 0/100 | 37.3 | 59.6 | 85.4 |
| 10/90 | 35.8 | 55.6 | 70.1 |
| 10/90 with UV exposure | 36.2 | 56.0 | 74.7 |
| 25/75 | 26.0 | 51.5 | 63.4 |
| 25/75 with UV exposure | 27.1 | 51.6 | 66.4 |
| 38/62 | 13.9 | 44.2 | 30.2 |
| 38/62 with UV exposure | 19.9 | 44.6 | 31.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tai, H.-T.; Lin, Y.-C.; Ma, J.-Y.; Lo, C.-T. Hydrogen Bonding-Induced Assembled Structures and Photoresponsive Behavior of Azobenzene Molecule/Polyethylene Glycol Complexes. Polymers 2019, 11, 1360. https://doi.org/10.3390/polym11081360
Tai H-T, Lin Y-C, Ma J-Y, Lo C-T. Hydrogen Bonding-Induced Assembled Structures and Photoresponsive Behavior of Azobenzene Molecule/Polyethylene Glycol Complexes. Polymers. 2019; 11(8):1360. https://doi.org/10.3390/polym11081360
Chicago/Turabian StyleTai, Hsin-Tzu, Yen-Chun Lin, Jing-Yao Ma, and Chieh-Tsung Lo. 2019. "Hydrogen Bonding-Induced Assembled Structures and Photoresponsive Behavior of Azobenzene Molecule/Polyethylene Glycol Complexes" Polymers 11, no. 8: 1360. https://doi.org/10.3390/polym11081360
APA StyleTai, H.-T., Lin, Y.-C., Ma, J.-Y., & Lo, C.-T. (2019). Hydrogen Bonding-Induced Assembled Structures and Photoresponsive Behavior of Azobenzene Molecule/Polyethylene Glycol Complexes. Polymers, 11(8), 1360. https://doi.org/10.3390/polym11081360