Thermal Cross Linking of Novel Azide Modified Polymers of Intrinsic Microporosity—Effect of Distribution and the Gas Separation Performance



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Monomer DMTTSBI and Trimer
2.2.1. Synthesis of DMTTSBI
2.2.2. Synthesis of Trimer
2.3. Synthesis of Homopolymer and Copolymers
2.3.1. Synthesis of Homopolymer and Randomly Distributed Copolymer
2.3.2. Synthesis of Alternating Copolymer
2.4. Modification of the Polymers
2.4.1. Synthesis of Brominated Homopolymer and Copolymers
2.4.2. Substitution of Bromine by Azide Group
2.5. Methods
3. Results and Discussion
3.1. Homopolymer and Copolymers
3.2. Brominated Homopolymer (PIM-Br-DMTTSBI-100) and Copolymers (PIM-Br-DMTTSBI-R50 and PIM-Br-DMTTSBI-A50)
3.3. Benzyl Azide Homopolymer (AZ-PIM-100) and Copolymers (AZ-PIM-R50 and AZ-PIM-A50)
3.4. Gas Separation Performance
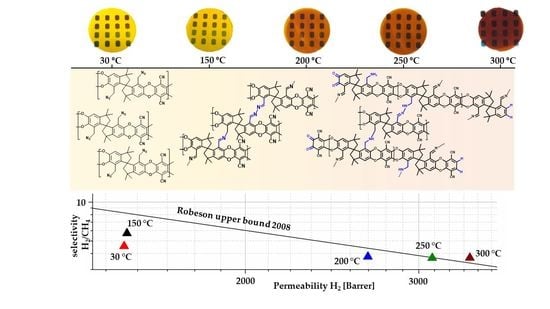
3.5. Aging of Cross Linked Azide PIMs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Methods
References
- Jiang, J.X.; Cooper, A.I. Microporous organic polymers: Design, synthesis, and function. In Functional Metal-Organic Frameworks: Gas Storage, Separation and Catalysis; Schröder, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 1–33. [Google Scholar]
- Chang, Z.; Zhang, D.-S.; Chen, Q.; Bu, X.-H. Microporous organic polymers for gas storage and separation applications. Phys. Chem. Chem. Phys. 2013, 15, 5430–5442. [Google Scholar] [CrossRef] [PubMed]
- Suh, M.P.; Park, H.J.; Prasad, T.K.; Lim, D.-W. Hydrogen storage in metal–organic frameworks. Chem. Rev. 2012, 112, 782–835. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Cao, D. Porous covalent–organic materials: Synthesis, clean energy application and design. J. Mater. Chem. A 2013, 1, 2691–2718. [Google Scholar] [CrossRef]
- Bernardo, P.; Drioli, E.; Golemme, G. Membrane gas separation: A review/state of the art. Ind. Eng. Chem. Res. 2009, 48, 4638–4663. [Google Scholar] [CrossRef]
- Yampolskii, Y. Polymeric gas separation membranes. Macromolecules 2012, 45, 3298–3311. [Google Scholar] [CrossRef]
- Robeson, L.M. The upper bound revisited. J. Membr. Sci. 2008, 320, 390–400. [Google Scholar] [CrossRef]
- McKeown, N.B.; Budd, P.M. Polymers of intrinsic microporosity (PIMs): Organic materials for membrane separations, heterogeneous catalysis and hydrogen storage. Chem. Soc. Rev. 2006, 35, 675–683. [Google Scholar] [CrossRef]
- Park, H.B.; Jung, C.H.; Lee, Y.M.; Hill, A.J.; Pas, S.J.; Mudie, S.T.; Van Wagner, E.; Freeman, B.D.; Cookson, D.J. Polymers with cavities tuned for fast selective transport of small molecules and ions. Science 2007, 318, 254–258. [Google Scholar] [CrossRef]
- Calle, M.; Chan, Y.; Jo, H.J.; Lee, Y.M. The relationship between the chemical structure and thermal conversion temperatures of thermally rearranged (tr) polymers. Polymer 2012, 53, 2783–2791. [Google Scholar] [CrossRef]
- Staiger, C.L.; Pas, S.J.; Hill, A.J.; Cornelius, C.J. Gas separation, free volume distribution, and physical aging of a highly microporous spirobisindane polymer. Chem. Mater. 2008, 20, 2606–2608. [Google Scholar] [CrossRef]
- Bernardo, P.; Bazzarelli, F.; Tasselli, F.; Clarizia, G.; Mason, C.R.; Maynard-Atem, L.; Budd, P.M.; Lanč, M.; Pilnáček, K.; Vopička, O. Effect of physical aging on the gas transport and sorption in PIM-1 membranes. Polymer 2017, 113, 283–294. [Google Scholar] [CrossRef]
- Du, N.; Park, H.B.; Dal-Cin, M.M.; Guiver, M.D. Advances in high permeability polymeric membrane materials for CO2 separations. Energy Environ. Sci. 2016, 9, 1863–1890. [Google Scholar]
- Du, N.; Robertson, G.P.; Song, J.; Pinnau, I.; Thomas, S.; Guiver, M.D. Polymers of intrinsic microporosity containing trifluoromethyl and phenylsulfone groups as materials for membrane gas separation. Macromolecules 2008, 41, 9656–9662. [Google Scholar] [CrossRef]
- Ghanem, B.S.; McKeown, N.B.; Budd, P.M.; Al-Harbi, N.M.; Fritsch, D.; Heinrich, K.; Starannikova, L.; Tokarev, A.; Yampolskii, Y. Synthesis, characterization, and gas permeation properties of a novel group of polymers with intrinsic microporosity: PIM-polyimides. Macromolecules 2009, 42, 7881–7888. [Google Scholar] [CrossRef]
- Ghanem, B.S.; Hashem, M.; Harris, K.D.M.; Msayib, K.J.; Xu, M.; Budd, P.M.; Chaukura, N.; Book, D.; Tedds, S.; Walton, A.; et al. Triptycene-based polymers of intrinsic microporosity: Organic materials that can be tailored for gas adsorption. Macromolecules 2010, 43, 5287–5294. [Google Scholar] [CrossRef]
- Patel, H.A.; Yavuz, C.T. Noninvasive functionalization of polymers of intrinsic microporosity for enhanced CO2 capture. Chem. Commun. 2012, 48, 9989–9991. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, B.S.; McKeown, N.B.; Budd, P.M.; Fritsch, D. Polymers of intrinsic microporosity derived from bis(phenazyl) monomers. Macromolecules 2008, 41, 1640–1646. [Google Scholar] [CrossRef]
- Carta, M.; Msayib, K.J.; Budd, P.M.; McKeown, N.B. Novel spirobisindanes for use as precursors to polymers of intrinsic microporosity. Org. Lett. 2008, 10, 2641–2643. [Google Scholar] [CrossRef] [PubMed]
- Du, N.; Robertson, G.P.; Dal-Cin, M.M.; Scoles, L.; Guiver, M.D. Polymers of intrinsic microporosity (PIMs) substituted with methyl tetrazole. Polymer 2012, 53, 4367–4372. [Google Scholar] [CrossRef]
- Mason, C.R.; Maynard-Atem, L.; Al-Harbi, N.M.; Budd, P.M.; Bernardo, P.; Bazzarelli, F.; Clarizia, G.; Jansen, J.C. Polymer of intrinsic microporosity incorporating thioamide functionality: Preparation and gas transport properties. Macromolecules 2011, 44, 6471–6479. [Google Scholar] [CrossRef]
- Khan, M.M.; Bengtson, G.; Neumann, S.; Rahman, M.M.; Abetz, V.; Filiz, V. Synthesis, characterization and gas permeation properties of anthracene maleimide-based polymers of intrinsic microporosity. RSC Adv. 2014, 4, 32148–32160. [Google Scholar] [CrossRef]
- Fuoco, A.; Comesaña-Gándara, B.; Longo, M.; Esposito, E.; Monteleone, M.; Rose, I.; Bezzu, C.G.; Carta, M.; McKeown, N.B.; Jansen, J.C. Temperature dependence of gas permeation and diffusion in triptycene-based ultrapermeable polymers of intrinsic microporosity. ACS Appl. Mater. Interfaces 2018, 10, 36475–36482. [Google Scholar] [CrossRef] [PubMed]
- Halder, K.; Neumann, S.; Bengtson, G.; Khan, M.M.; Filiz, V.; Abetz, V. Polymers of intrinsic microporosity postmodified by vinyl groups for membrane applications. Macromolecules 2018, 51, 7309–7319. [Google Scholar] [CrossRef]
- Li, F.Y.; Xiao, Y.; Ong, Y.K.; Chung, T.-S. UV-rearranged PIM-1 polymeric membranes for advanced hydrogen purification and production. Adv. Energy Mater. 2012, 2, 1456–1466. [Google Scholar] [CrossRef]
- Li, F.Y.; Xiao, Y.; Chung, T.-S.; Kawi, S. High-performance thermally self-cross-linked polymer of intrinsic microporosity (PIM-1) membranes for energy development. Macromolecules 2012, 45, 1427–1437. [Google Scholar] [CrossRef]
- Song, Q.; Cao, S.; Zavala-Rivera, P.; Ping Lu, L.; Li, W.; Ji, Y.; Al-Muhtaseb, S.A.; Cheetham, A.K.; Sivaniah, E. Photo-oxidative enhancement of polymeric molecular sieve membranes. Nat. Commun. 2013, 4, 1918. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Bengtson, G.; Shishatskiy, S.; Gacal, B.N.; Rahman, M.M.; Neumann, S.; Filiz, V.; Abetz, V. Cross-linking of polymer of intrinsic microporosity (PIM-1) via nitrene reaction and its effect on gas transport property. Eur. Polym. J. 2013, 49, 4157–4166. [Google Scholar] [CrossRef]
- Du, N.; Cin, M.M.D.; Pinnau, I.; Nicalek, A.; Robertson, G.P.; Guiver, M.D. Azide-based cross-linking of polymers of intrinsic microporosity (PIMs) for condensable gas separation. Macromol. Rapid Commun. 2011, 32, 631–636. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.O.; Akhtar, R.; Lau, C.H.; Ratvijitvech, T.; Cheng, G.; Clowes, R.; Adams, D.J.; Hasell, T.; Cooper, A.I. Using intermolecular interactions to crosslink PIM-1 and modify its gas sorption properties. J. Mater. Chem. A 2015, 3, 4855–4864. [Google Scholar] [CrossRef]
- Song, Q.; Cao, S.; Pritchard, R.H.; Ghalei, B.; Al-Muhtaseb, S.A.; Terentjev, E.M.; Cheetham, A.K.; Sivaniah, E. Controlled thermal oxidative crosslinking of polymers of intrinsic microporosity towards tunable molecular sieve membranes. Nat. Commun. 2014, 5, 4813. [Google Scholar] [CrossRef]
- Du, N.; Dal-Cin, M.M.; Robertson, G.P.; Guiver, M.D. Decarboxylation-induced cross-linking of polymers of intrinsic microporosity (PIMs) for membrane gas separation. Macromolecules 2012, 45, 5134–5139. [Google Scholar] [CrossRef]
- McKeown, N.B.; Budd, P.M.; Msayib, K.J.; Ghanem, B.S.; Kingston, H.J.; Tattershall, C.E.; Makhseed, S.; Reynolds, K.J.; Fritsch, D. Polymers of intrinsic microporosity (PIMs): Bridging the void between microporous and polymeric materials. Chem. A Eur. J. 2005, 11, 2610–2620. [Google Scholar] [CrossRef] [PubMed]
- Budd, P.M.; Msayib, K.J.; Tattershall, C.E.; Ghanem, B.S.; Reynolds, K.J.; McKeown, N.B.; Fritsch, D. Gas separation membranes from polymers of intrinsic microporosity. J. Membr. Sci. 2005, 251, 263–269. [Google Scholar] [CrossRef]
- Fritsch, D.; Bengtson, G.; Carta, B.G.; McKeown, N.B. Synthesis and gas permeation properties of spirochroman-based polymers of intrinsic microporosity. Macromol. Chem. Phys. 2011, 212, 1137–1146. [Google Scholar] [CrossRef]
- Fritsch, D.; Heinrich, K.; Bengtson, G. Polymers of intrinsic microporosity: Copolymers, improved synthesis and applications as membrane separation material. PMSE Repr. 2009, 761–762. [Google Scholar]
- Zhang, J.; Jin, J.; Cooney, R.; Fu, Q.; Qiao, G.G.; Thomas, S.; Merkel, T.C. Synthesis of perfectly alternating copolymers for polymers of intrinsic microporosity. Polym. Chem. 2015, 6, 5003–5008. [Google Scholar] [CrossRef]
- Mei, W.; Lü, C.; Yan, J.; Wang, Z. Anion exchange membranes by bromination of benzylmethyl-containing poly(fluorene ether sulfone)s. RSC Adv. 2014, 4, 27502–27509. [Google Scholar] [CrossRef]
- Weiber, E.A.; Jannasch, P. Polysulfones with highly localized imidazolium groups for anion exchange membranes. J. Membr. Sci. 2015, 481, 164–171. [Google Scholar] [CrossRef]
- Bengtson, G.; Neumann, S.; Filiz, V. Membranes of polymers of intrinsic microporosity (PIM-1) modified by poly(ethylene glycol). Membranes 2017, 7, 28. [Google Scholar] [CrossRef]
- Eroǧlu, M.S.; Güven, O. Thermal decomposition of poly(glycidyl azide) as studied by high-temperature ftir and thermogravimetry. J. Appl. Polym. Sci. 1996, 61, 201–206. [Google Scholar] [CrossRef]
- Wang, T.; Li, S.; Yang, B.; Huang, C.; Li, Y. Thermal decomposition of glycidyl azide polymer studied by synchrotron photoionization mass spectrometry. J. Phys. Chem. B 2007, 111, 2449–2455. [Google Scholar] [CrossRef] [PubMed]
- Pisharath, S.; Ang, H.G. Synthesis and thermal decomposition of GAP–poly(bamo) copolymer. Polym. Degrad. Stab. 2007, 92, 1365–1377. [Google Scholar] [CrossRef]
- Arisawa, H.; Brill, T.B. Thermal decomposition of energetic materials 71: Structure-decomposition and kinetic relationships in flash pyrolysis of glycidyl azide polymer (GAP). Combust. Flame 1998, 112, 533–544. [Google Scholar] [CrossRef]
- Schuh, K.; Prucker, O.; Rühe, J. Surface attached polymer networks through thermally induced cross-linking of sulfonyl azide group containing polymers. Macromolecules 2008, 41, 9284–9289. [Google Scholar] [CrossRef]
- Pinto, R.M.; Guerra, M.; Copeland, G.; Olariu, R.I.; Rodrigues, P.; Barros, M.T.; Costa, M.L.; Dias, A.A. The mechanism of pyrolysis of benzyl azide: Spectroscopic evidence for benzenemethanimine formation. J. Phys. Chem. A 2015, 119, 4118–4126. [Google Scholar] [CrossRef]
- Mason, C.R.; Maynard-Atem, L.; Heard, K.W.J.; Satilmis, B.; Budd, P.M.; Friess, K.; Lanč, M.; Bernardo, P.; Clarizia, G.; Jansen, J.C. Enhancement of CO2 affinity in a polymer of intrinsic microporosity by amine modification. Macromolecules 2014, 47, 1021–1029. [Google Scholar] [CrossRef]
- Jeon, J.W.; Kim, D.-G.; Sohn, E.-H.; Yoo, Y.; Kim, Y.S.; Kim, B.G.; Lee, J.-C. Highly carboxylate-functionalized polymers of intrinsic microporosity for CO2-selective polymer membranes. Macromolecules 2017, 50, 8019–8027. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Burnham, A.K.; Criado, J.M.; Pérez-Maqueda, L.A.; Popescu, C.; Sbirrazzuoli, N. Ictac kinetics committee recommendations for performing kinetic computations on thermal analysis data. Thermochim. Acta 2011, 520, 1–19. [Google Scholar] [CrossRef]
- Morancho, J.M.; Salla, J.M.; Cadenato, A.; Fernández-Francos, X.; Ramis, X.; Colomer, P.; Calventus, Y.; Ruíz, R. Kinetic studies of the degradation of poly(vinyl alcohol)-based proton-conducting membranes at low temperatures. Thermochim. Acta 2011, 521, 139–147. [Google Scholar] [CrossRef]
- Nguyen, X.Q.; Brož, Z.; Uchytil, P.; Nguyen, Q.T. Methods for the determination of transport parameters of gases in membranes. J. Chem. Soc. Faraday Trans. 1992, 88, 3553–3560. [Google Scholar] [CrossRef]
- Rahman, M.M.; Filiz, V.; Shishatskiy, S.; Abetz, C.; Neumann, S.; Bolmer, S.; Khan, M.M.; Abetz, V. Pebax® with PEG functionalized POSS as nanocomposite membranes for CO2 separation. J. Membr. Sci. 2013, 437, 286–297. [Google Scholar] [CrossRef]
- Wijmans, J.G.; Baker, R.W. The solution-diffusion model: A review. J. Membr. Sci. 1995, 107, 1–21. [Google Scholar] [CrossRef]
- Shishatskiy, S.; Yampol’skii, Y.P.; Peinemann, K.V. Effects of film thickness on density and gas permeation parameters of glassy polymers. J. Membr. Sci. 1996, 112, 275–285. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | DSC | TGA | ||||||
|---|---|---|---|---|---|---|---|---|
| TOnset [°C] a | TPeak [°C] a | TOnset/TInflection [°C] b | Weight Loss [%] b | Weight Loss [%] c | Weight Loss [%] d | Residual Mass [%] | ||
| AZ-PIM-100 | Argon | 225 | 257 | 223/243 | 9.4 | 29.3 | 60.5 | |
| Air | 225/245 | 9.2 | 24.5 | 65.2 | 1.0 | |||
| AZ-PIM-R50 | Argon | 224 | 258 | 224/246 | 5.1 | 32.6 | 61.5 | |
| Air | 224/245 | 4.9 | 23.9 | 69.6 | 1.5 | |||
| AZ-PIM-A50 | Argon | 224 | 258 | 224/245 | 5.0 | 39.4 | 53.8 | |
| Air | 226/246 | 4.7 | 27.8 | 65.1 | 1.7 | |||
| Polymer | T [°C] | |||||
|---|---|---|---|---|---|---|
| 30 | 150 | 200 | 250 | 300 | ||
| AZ-PIM-100 | P(CH4) | 245 | 116 | 308 | 298 | 351 |
| P(CO2) | 2894 | 1483 | 4239 | 4551 | 4977 | |
| D(CH4) | 0.20 | 0.10 | 0.19 | 0.17 | 0.20 | |
| D(CO2) | 0.50 | 0.27 | 0.60 | 0.59 | 0.64 | |
| S(CH4) | 0.13 | 0.12 | 0.16 | 0.18 | 0.18 | |
| S(CO2) | 0.58 | 0.56 | 0.71 | 0.78 | 0.78 | |
| AZ-PIM-R50 | P(CH4) | 384 | 126 | 368 | 476 | 479 |
| P(CO2) | 4246 | 1846 | 4709 | 5860 | 6042 | |
| D(CH4) | 0.23 | 0.11 | 0.22 | 0.25 | 0.26 | |
| D(CO2) | 0.60 | 0.31 | 0.62 | 0.71 | 0.76 | |
| S(CH4) | 0.17 | 0.11 | 0.17 | 0.20 | 0.19 | |
| S(CO2) | 0.71 | 0.61 | 0.76 | 0.83 | 0.80 | |
| AZ-PIM-A50 | P(CH4) | 331 | 264 | 704 | 839 | 911 |
| P(CO2) | 3879 | 3422 | 7206 | 8712 | 8868 | |
| D(CH4) | 0.23 | 0.18 | 0.44 | 0.50 | 0.49 | |
| D(CO2) | 0.60 | 0.53 | 1.07 | 1.24 | 1.23 | |
| S(CH4) | 0.15 | 0.15 | 0.16 | 0.17 | 0.19 | |
| S(CO2) | 0.64 | 0.65 | 0.67 | 0.70 | 0.72 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neumann, S.; Bengtson, G.; Meis, D.; Filiz, V. Thermal Cross Linking of Novel Azide Modified Polymers of Intrinsic Microporosity—Effect of Distribution and the Gas Separation Performance. Polymers 2019, 11, 1241. https://doi.org/10.3390/polym11081241
Neumann S, Bengtson G, Meis D, Filiz V. Thermal Cross Linking of Novel Azide Modified Polymers of Intrinsic Microporosity—Effect of Distribution and the Gas Separation Performance. Polymers. 2019; 11(8):1241. https://doi.org/10.3390/polym11081241
Chicago/Turabian StyleNeumann, Silvio, Gisela Bengtson, David Meis, and Volkan Filiz. 2019. "Thermal Cross Linking of Novel Azide Modified Polymers of Intrinsic Microporosity—Effect of Distribution and the Gas Separation Performance" Polymers 11, no. 8: 1241. https://doi.org/10.3390/polym11081241
APA StyleNeumann, S., Bengtson, G., Meis, D., & Filiz, V. (2019). Thermal Cross Linking of Novel Azide Modified Polymers of Intrinsic Microporosity—Effect of Distribution and the Gas Separation Performance. Polymers, 11(8), 1241. https://doi.org/10.3390/polym11081241