ATRP of Methyl Acrylate by Continuous Feeding of Activators Giving Polymers with Predictable End-Group Fidelity


Abstract

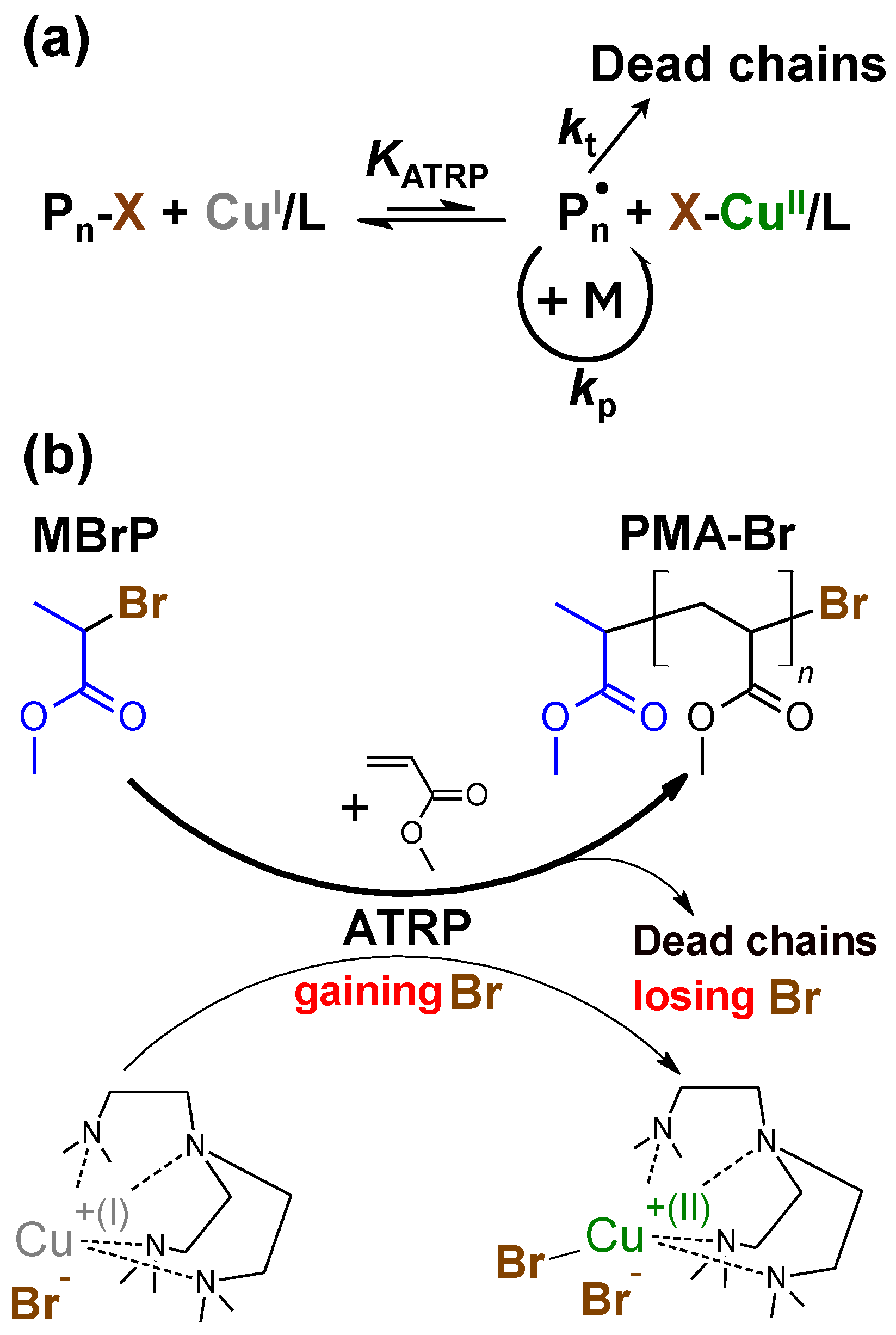
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
2.3. Preparation of CuBr Stock Solution
2.4. General Procedure for Polymerization
2.5. Chain Extension Experiment
3. Results and Discussion
4. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Matyjaszewski, K.; Xia, J. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef] [PubMed]
- Tsarevsky, N.V.; Matyjaszewski, K. “Green” Atom Transfer Radical Polymerization: From Process Design to Preparation of Well-Defined Environmentally Friendly Polymeric Materials. Chem. Rev. 2007, 107, 2270–2299. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Fantin, M.; Yuan, F.; Matyjaszewski, K. Externally controlled atom transfer radical polymerization. Chem. Soc. Rev. 2018, 47, 5457–5490. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Fantin, M.; Pan, X.; de Fiebre, K.; Coote, M.L.; Matyjaszewski, K.; Liu, P. Mechanistically Guided Predictive Models for Ligand and Initiator Effects in Copper-Catalyzed Atom Transfer Radical Polymerization (Cu-ATRP). J. Am. Chem. Soc. 2019, 141, 7486–7497. [Google Scholar] [CrossRef] [PubMed]
- Coessens, V.; Pintauer, T.; Matyjaszewski, K. Functional polymers by atom transfer radical polymerization. Prog. Polym. Sci. 2001, 26, 337–377. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Tsarevsky, N.V. Macromolecular Engineering by Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2014, 136, 6513–6533. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Advanced Materials by Atom Transfer Radical Polymerization. Adv. Mater. 2018, 30, 1706441. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Zhong, M.; Matyjaszewski, K. How Fast Can a CRP Be Conducted with Preserved Chain End Functionality? Macromolecules 2011, 44, 2668–2677. [Google Scholar] [CrossRef]
- Lunn, D.J.; Discekici, E.H.; Read de Alaniz, J.; Gutekunst, W.R.; Hawker, C.J. Established and emerging strategies for polymer chain-end modification. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 2903–2914. [Google Scholar] [CrossRef]
- Anastasaki, A.; Willenbacher, J.; Fleischmann, C.; Gutekunst, W.R.; Hawker, C.J. End group modification of poly(acrylates) obtained via ATRP: A user guide. Polym. Chem. 2017, 8, 689–697. [Google Scholar] [CrossRef]
- Gutekunst, W.R.; Anastasaki, A.; Lunn, D.J.; Truong, N.P.; Whitfield, R.; Jones, G.R.; Treat, N.J.; Abdilla, A.; Barton, B.E.; Clark, P.G.; et al. Practical Chain-End Reduction of Polymers Obtained with ATRP. Macromol. Chem. Phys. 2017, 218, 1700107. [Google Scholar] [CrossRef]
- Whitfield, R.; Anastasaki, A.; Truong, N.P.; Wilson, P.; Kempe, K.; Burns, J.A.; Davis, T.P.; Haddleton, D.M. Well-Defined PDMAEA Stars via Cu(0)-Mediated Reversible Deactivation Radical Polymerization. Macromolecules 2016, 49, 8914–8924. [Google Scholar] [CrossRef]
- Barbon, S.M.; Rolland, M.; Anastasaki, A.; Truong, N.P.; Schulze, M.W.; Bates, C.M.; Hawker, C.J. Macrocyclic Side-Chain Monomers for Photoinduced ATRP: Synthesis and Properties versus Long-Chain Linear Isomers. Macromolecules 2018, 51, 6901–6910. [Google Scholar] [CrossRef]
- Pospiech, D.; Jehnichen, D.; Eckstein, K.; Scheibe, P.; Komber, H.; Sahre, K.; Janke, A.; Reuter, U.; Häußler, L.; Schellkopf, L.; et al. Semifluorinated PMMA Block Copolymers: Synthesis, Nanostructure, and Thin Film Properties. Macromol. Chem. Phys. 2017, 218, 1600599. [Google Scholar] [CrossRef]
- Elupula, R.; Oh, J.; Haque, F.M.; Chang, T.; Grayson, S.M. Determining the Origins of Impurities during Azide–Alkyne Click Cyclization of Polystyrene. Macromolecules 2016, 49, 4369–4372. [Google Scholar] [CrossRef]
- Oh, J.; Kuk, J.; Lee, T.; Ye, J.; Paik, H.J.; Lee, H.W.; Chang, T. Molecular Weight Distribution of Living Chains in Polystyrene Prepared by Atom Transfer Radical Polymerization. ACS Macro Lett. 2017, 6, 758–761. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Levere, M.E.; Kulis, J.; Monteiro, M.J.; Percec, V. Analysis of the Cu(0)-Catalyzed Polymerization of Methyl Acrylate in Disproportionating and Nondisproportionating Solvents. Macromolecules 2012, 45, 4606–4622. [Google Scholar] [CrossRef]
- Lligadas, G.; Rosen, B.M.; Monteiro, M.J.; Percec, V. Solvent Choice Differentiates SET-LRP and Cu-Mediated Radical Polymerization with Non-First-Order Kinetics. Macromolecules 2008, 41, 8360–8364. [Google Scholar] [CrossRef]
- Nyström, F.; Soeriyadi, A.H.; Boyer, C.; Zetterlund, P.B.; Whittaker, M.R. End-group fidelity of copper(0)-meditated radical polymerization at high monomer conversion: An ESI-MS investigation. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 5313–5321. [Google Scholar] [CrossRef]
- Town, J.S.; Jones, G.R.; Haddleton, D.M. MALDI-LID-ToF/ToF analysis of statistical and diblock polyacrylate copolymers. Polym. Chem. 2018, 9, 4631–4641. [Google Scholar] [CrossRef]
- Wang, Y.; Zhong, M.; Zhang, Y.; Magenau, A.J.D.; Matyjaszewski, K. Halogen Conservation in Atom Transfer Radical Polymerization. Macromolecules 2012, 45, 8929–8932. [Google Scholar] [CrossRef]
- Tang, W.; Tsarevsky, N.V.; Matyjaszewski, K. Determination of Equilibrium Constants for Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2006, 128, 1598–1604. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Matyjaszewski, K. Effect of Ligand Structure on Activation Rate Constants in ATRP. Macromolecules 2006, 39, 4953–4959. [Google Scholar] [CrossRef]
- Tang, W.; Matyjaszewski, K. Effects of Initiator Structure on Activation Rate Constants in ATRP. Macromolecules 2007, 40, 1858–1863. [Google Scholar] [CrossRef]
- Tang, W.; Kwak, Y.; Braunecker, W.; Tsarevsky, N.V.; Coote, M.L.; Matyjaszewski, K. Understanding Atom Transfer Radical Polymerization: Effect of Ligand and Initiator Structures on the Equilibrium Constants. J. Am. Chem. Soc. 2008, 130, 10702–10713. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kwak, Y.; Buback, J.; Buback, M.; Matyjaszewski, K. Determination of ATRP equilibrium constants under polymerization conditions. ACS Macro Lett. 2012, 1, 1367–1370. [Google Scholar] [CrossRef]
- Jakubowski, W. Adapting Atom Transfer Radical Polymerization to Industrial Scale Production: The Ultimate ATRP SM Technology; Oxford University Press, Inc.: Danvers, MA, USA, 2012; pp. 203–216. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Tsarevsky, N.V.; Braunecker, W.A.; Dong, H.; Huang, J.; Jakubowski, W.; Kwak, Y.; Nicolay, R.; Tang, W.; Yoon, J.A. Role of Cu 0 in Controlled/“Living” Radical Polymerization. Macromolecules 2007, 40, 7795–7806. [Google Scholar] [CrossRef]
- Schröder, K.; Konkolewicz, D.; Poli, R.; Matyjaszewski, K. Formation and Possible Reactions of Organometallic Intermediates with Active Copper(I) Catalysts in ATRP. Organometallics 2012, 31, 7994–7999. [Google Scholar] [CrossRef]
- Wang, Y.; Soerensen, N.; Zhong, M.; Schroeder, H.; Buback, M.; Matyjaszewski, K. Improving the “Livingness” of ATRP by Reducing Cu Catalyst Concentration. Macromolecules 2013, 46, 683–691. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Russell, G.T. Chain-length-dependent termination in radical polymerization: Subtle revolution in tackling a long-standing challenge. Prog. Polym. Sci. 2009, 34, 1211–1259. [Google Scholar] [CrossRef]
- Buback, M.; Kurz, C.H.; Schmaltz, C. Pressure dependence of propagation rate coefficients in free-radical homopolymerizations of methyl acrylate and dodecyl acrylate. Macromol. Chem. Phys. 1998, 199, 1721–1727. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Buback, M.; Egorov, M.; Fukuda, T.; Goto, A.; Olaj, O.F.; Russell, G.T.; Vana, P.; Yamada, B.; Zetterlund, P.B. Critically evaluated termination rate coefficients for free-radical polymerization: Experimental methods. Prog. Polym. Sci. 2005, 30, 605–643. [Google Scholar] [CrossRef]
- Ribelli, T.G.; Wahidur Rahaman, S.M.; Daran, J.C.; Krys, P.; Matyjaszewski, K.; Poli, R. Effect of Ligand Structure on the Cu II –R OMRP Dormant Species and Its Consequences for Catalytic Radical Termination in ATRP. Macromolecules 2016, 49, 7749–7757. [Google Scholar] [CrossRef]
- Fantin, M.; Lorandi, F.; Ribelli, T.G.; Szczepaniak, G.; Enciso, A.E.; Fliedel, C.; Thevenin, L.; Isse, A.A.; Poli, R.; Matyjaszewski, K. Impact of Organometallic Intermediates on Copper-Catalyzed Atom Transfer Radical Polymerization. Macromolecules 2019, 52, 4079–4090. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a | b | c | ||||||
|---|---|---|---|---|---|---|---|---|
| 50 | 2 h | 86% | 3900 | 3700 | 1.15 | 26 | 2% | 3% |
| 100 | 2 h | 92% | 8100 | 8200 | 1.13 | 16 | 4% | 5% |
| 100 | 4 h | 87% | 7700 | 7900 | 1.10 | 49 | 4% | 6% |
| 100 | 8 h | 94% | 8200 | 9600 | 1.10 | 52 | 4% | 4% |
| 200 | 2 h | 84% | 14,600 | 15,100 | 1.10 | 30 | 8% | 6% |
| 50 d | 2 h | 73% | 6500 | 7300 | 1.09 | 60 | 6% |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y. ATRP of Methyl Acrylate by Continuous Feeding of Activators Giving Polymers with Predictable End-Group Fidelity. Polymers 2019, 11, 1238. https://doi.org/10.3390/polym11081238
Wang Y. ATRP of Methyl Acrylate by Continuous Feeding of Activators Giving Polymers with Predictable End-Group Fidelity. Polymers. 2019; 11(8):1238. https://doi.org/10.3390/polym11081238
Chicago/Turabian StyleWang, Yu. 2019. "ATRP of Methyl Acrylate by Continuous Feeding of Activators Giving Polymers with Predictable End-Group Fidelity" Polymers 11, no. 8: 1238. https://doi.org/10.3390/polym11081238
APA StyleWang, Y. (2019). ATRP of Methyl Acrylate by Continuous Feeding of Activators Giving Polymers with Predictable End-Group Fidelity. Polymers, 11(8), 1238. https://doi.org/10.3390/polym11081238