Lignin Biopolymers in the Age of Controlled Polymerization


Abstract
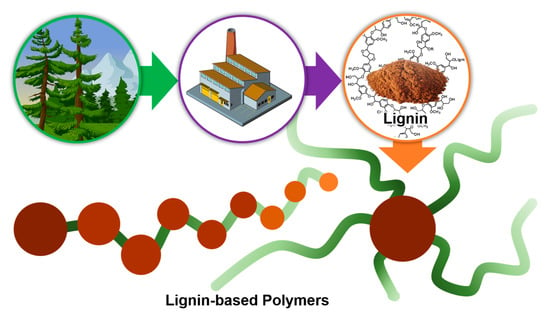
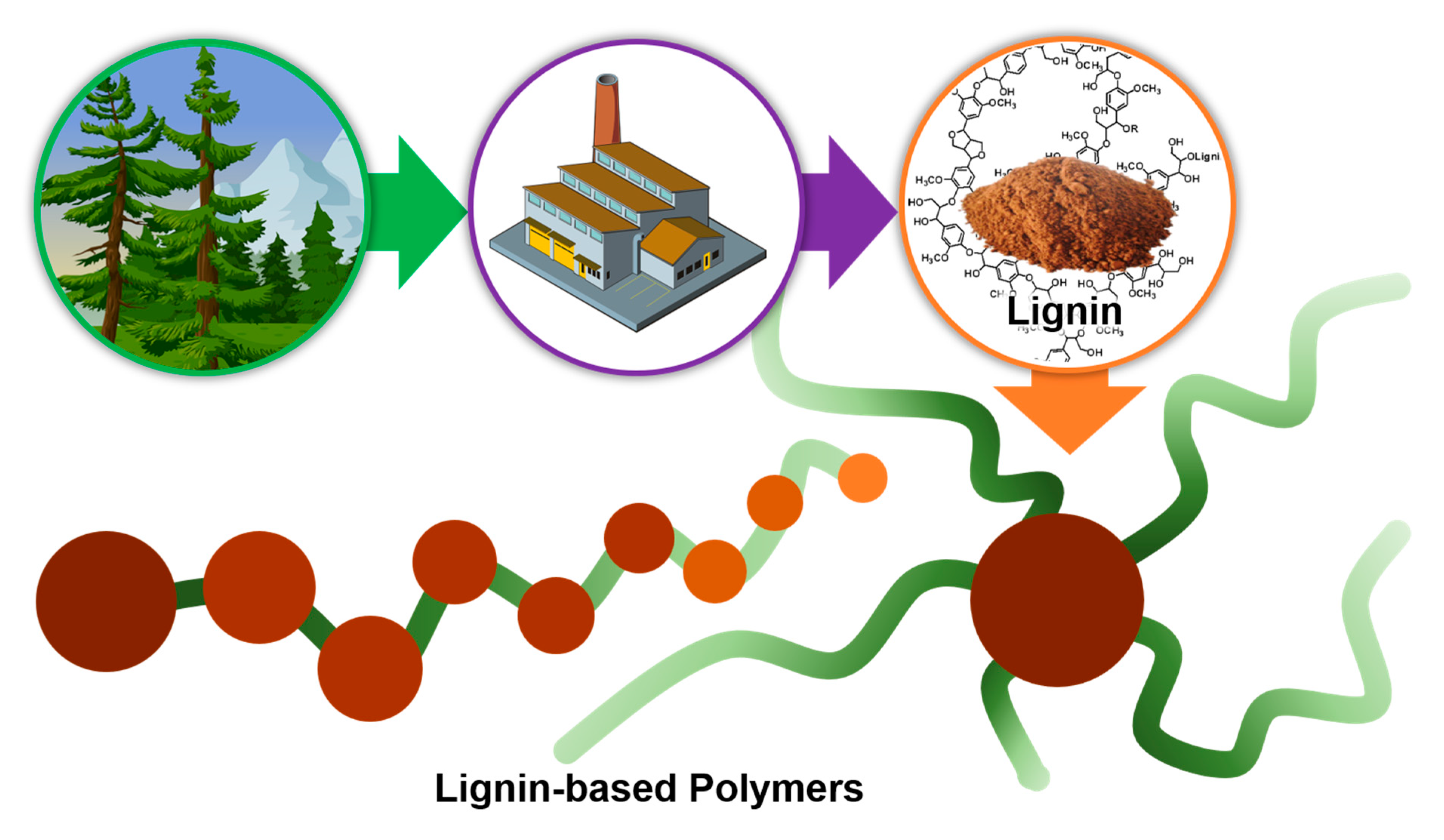
1. Introduction
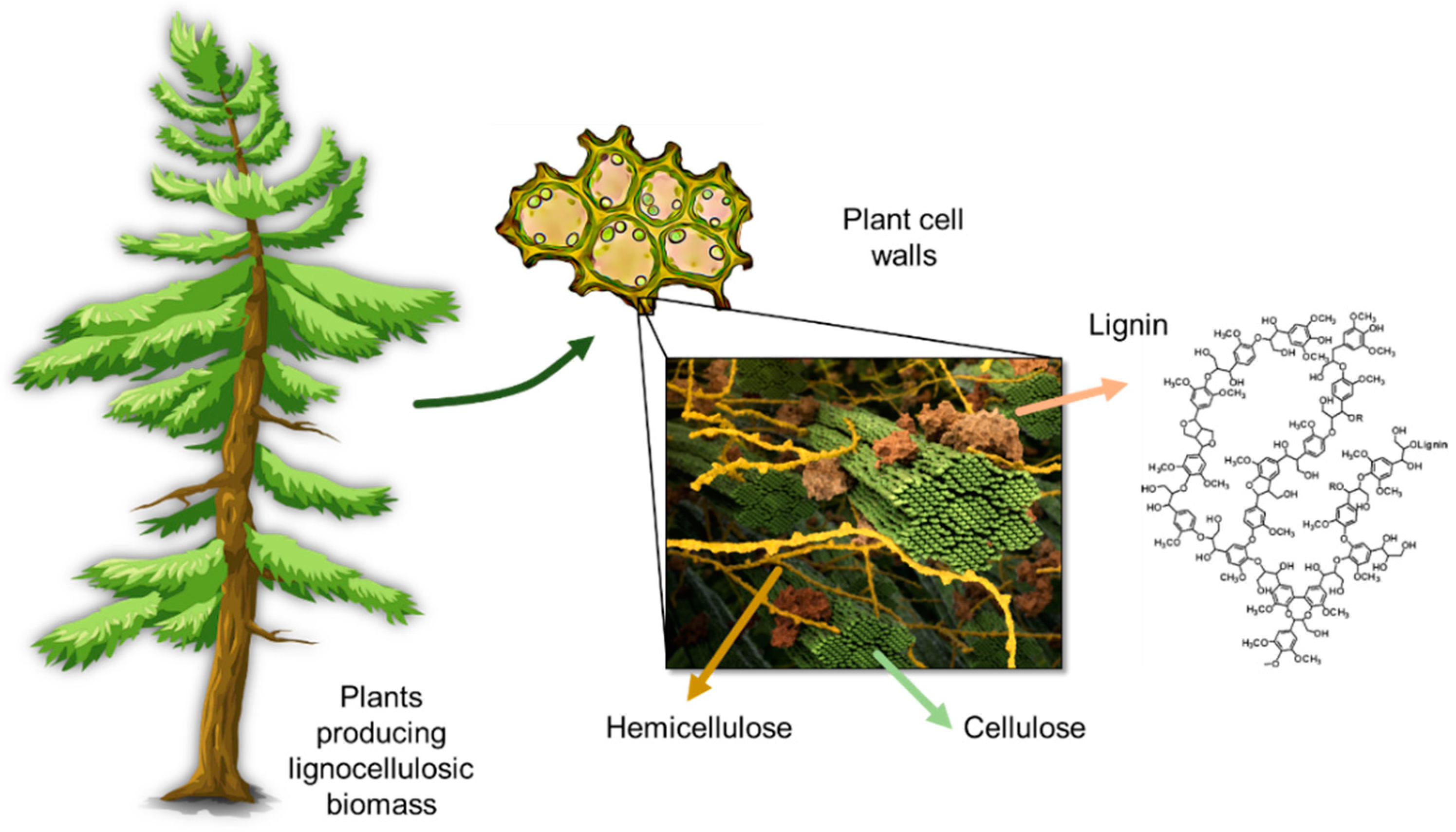
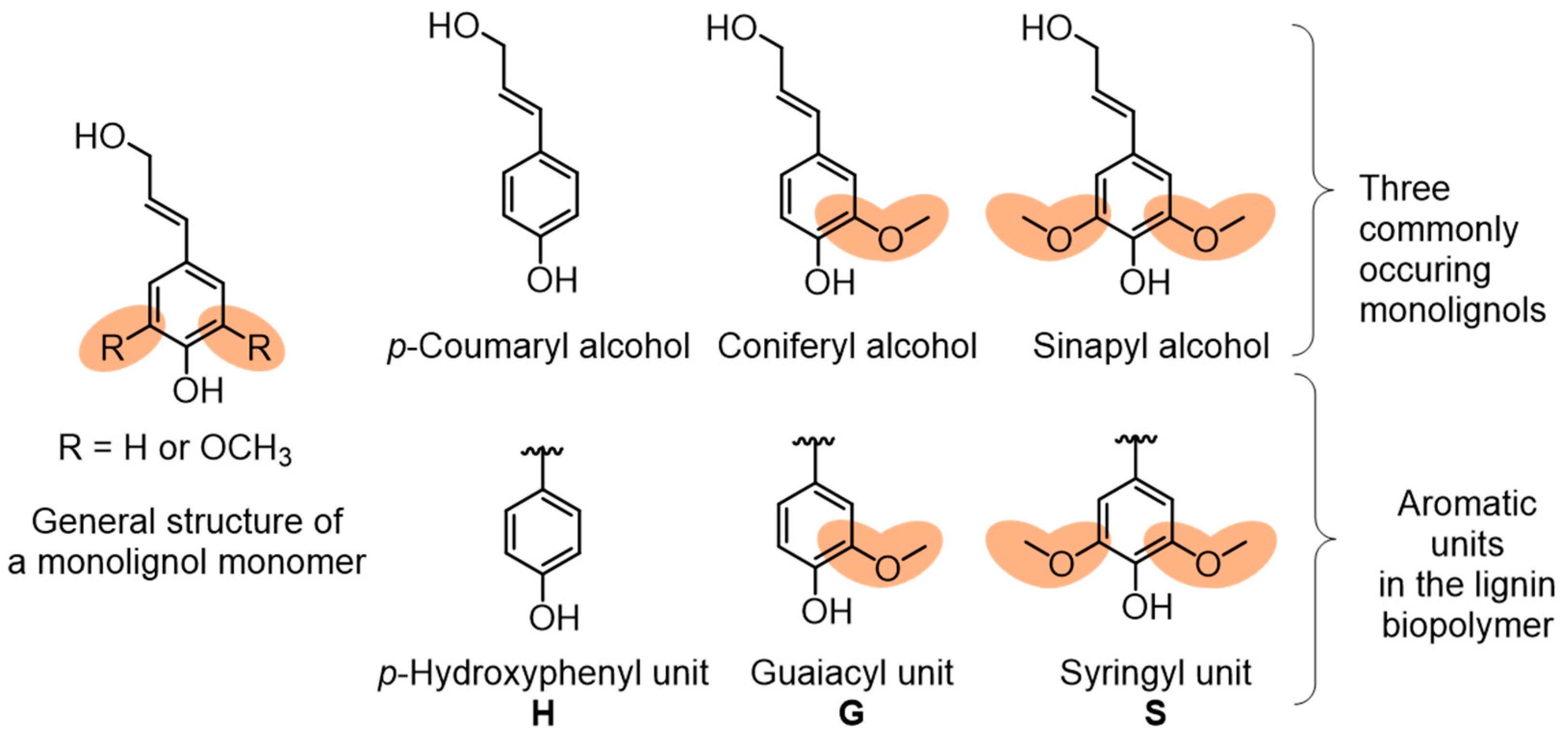
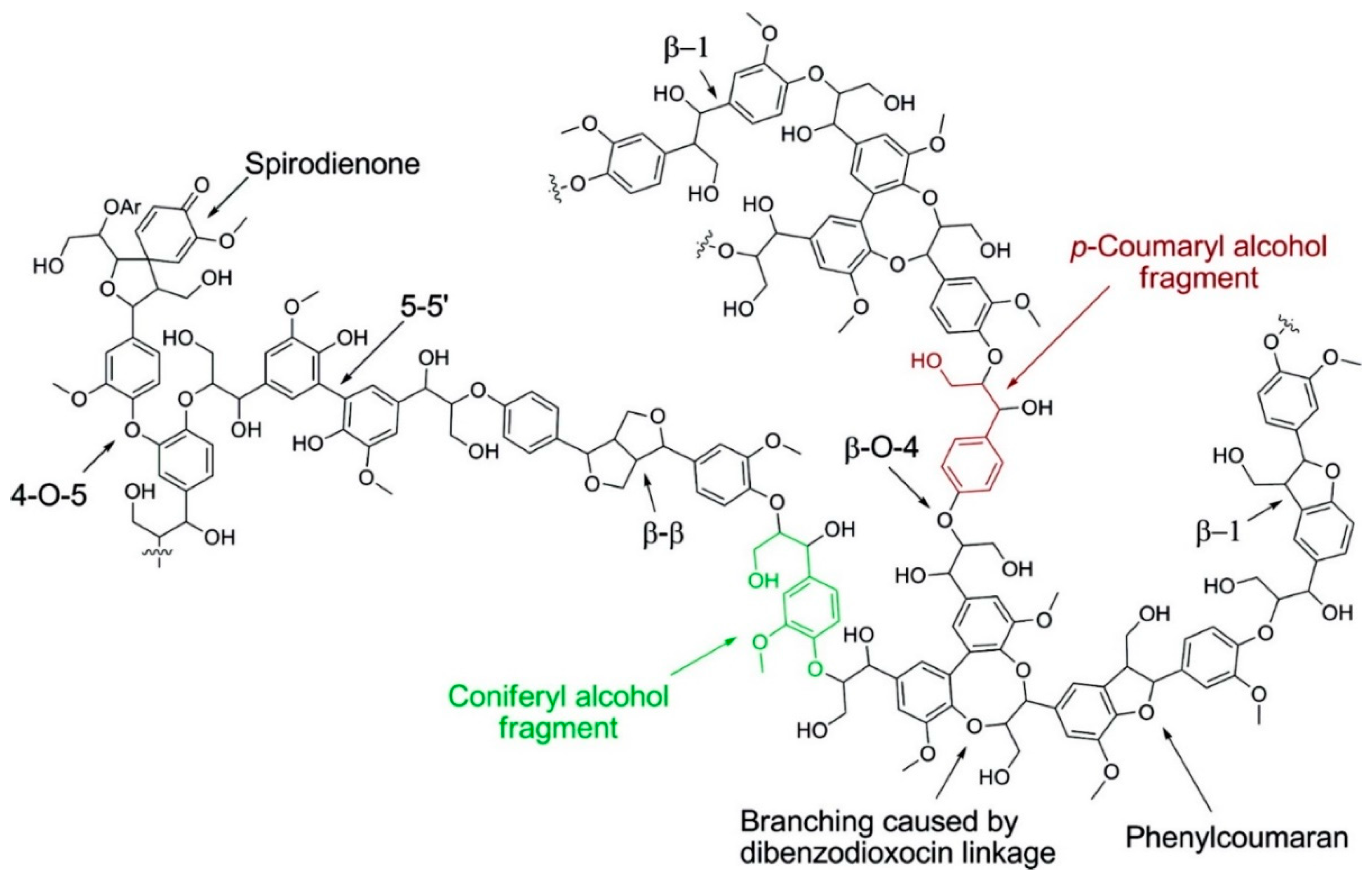
2. Lignin Structure and Composition
3. Extraction of Lignin
3.1. Ionic Liquids for Lignin Extraction
3.2. Lignin-First Method and Biorefinery Concepts
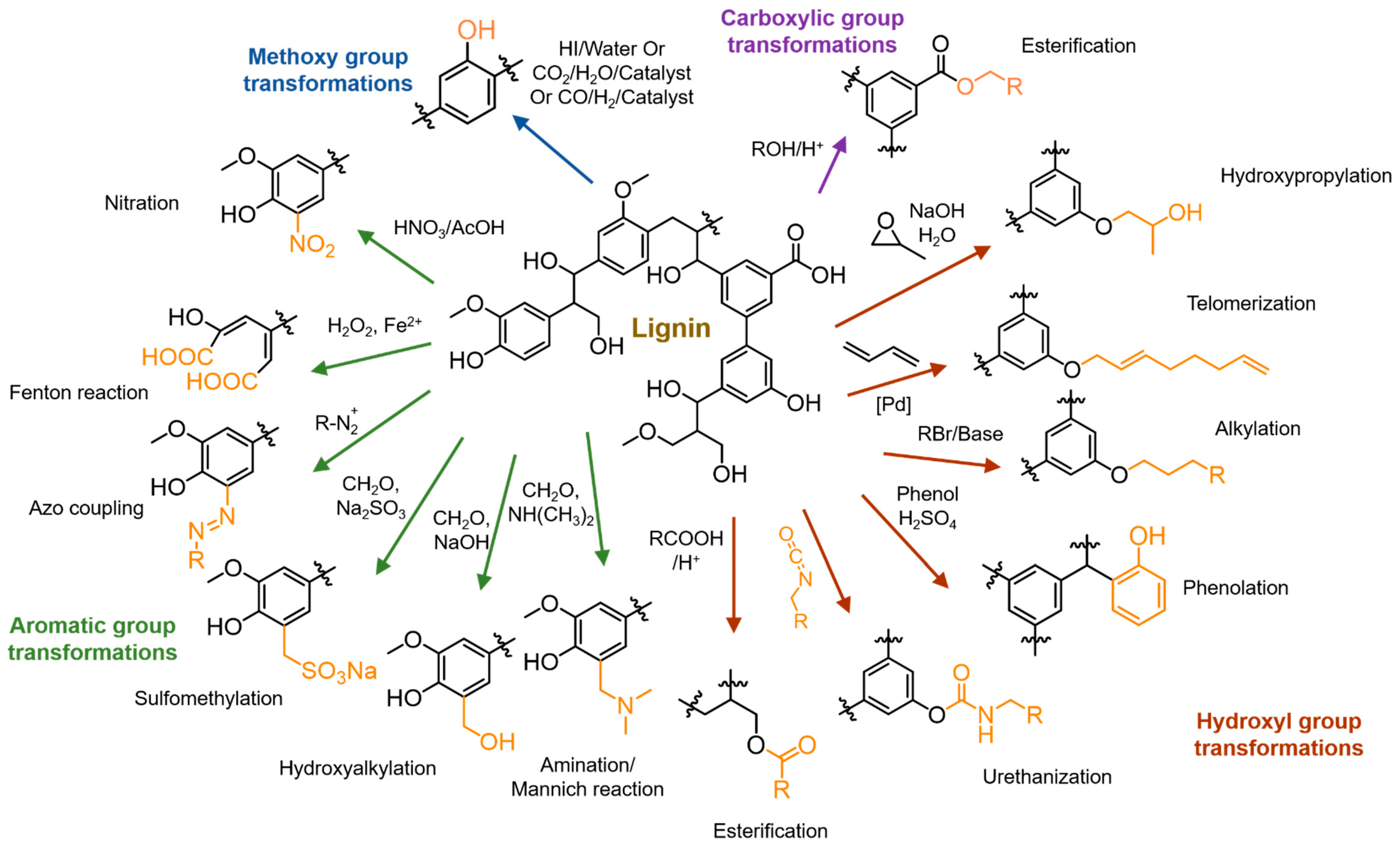
4. Chemical Modifications of Lignin
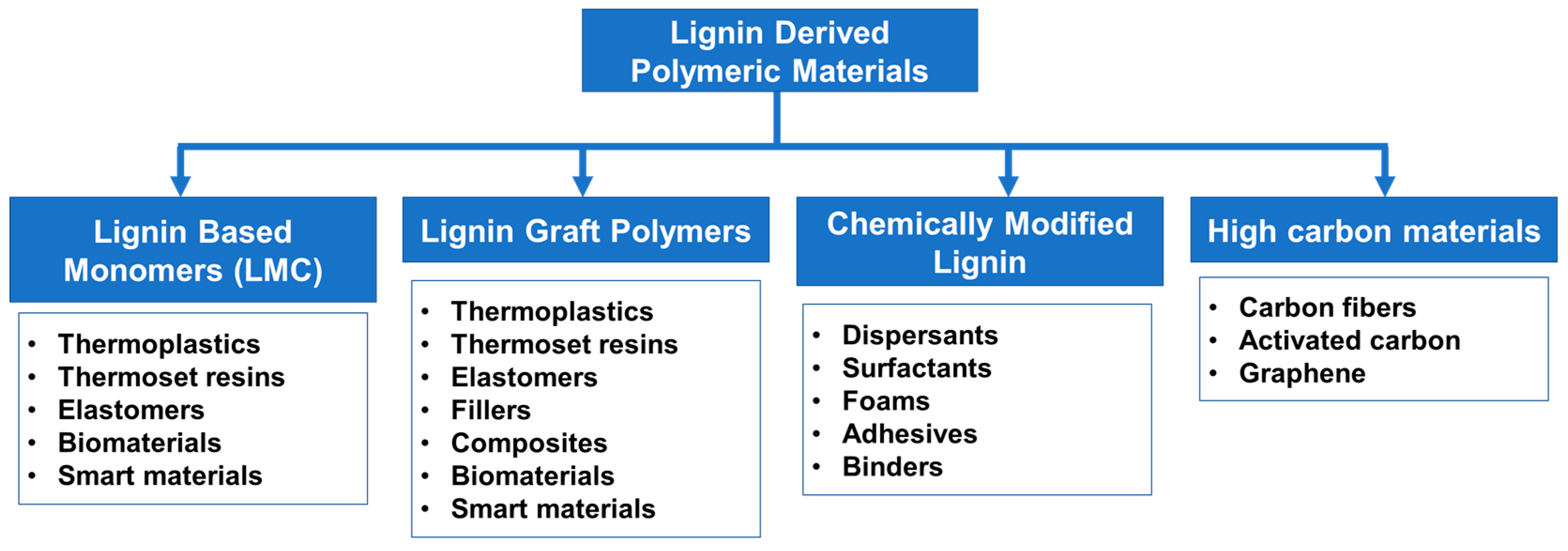
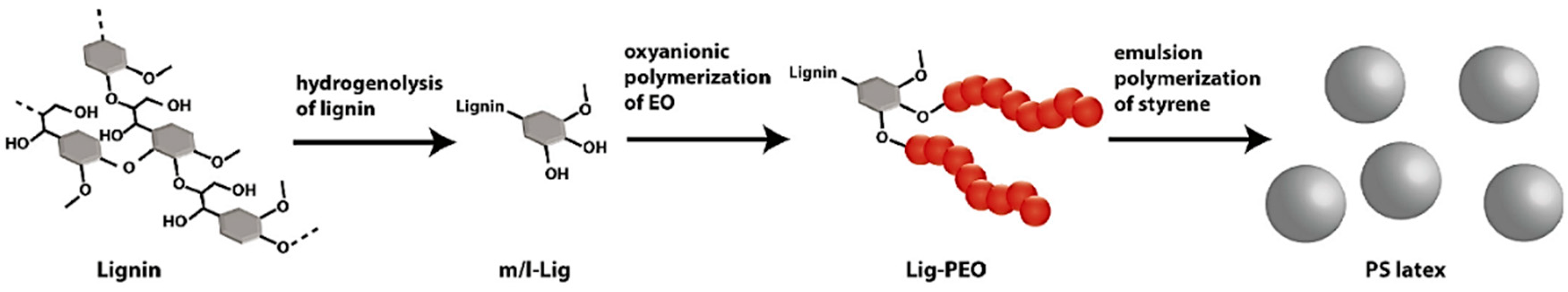
5. Lignin-Derived Polymers
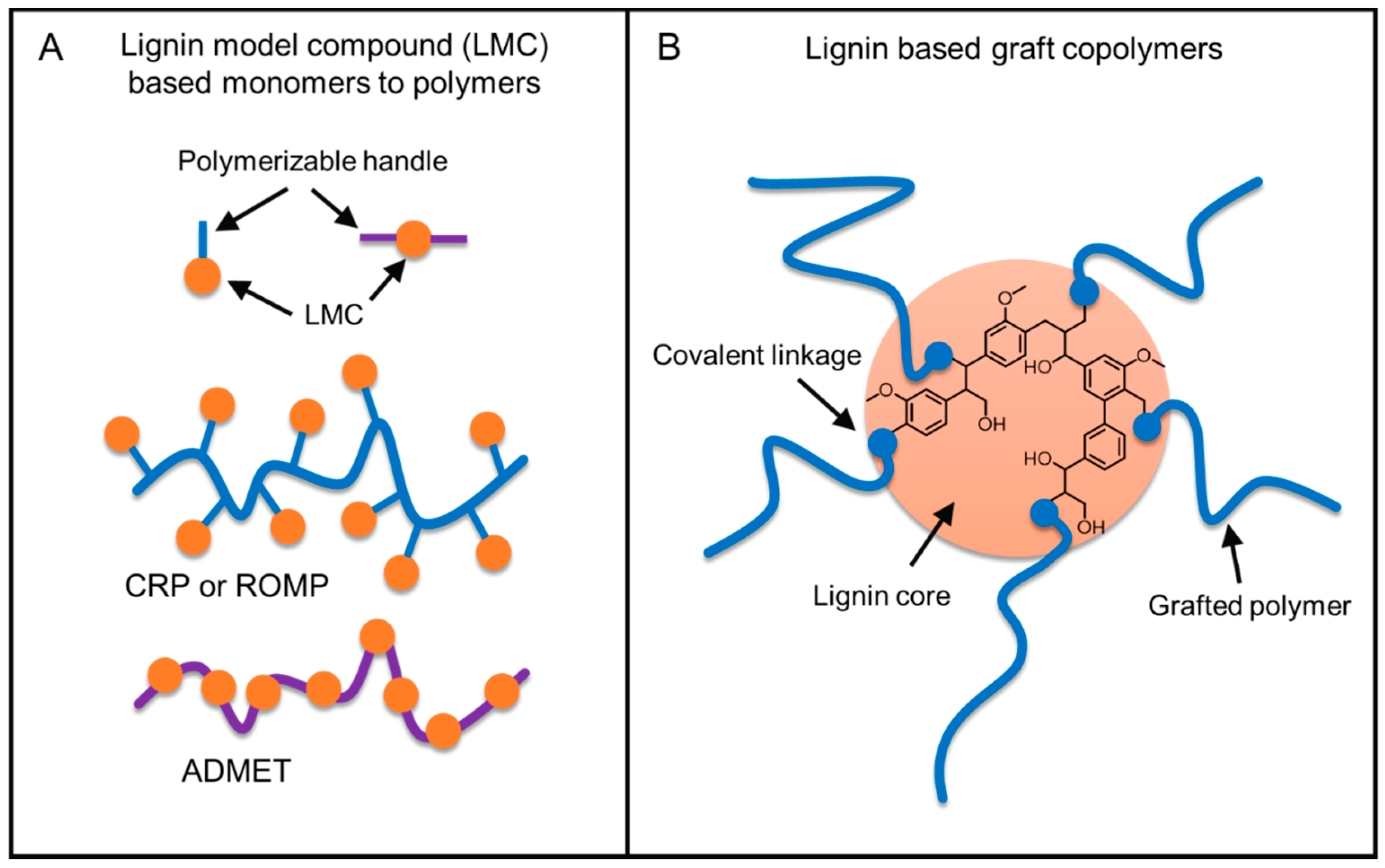
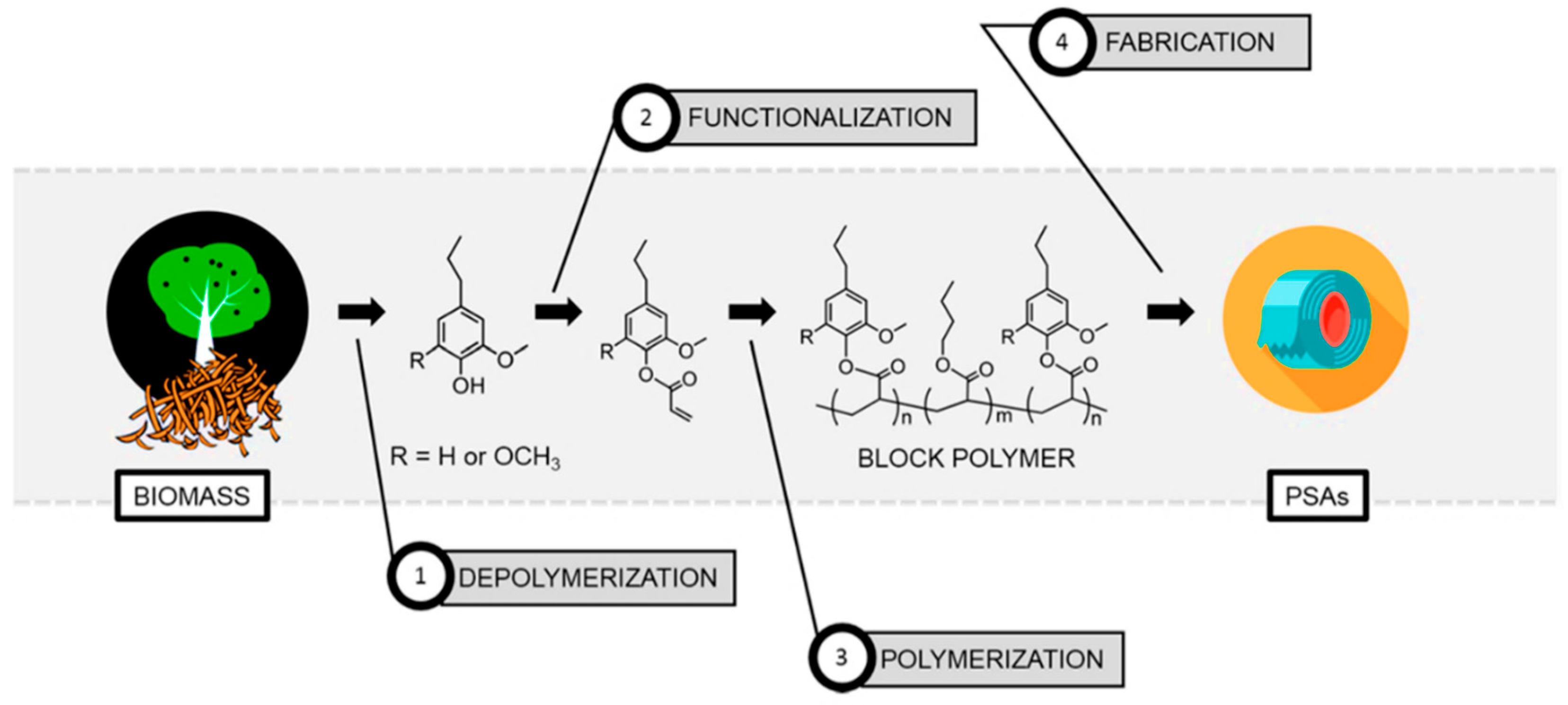
5.1. Overview
5.2. Strategies on Lignin-Based Polymer Synthesis
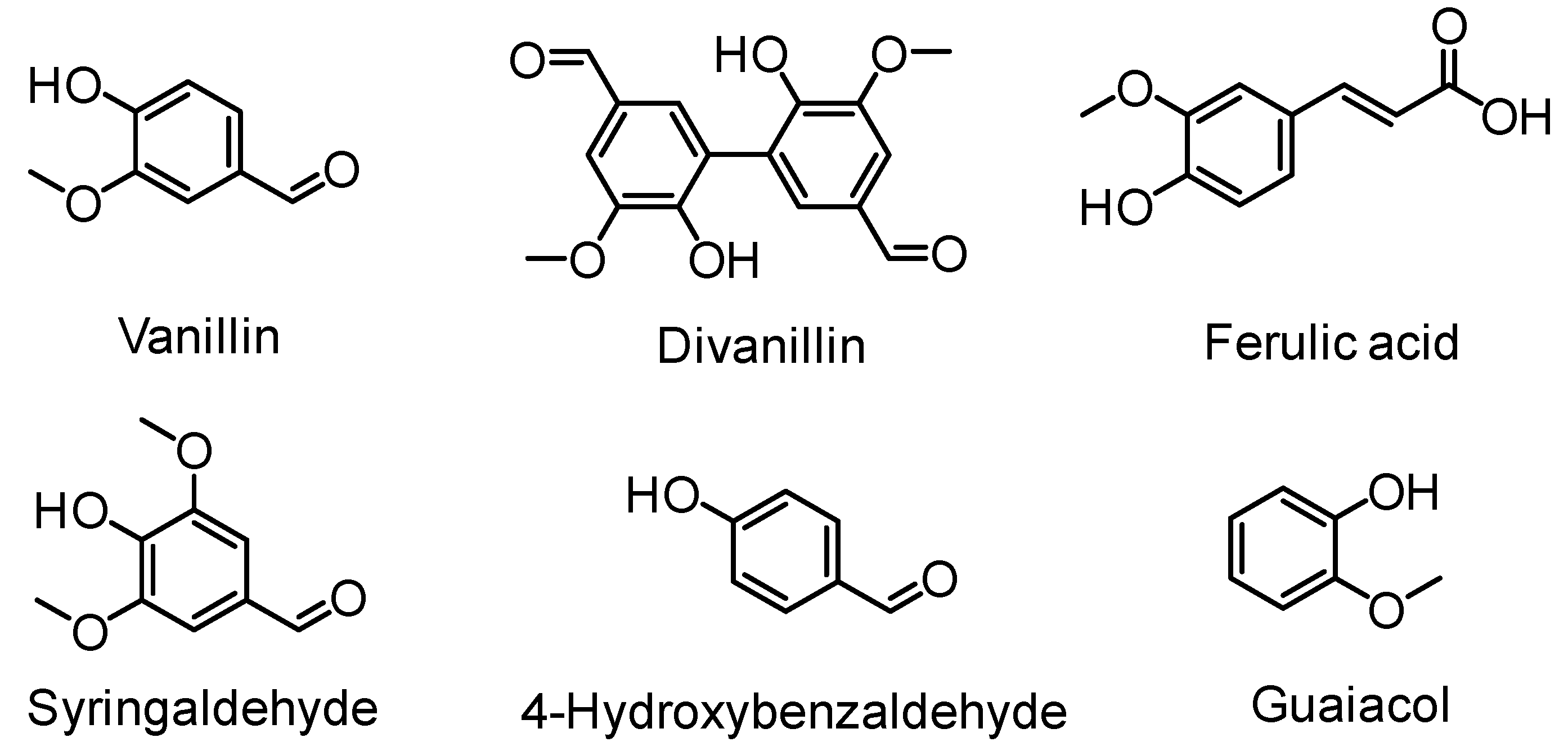
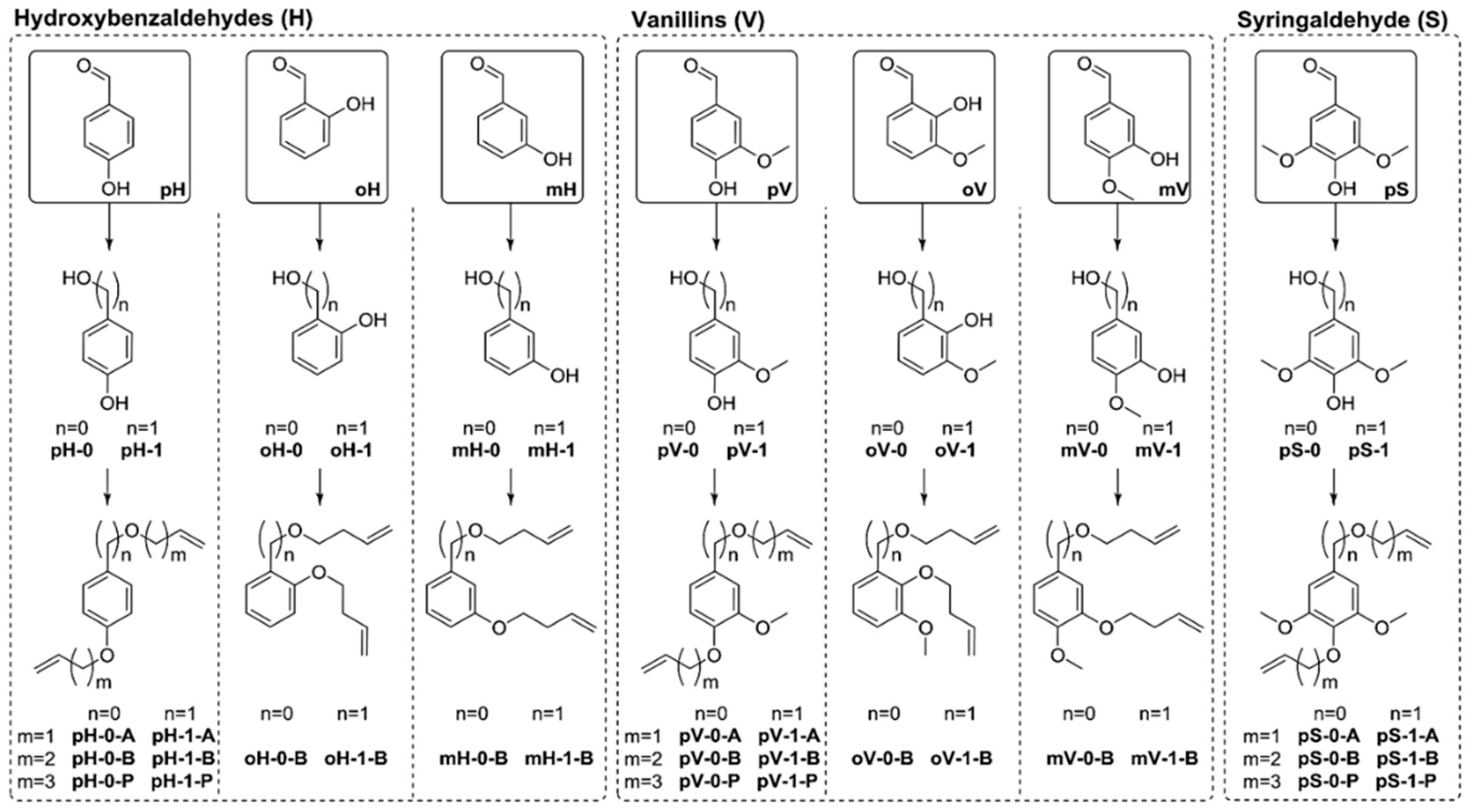
5.3. From Lignin Model Compounds (LMC) to Novel Biobased Polymers
5.3.1. Radical Polymerization Routes (FRP and RAFT)
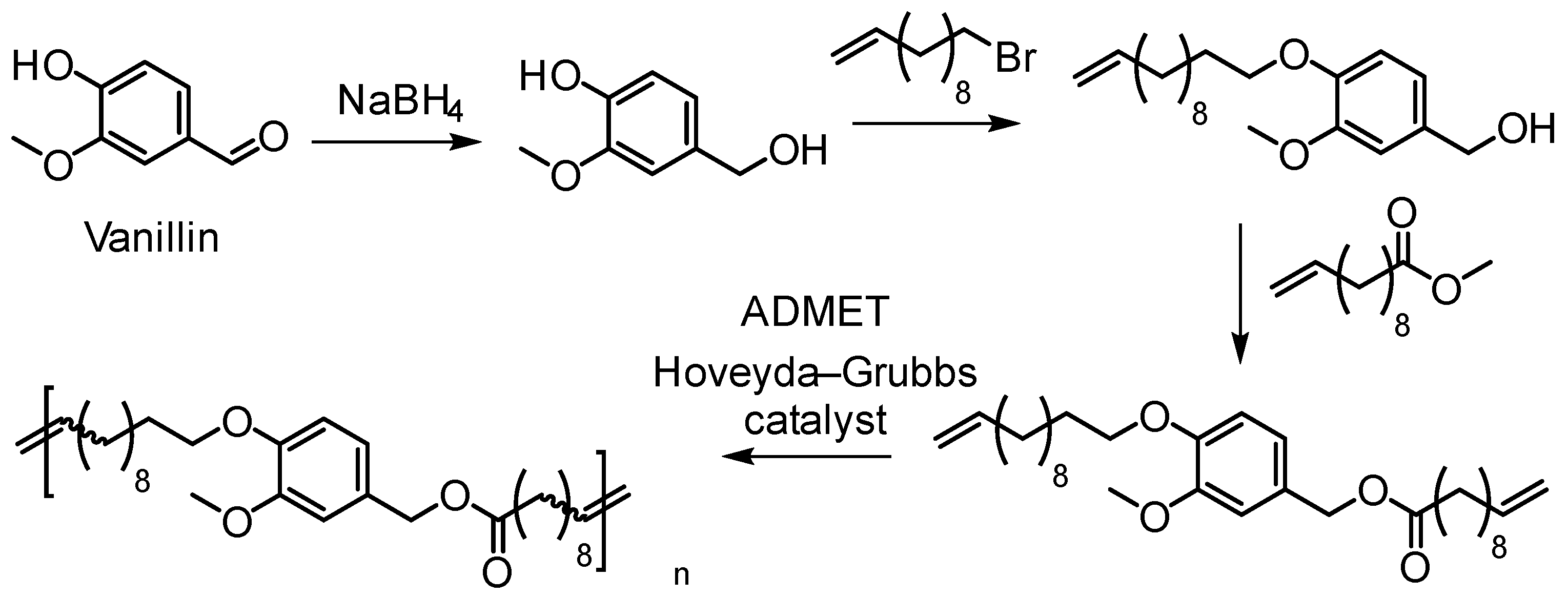
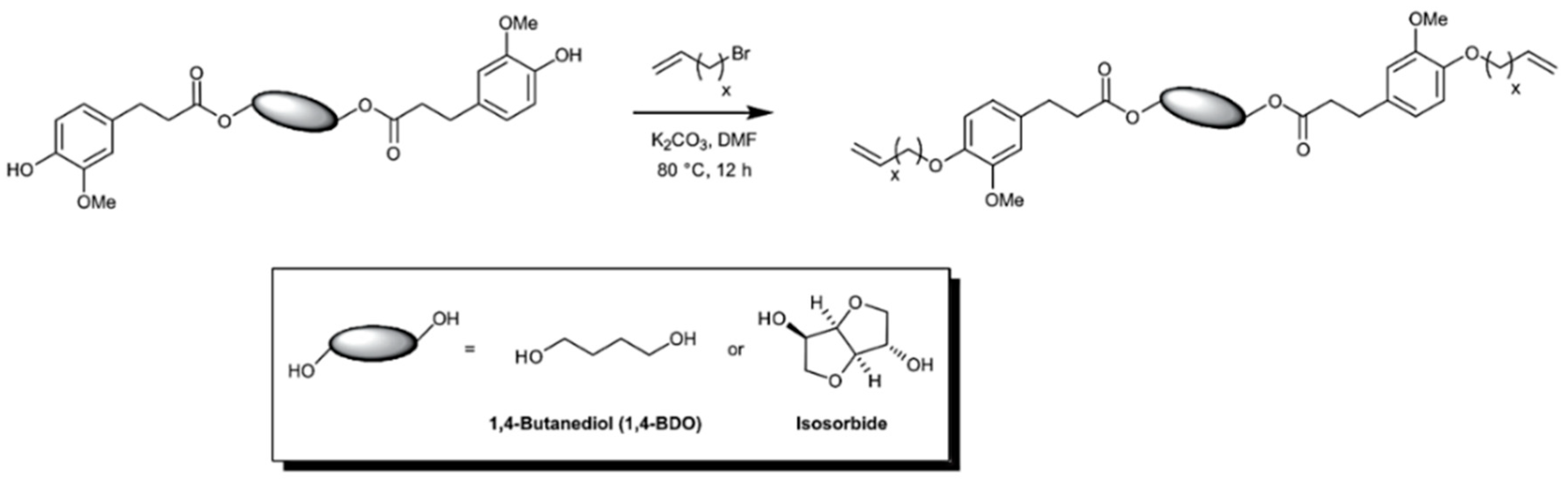
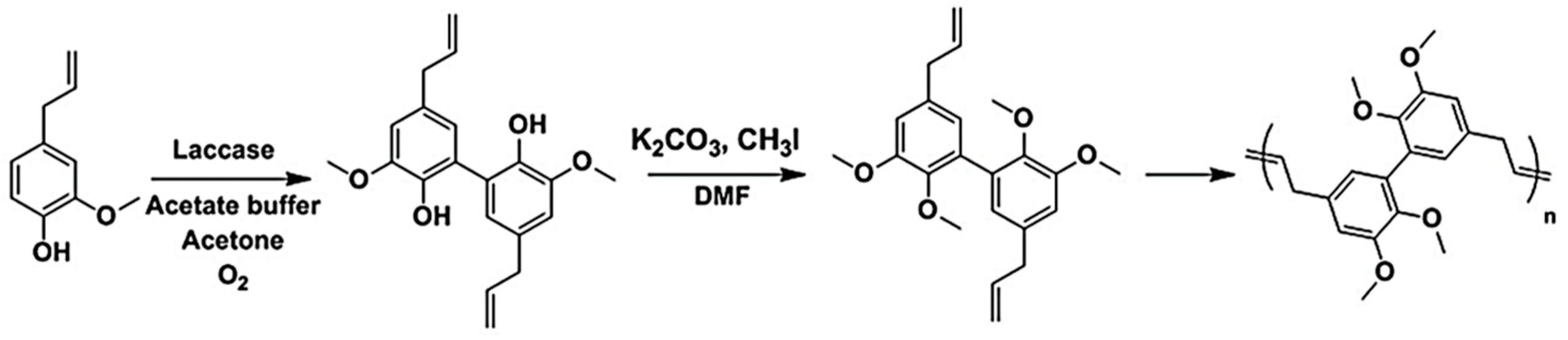
5.3.2. Acyclic Diene Metathesis (ADMET) Polymerization
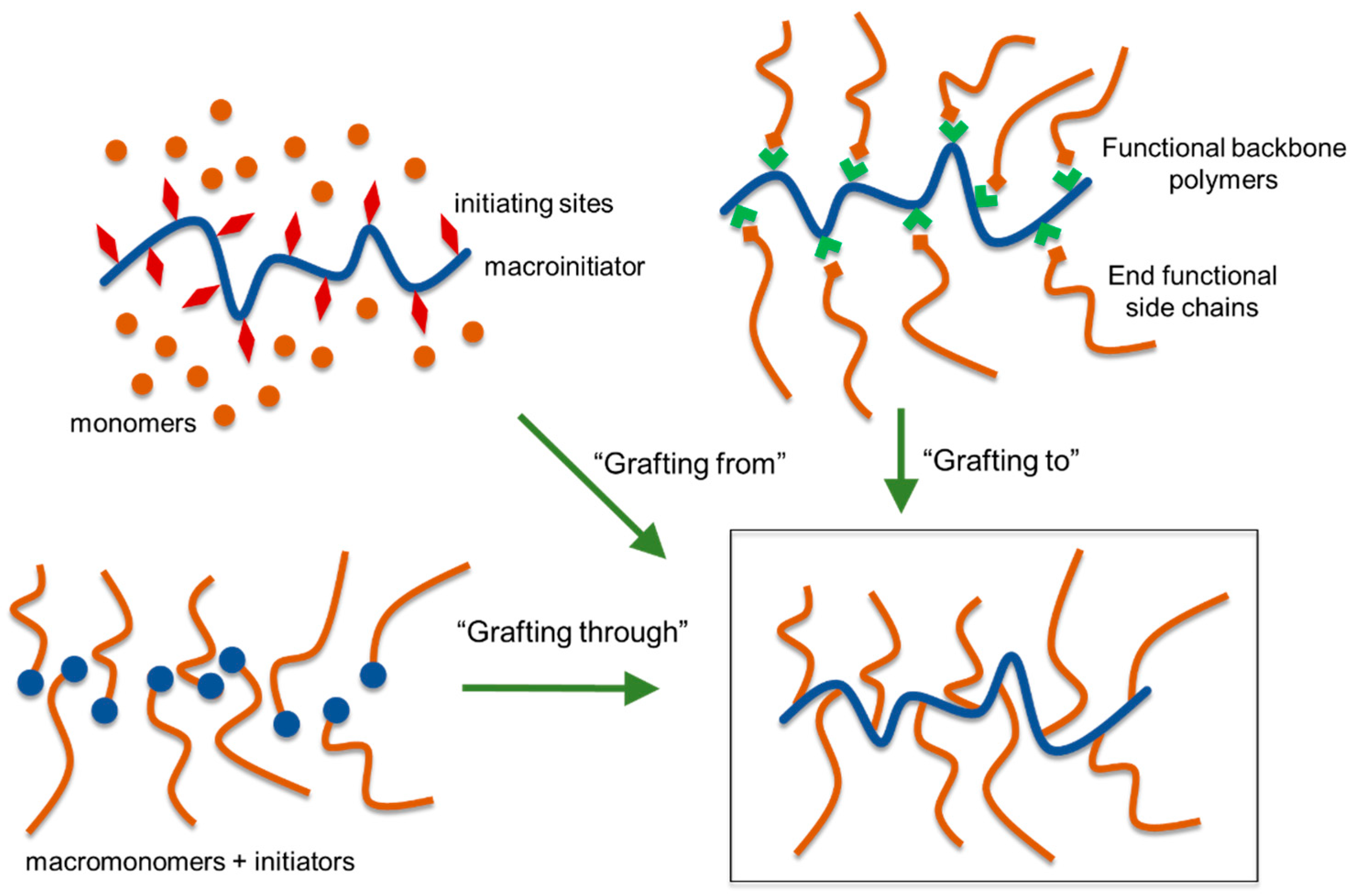
5.4. Lignin Graft Copolymers
5.4.1. “Grafting Through” Method
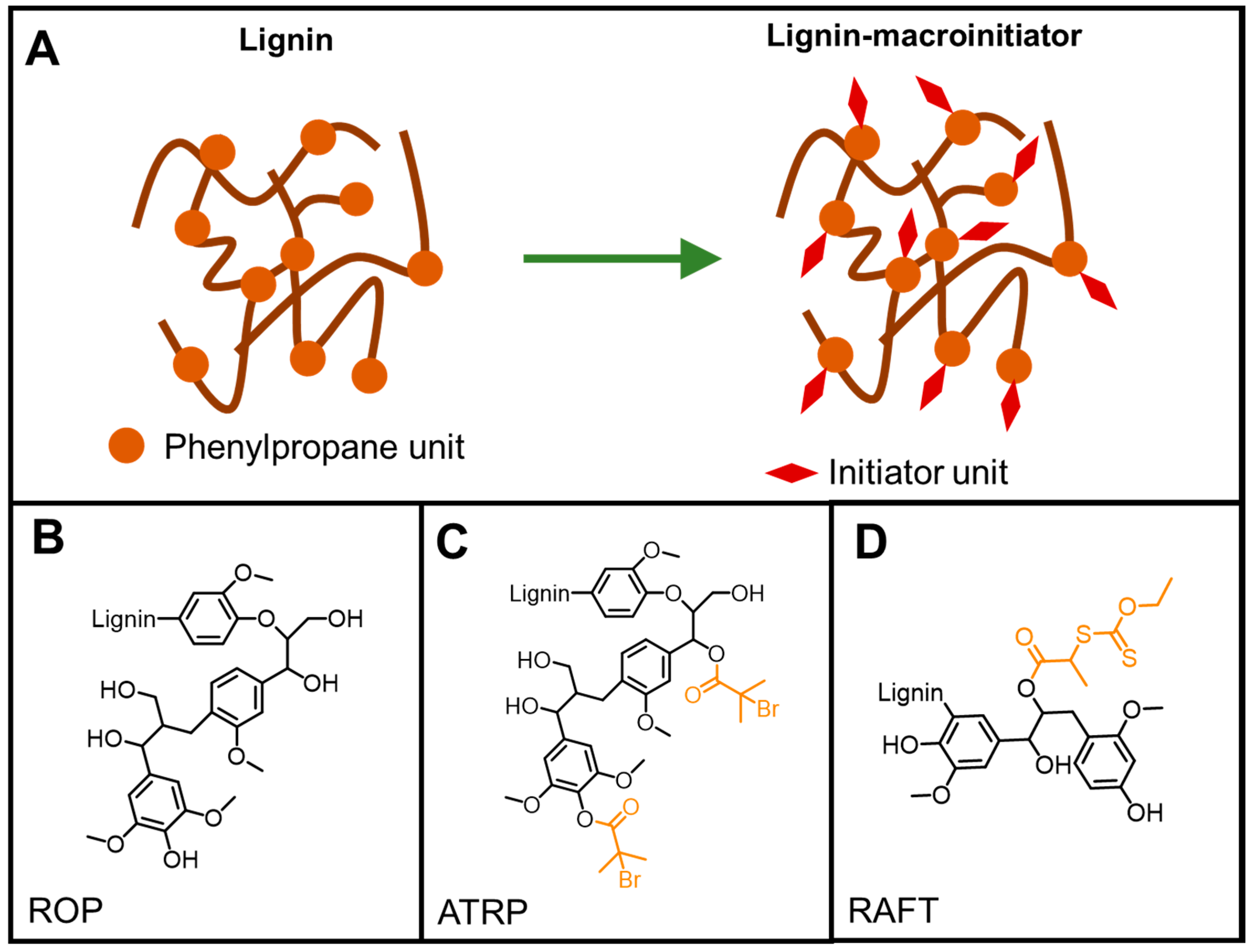
5.4.2. “Grafting From” Method
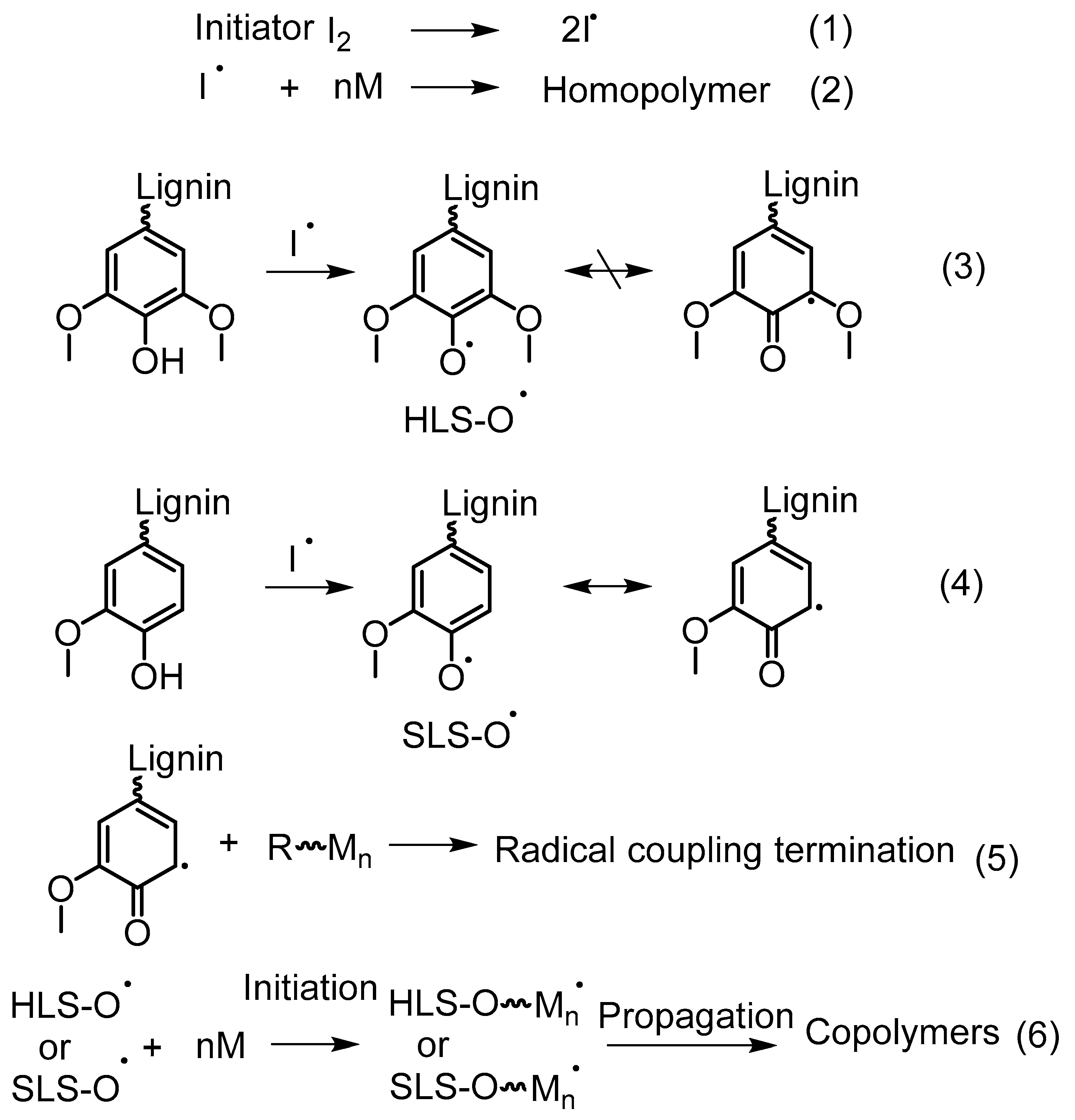
5.4.3. Free Radical Graft Polymers
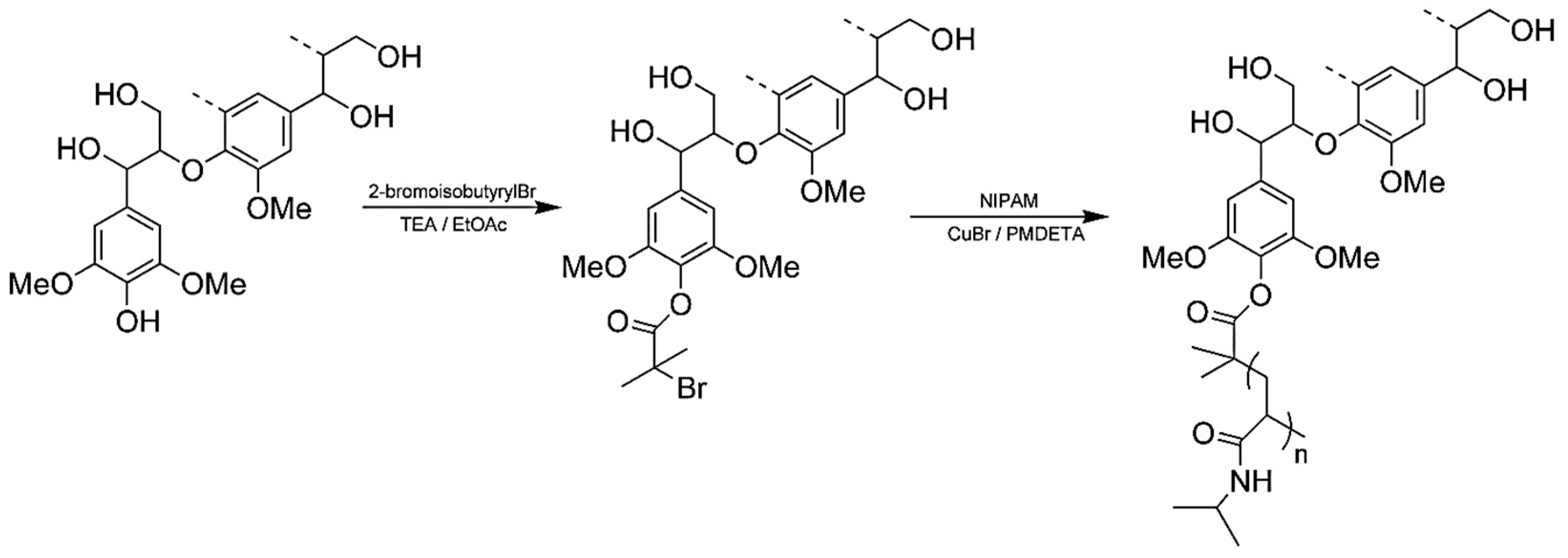
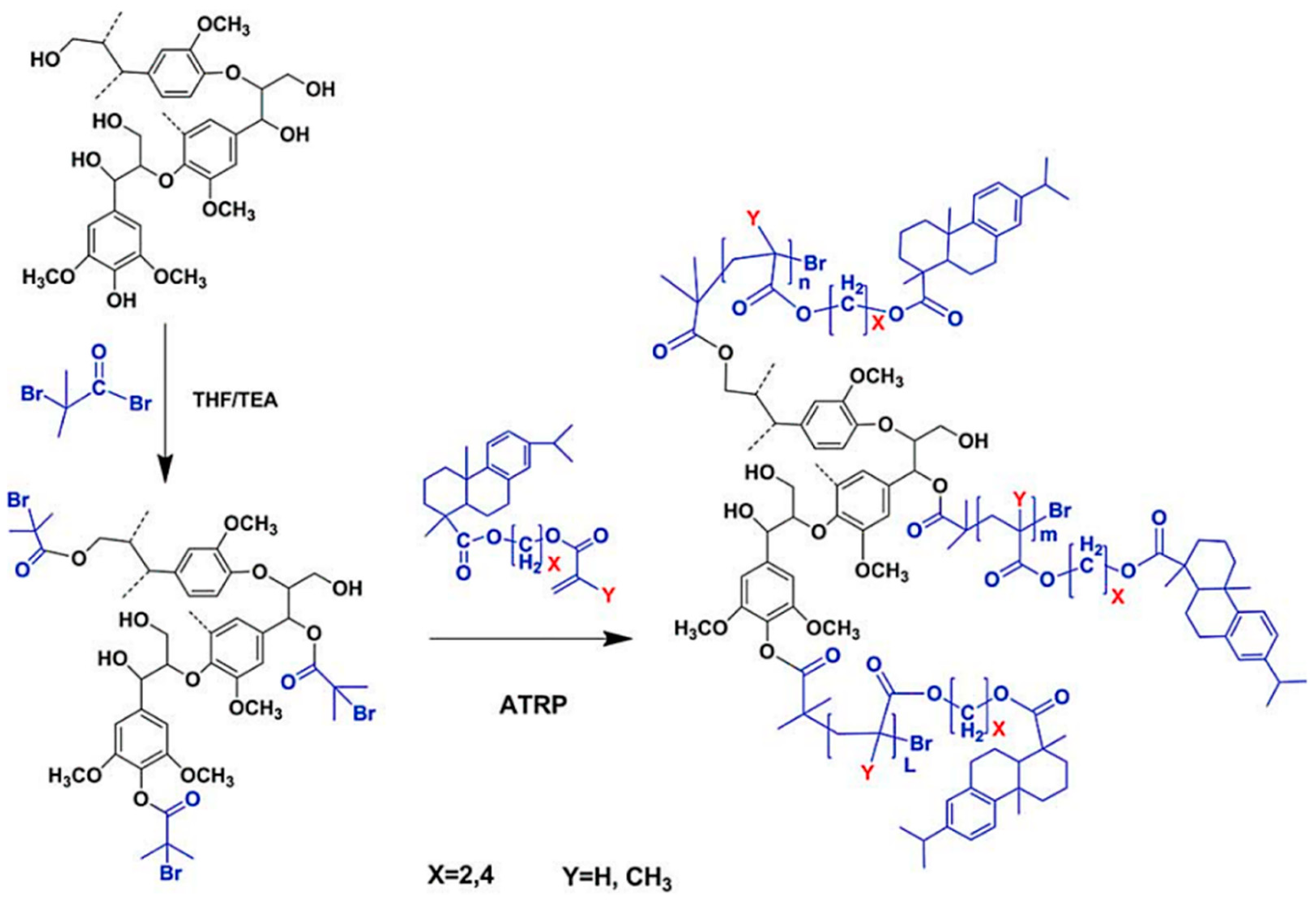
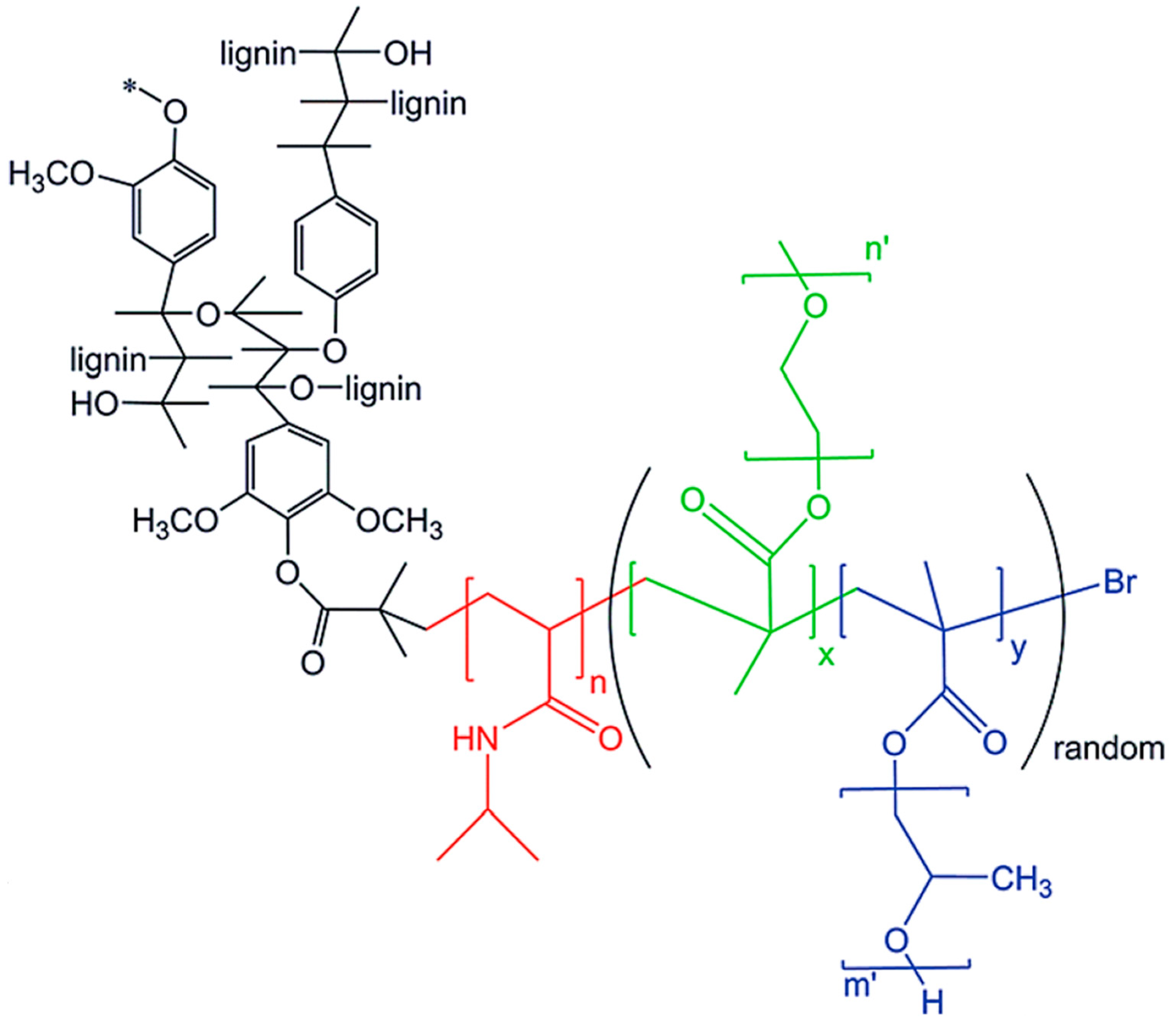
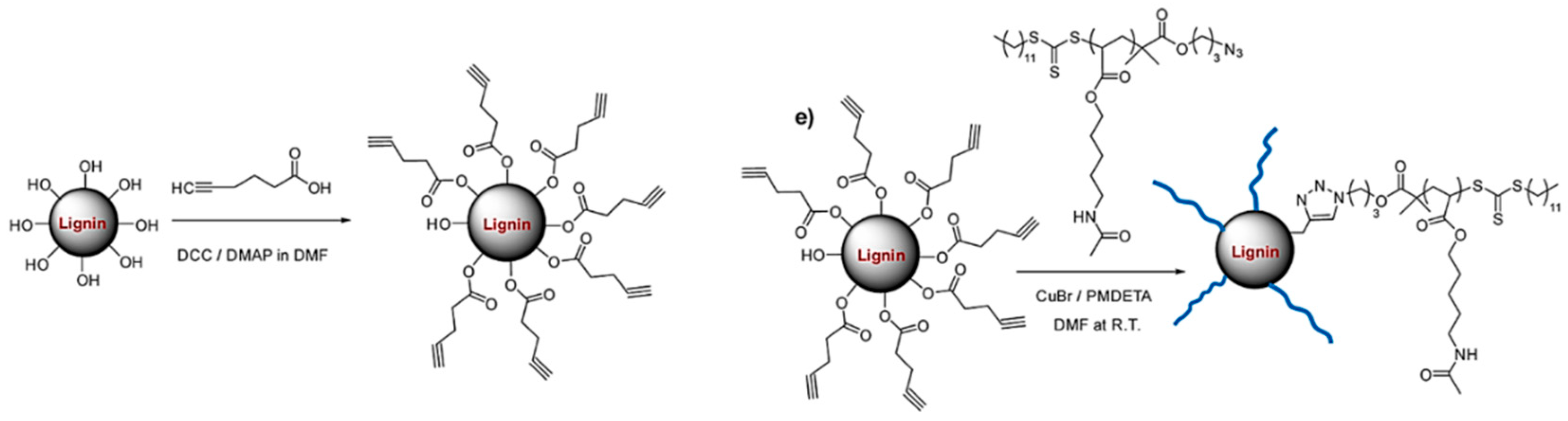
5.4.4. ATRP
RAFT Polymerization
ROP
ROMP
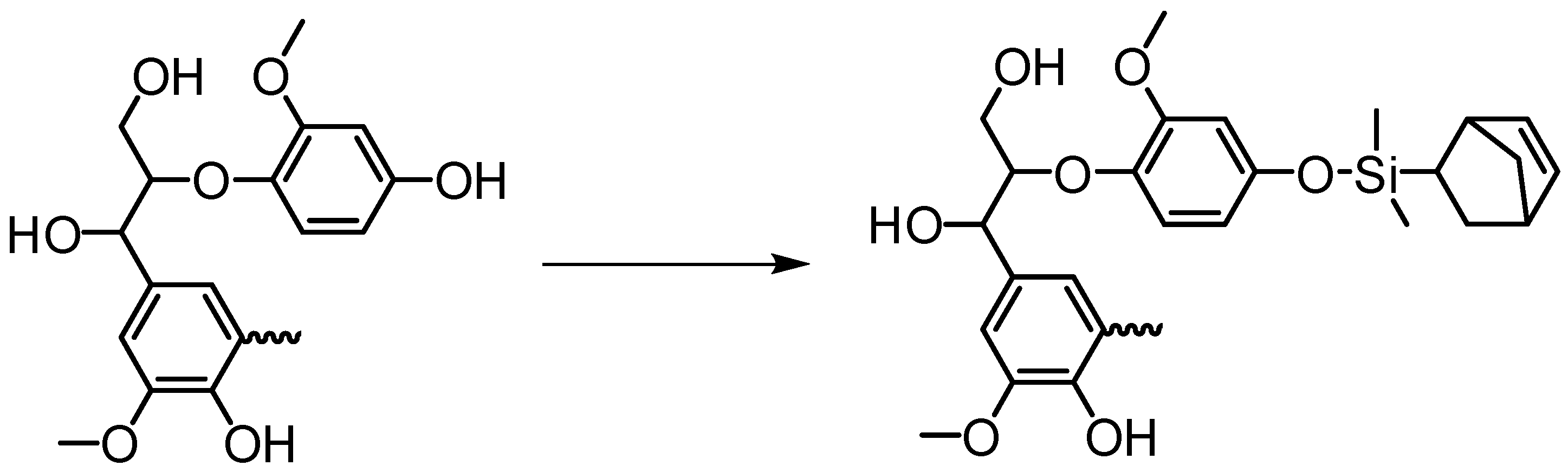
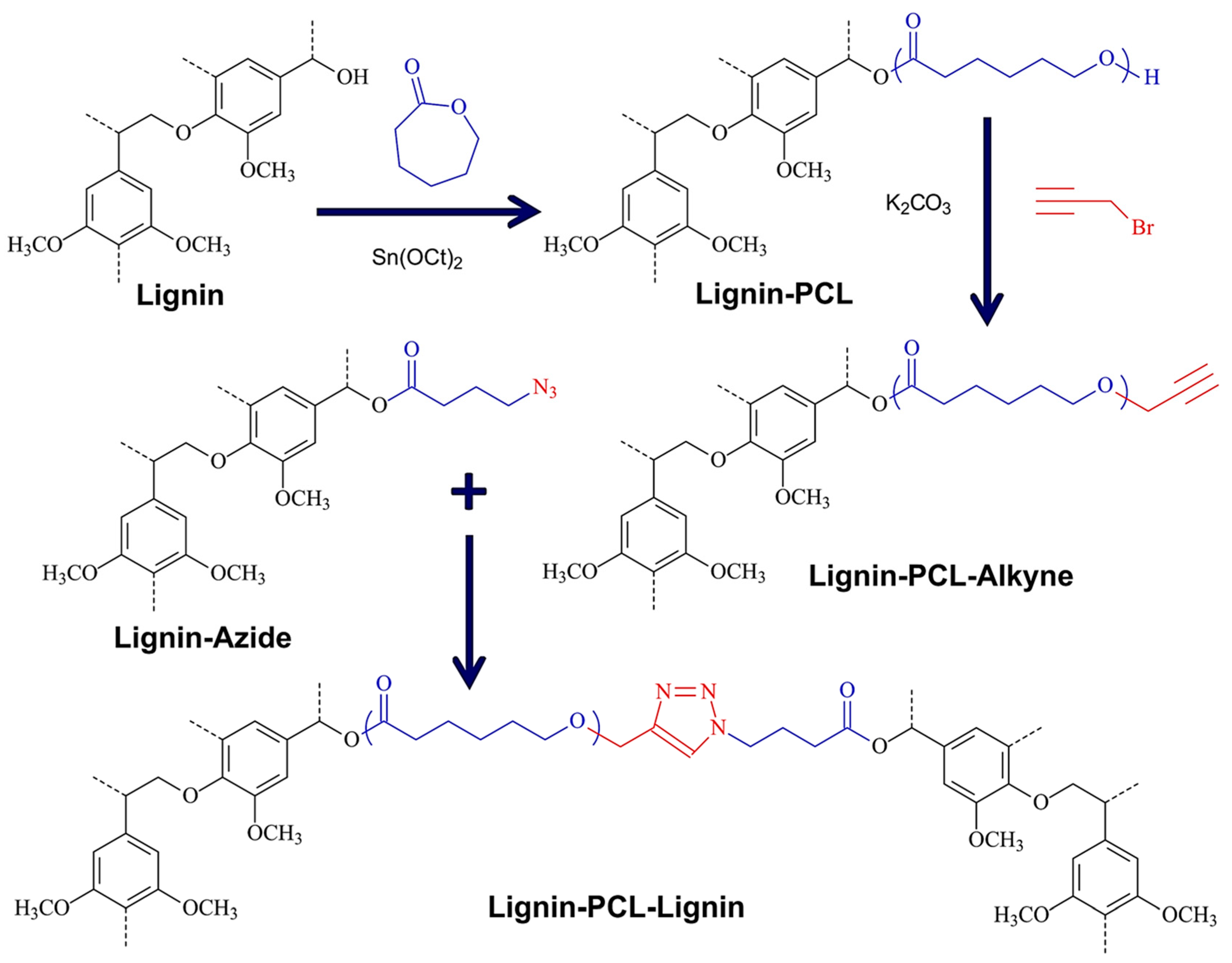
5.4.5. “Grafting To” Method
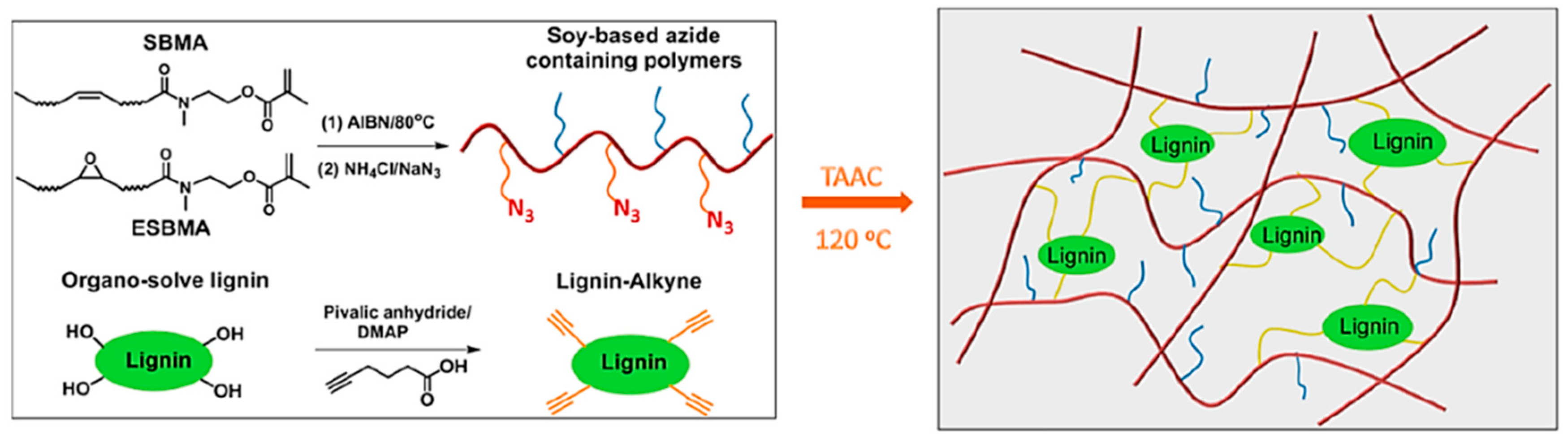
Azide–Alkyne Cycloaddition
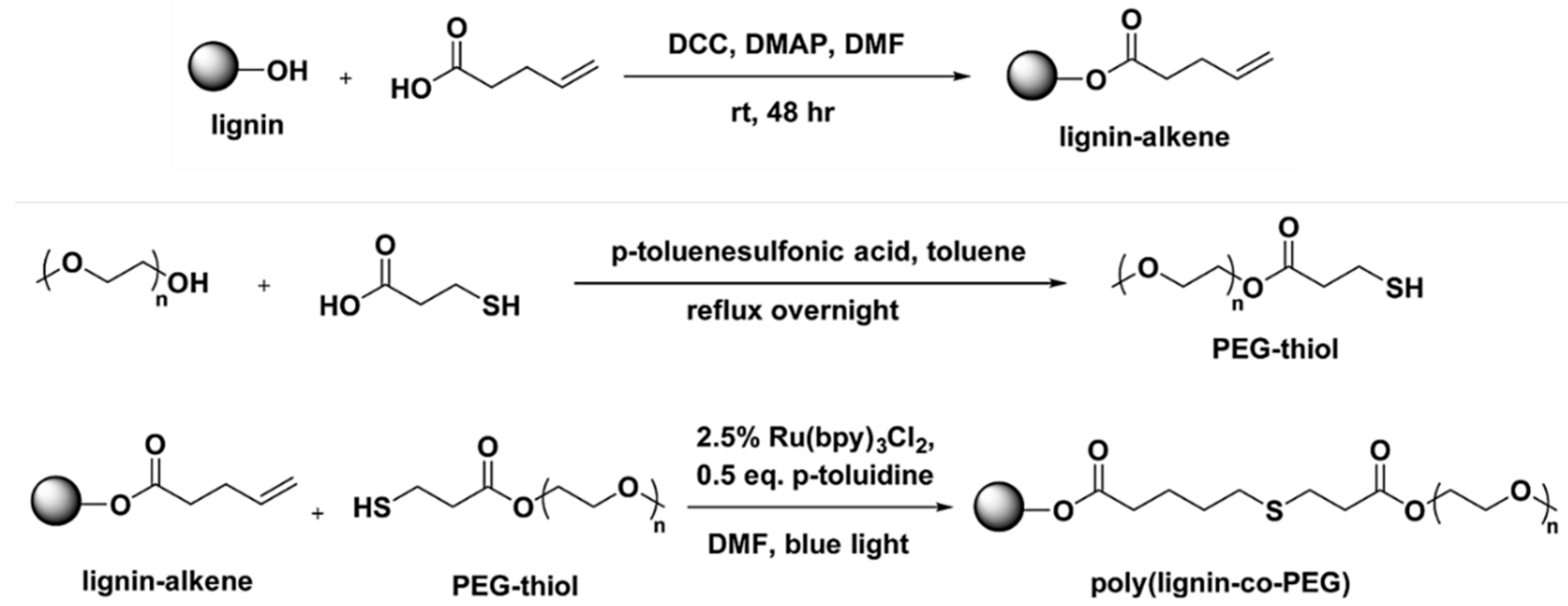
Thiol-Based Reactions
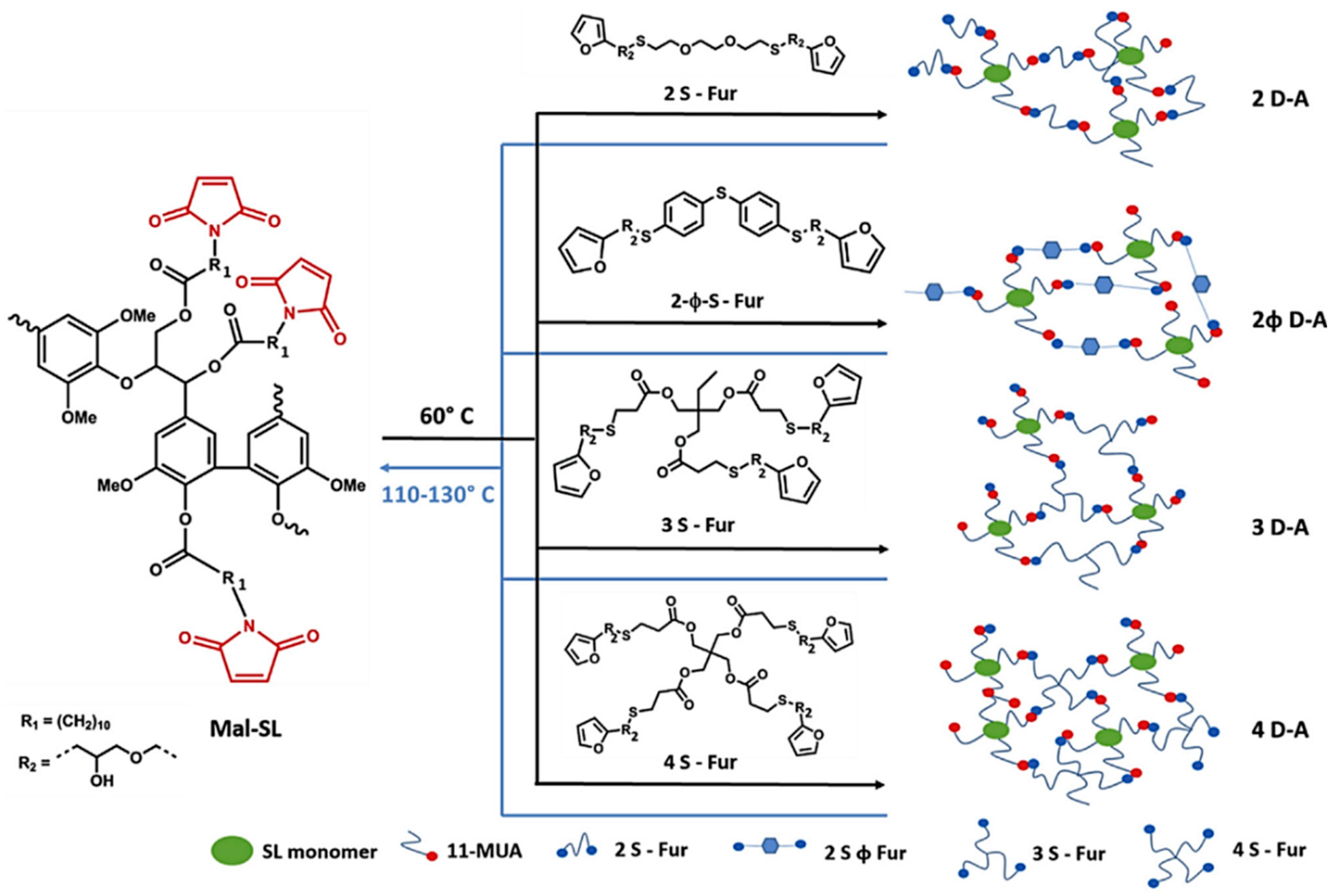
Diels-Alder Reaction
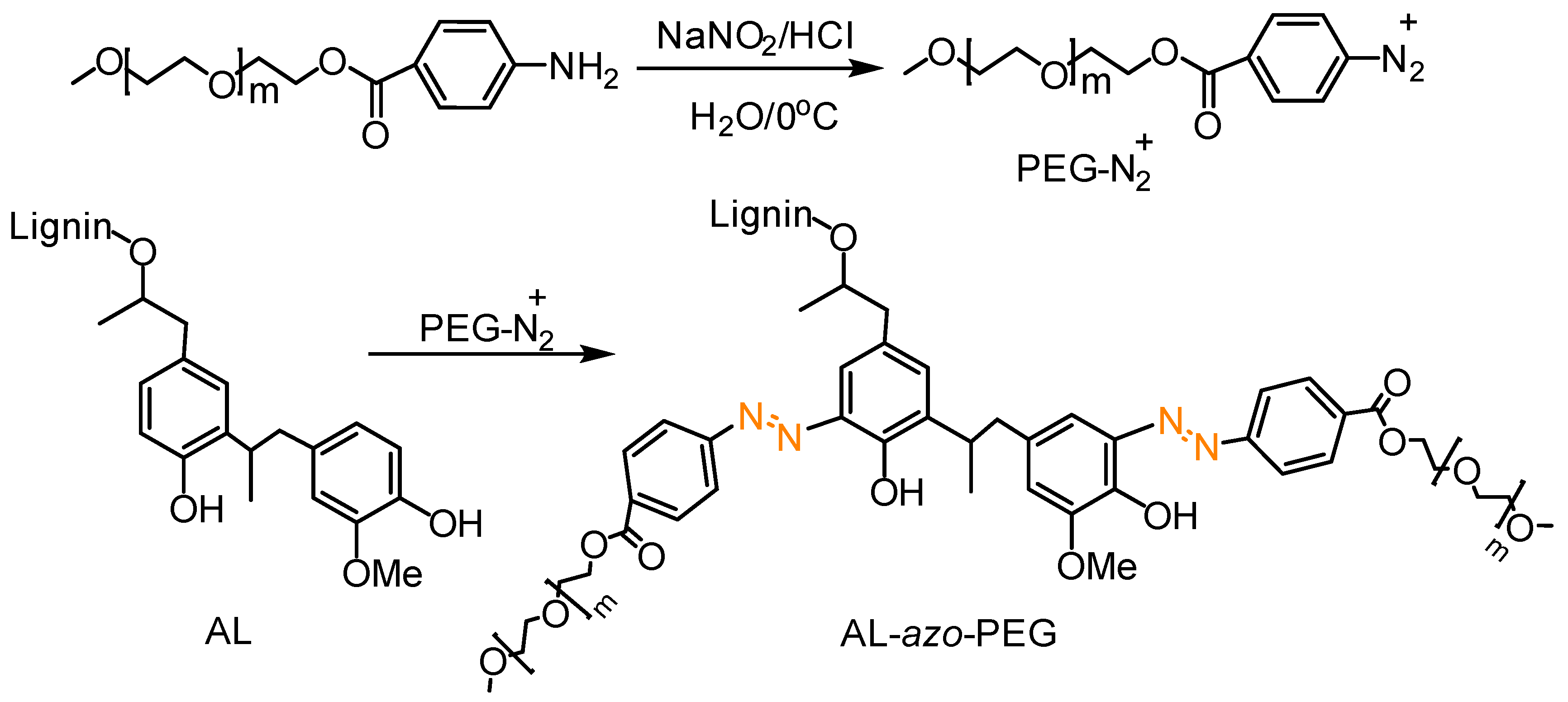
Azo Coupling Reactions
Etherification
Esterification
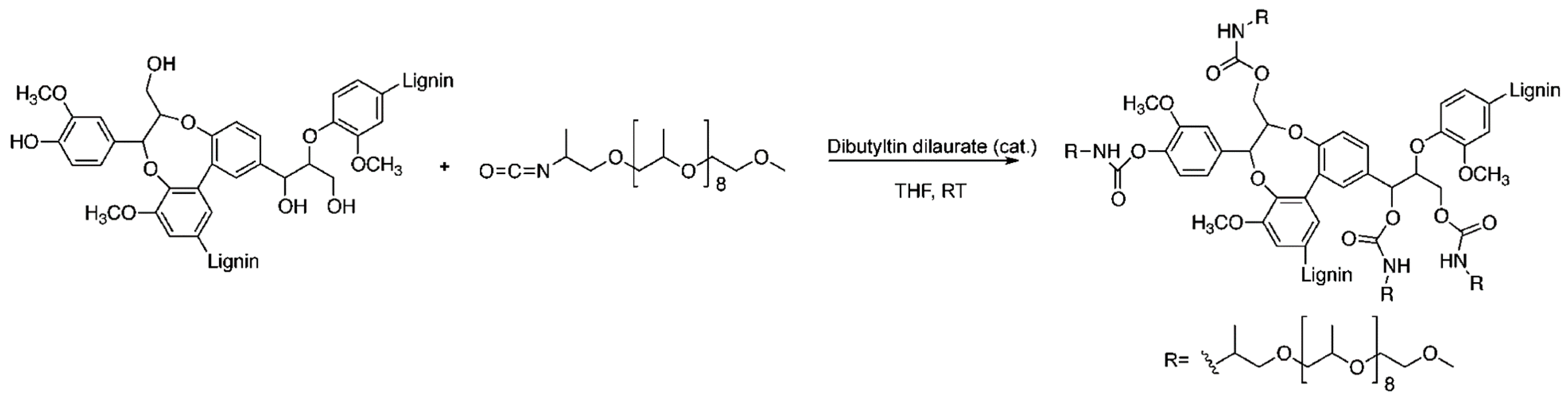
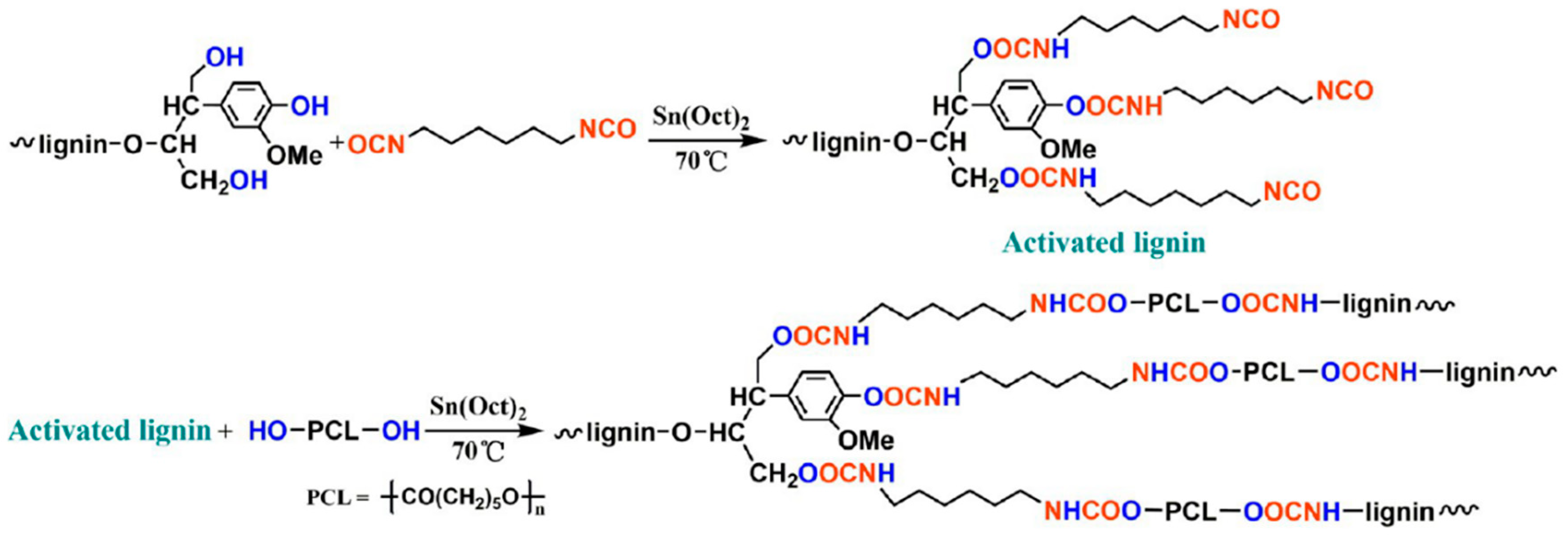
Urethane Linkages
6. Conclusions and Future Prospects
Acknowledgments
Conflicts of Interest
Abbreviations
| ADMET | Acyclic diene metathesis |
| AGET ATRP | Activators generated by electron transfer atom transfer radical polymerization |
| AIBN | Azobisisobutyronitrile |
| ATRP | Atom transfer radical polymerization |
| BBL | Biobutanol lignin |
| BiBB | 2-Bromoisobutyryl bromide |
| CTA | Chain transfer agent |
| CRP | Controlled radical polymerization |
| DEAEMA | 2-(Diethylamino)ethyl methacrylate |
| DMAEMA | 2-(Dimethylamino)ethyl methacrylate |
| DMPA | 2,2-Dimethoxy-2-phenylacetophenone |
| FRP | Free radical polymerization |
| HLS | Hardwood lignosulfonates |
| KF-3CR | Kabachnik-Fields three-component reaction |
| LCST | Lower critical solution temperature |
| LMC | Lignin model compound |
| LP | Lignophenol |
| MCR | Multi-component reaction |
| NIPAM | N-isopropylacrylamide |
| NMP | Nitroxide mediated polymerization |
| PAA | Poly(5-acetylaminopentyl acrylate) |
| PC | Polycarbonate |
| PCL | Polycaprolactone |
| PEG | Poly(ethylene glycol) |
| PET | Poly(ethylene terephthalate) |
| PHA | Polyhydroxyalkanoate |
| PHB | Poly(3-hydroxybutyrate) |
| PLLA | Poly(l-lactide) |
| PNIPAAm | Poly(N-isopropylacrylamide) |
| PPG | Poly(propylene glycol) |
| PSA | Pressure-sensitive adhesives |
| PSt | Polystyrene |
| PVC | Poly(vinyl chloride) |
| PVP | Polyvinylpyrrolidone |
| RAFT | Reversible addition fragmentation chain transfer |
| ROMP | Ring-opening metathesis polymerization |
| ROP | Ring-opening polymerization |
| SI-ATRP | Surface-initiated ATRP |
| SLS | Softwood lignosulfonates |
| TAAC | Thermal azide−alkyne cycloaddition reaction |
| TBD | Triazabicyclodecene |
| TGA | Thermal gravimetric analysis |
| TPE | Thermoplastic elastomers |
Appendix A



References
- Weng, J.K.; Chapple, C. The origin and evolution of lignin biosynthesis. New Phytol. 2010, 187, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Ryu, C.Y. Sustainable Polymers from Biomass; Tang, C., Ryu, C.Y., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017. [Google Scholar]
- Cazacu, G.; Capraru, M.; Popa, V.I. Advances concerning lignin utilization in new materials. In Advances in Natural Polymers; Springer: Berlin, Germany, 2013; Volume 18, pp. 255–312. [Google Scholar]
- Ľudmila, H.; Michal, J.; Andrea, Š.; Aleš, H. Lignin, potential products and their market value. Wood Res. 2015, 60, 973–986. [Google Scholar]
- Ragauskas, A.J.; Beckham, G.T.; Biddy, M.J.; Chandra, R.; Chen, F.; Davis, M.F.; Davison, B.H.; Dixon, R.A.; Gilna, P.; Keller, M. Lignin valorization: Improving lignin processing in the biorefinery. Science 2014, 344, 1246843. [Google Scholar] [CrossRef] [PubMed]
- Zion Research. Lignin Market (Lignosulfonates, Kraft Lignin Andothers) for Concrete Additive, Animal Feed, Dye Stuff, and Other Applications: Global Industry Perspective, Comprehensive Analysis and Forecast 2014–2020; Market Research Store: Deerfield Beach, FL, USA, 2015. [Google Scholar]
- Grand View Research. Lignin Market Size, Share & Trends Analysis Report by Product (Ligno-Sulphonates, Kraft, Organosolv), by Application (Macromolecule, Aromatic), by Region (Apac, Mea, North America, Europe), and Segment Forecasts 2019–2025; Research and Markets: Dublin, Ireland, 2019. [Google Scholar]
- State of the Market Report. Finding Untapped Value: Converting Lignin to Higher Value Chemicals; Lux Reasearch: Boston, MA, USA, 2014. [Google Scholar]
- McCoy, M. Has lignin’s time finally come? Chem. Eng. News 2016, 94, 35–37. [Google Scholar]
- Grossman, A.; Wilfred, V. Lignin-based polymers and nanomaterials. Curr. Opin. Biotechnol. 2019, 56, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yuan, L.; Tang, C. Sustainable elastomers from renewable biomass. Acc. Chem. Res. 2017, 50, 1762–1773. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef]
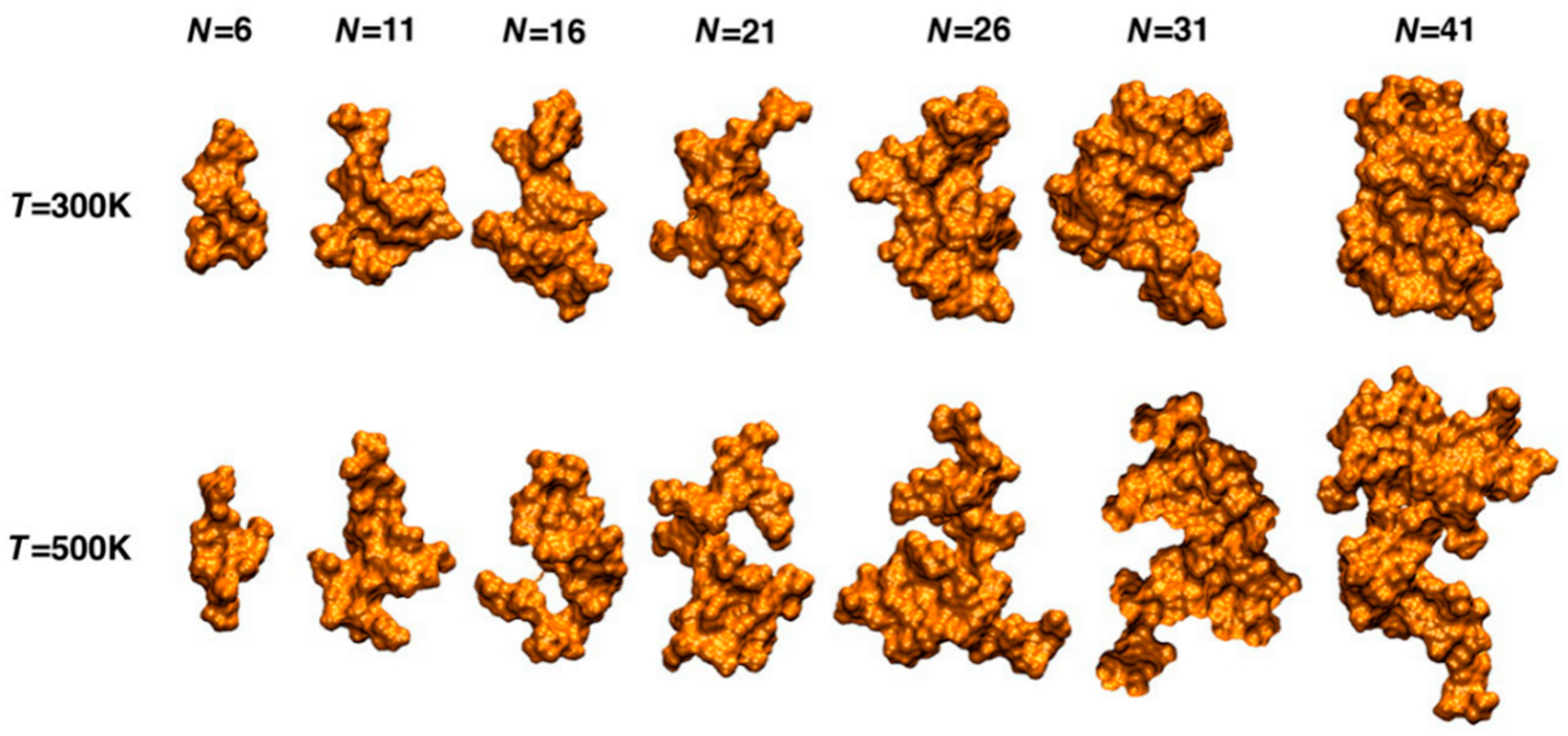
- Petridis, L.; Smith, J.C. Conformations of low-molecular-weight lignin polymers in water. ChemSusChem 2016, 9, 289–295. [Google Scholar] [CrossRef]
- Petridis, L.; Schulz, R.; Smith, J.C. Simulation analysis of the temperature dependence of lignin structure and dynamics. J. Am. Chem. Soc. 2011, 133, 20277–20287. [Google Scholar] [CrossRef]
- Wang, H.; Pu, Y.; Ragauskas, A.; Yang, B. From lignin to valuable products-strategies, challenges, and prospects. Bioresour. Technol. 2019, 271, 449–461. [Google Scholar] [CrossRef]
- Schutyser, W.; Renders, T.; Van den Bosch, S.; Koelewijn, S.-F.; Beckham, G.; Sels, B.F. Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Fridrich, B.; de Santi, A.; Elangovan, S.; Barta, K. Bright side of lignin depolymerization: Toward new platform chemicals. Chem. Rev. 2018, 118, 614–678. [Google Scholar] [CrossRef] [PubMed]
- Kai, D.; Tan, M.J.; Chee, P.L.; Chua, Y.K.; Yap, Y.L.; Loh, X.J. Towards lignin-based functional materials in a sustainable world. Green Chem. 2016, 18, 1175–1200. [Google Scholar] [CrossRef]
- Gillet, S.; Aguedo, M.; Petitjean, L.; Morais, A.; da Costa Lopes, A.; Łukasik, R.; Anastas, P. Lignin transformations for high value applications: Towards targeted modifications using green chemistry. Green Chem. 2017, 19, 4200–4233. [Google Scholar] [CrossRef]
- Figueiredo, P.; Lintinen, K.; Hirvonen, J.T.; Kostiainen, M.A.; Santos, H.A. Properties and chemical modifications of lignin: Towards lignin-based nanomaterials for biomedical applications. Prog. Mater. Sci. 2018, 93, 233–269. [Google Scholar] [CrossRef]
- Chakar, F.S.; Ragauskas, A.J. Review of current and future softwood kraft lignin process chemistry. Ind. Crops Prod. 2004, 20, 131–141. [Google Scholar] [CrossRef]
- Arthur, P.; Keilen, J.J.J.; Drum, L.F. Method of Producing Lignin from Black Liquor. Patent No. US2464828A, 1949. [Google Scholar]
- Tomani, P. The lignoboost process. Cell. Chem. Technol. 2010, 44, 53. [Google Scholar]
- Kouisni, L.; Holt-Hindle, P.; Maki, K.; Paleologou, M. The lignoforce system: A new process for the production of high-quality lignin from black liquor. J. Sci. Technol. For. Prod. Processes 2012, 2, 6–10. [Google Scholar]
- Yinghuai, Z.; Yuanting, K.T.; Hosmane, N.S. Applications of ionic liquids in lignin chemistry. In Ionic Liquids-New Aspects for the Future; Kadokawa, J., Ed.; IntechOpen: London, UK, 2013; pp. 315–346. [Google Scholar]
- Hossain, M.M.; Aldous, L. Ionic liquids for lignin processing: Dissolution, isolation, and conversion. Aust. J. Chem. 2012, 65, 1465–1477. [Google Scholar] [CrossRef]
- Renders, T.; Van den Bosch, S.; Koelewijn, S.-F.; Schutyser, W.; Sels, B. Lignin-first biomass fractionation: The advent of active stabilisation strategies. Energ. Environ. Sci. 2017, 10, 1551–1557. [Google Scholar] [CrossRef]
- Laurichesse, S.; Avérous, L. Chemical modification of lignins: Towards biobased polymers. Prog. Polym. Sci. 2014, 39, 1266–1290. [Google Scholar] [CrossRef]
- Dumont, C.; Gauvin, R.M.; Belva, F.; Sauthier, M. Palladium-catalyzed functionalization of kraft lignin: Ether linkages through the telomerization reaction. ChemSusChem 2018, 11, 1649–1655. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, B.; Li, S.; Sinawang, G.; Wang, X.; He, Y. Synthesis and characterization of photoprocessable lignin-based azo polymer. ACS Sustain. Chem. Eng. 2016, 4, 4036–4042. [Google Scholar] [CrossRef]
- Deng, Y.; Liu, Y.; Qian, Y.; Zhang, W.; Qiu, X. Preparation of photoresponsive azo polymers based on lignin, a renewable biomass resource. ACS Sustain. Chem. Eng. 2015, 3, 1111–1116. [Google Scholar] [CrossRef]
- Kent, M.S.; Zeng, J.; Rader, N.; Avina, I.C.; Simoes, C.T.; Brenden, C.K.; Busse, M.L.; Watt, J.; Giron, N.H.; Alam, T.M. Efficient conversion of lignin into a water-soluble polymer by a chelator-mediated Fenton reaction: Optimization of H2O2 use and performance as a dispersant. Green Chem. 2018, 20, 3024–3037. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Xia, J. Atom transfer radical polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef] [PubMed]
- Hawker, C.J.; Bosman, A.W.; Harth, E. New polymer synthesis by nitroxide mediated living radical polymerizations. Chem. Rev. 2001, 101, 3661–3688. [Google Scholar] [CrossRef] [PubMed]
- Chiefari, J.; Chong, Y.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.; Mayadunne, R.T.; Meijs, G.F.; Moad, C.L.; Moad, G. Living free-radical polymerization by reversible addition−fragmentation chain transfer: The RAFT process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Bielawski, C.W.; Grubbs, R.H. Living ring-opening metathesis polymerization. Prog. Polym. Sci. 2007, 32, 1–29. [Google Scholar] [CrossRef]
- Atallah, P.; Wagener, K.B.; Schulz, M.D. ADMET: The Future Revealed. Macromolecules 2013, 46, 4735–4741. [Google Scholar] [CrossRef]
- Klapiszewski, Ł.; Szalaty, T.J.; Jesionowski, T. Depolymerization and Activation of Lignin: Current State of Knowledge and Perspectives. In Lignin-Trends and Applications; Poletto, M., Ed.; IntechOpen: London, UK, 2017; pp. 1–27. [Google Scholar]
- Da Silva, E.B.; Zabkova, M.; Araújo, J.; Cateto, C.; Barreiro, M.; Belgacem, M.; Rodrigues, A. An integrated process to produce vanillin and lignin-based polyurethanes from Kraft lignin. Chem. Eng. Res. Des. 2009, 87, 1276–1292. [Google Scholar] [CrossRef]
- Fache, M.; Boutevin, B.; Caillol, S. Vanillin, a key-intermediate of biobased polymers. Eur. Polym. J. 2015, 68, 488–502. [Google Scholar] [CrossRef]
- Llevot, A.; Grau, E.; Carlotti, S.; Grelier, S.; Cramail, H. From lignin-derived aromatic compounds to novel biobased polymers. Macromol. Rapid Commun. 2016, 37, 9–28. [Google Scholar] [CrossRef] [PubMed]
- Kristufek, S.L.; Wacker, K.T.; Tsao, Y.-Y.T.; Su, L.; Wooley, K.L. Monomer design strategies to create natural product-based polymer materials. Nat. Prod. Rep. 2017, 34, 433–459. [Google Scholar] [CrossRef] [PubMed]
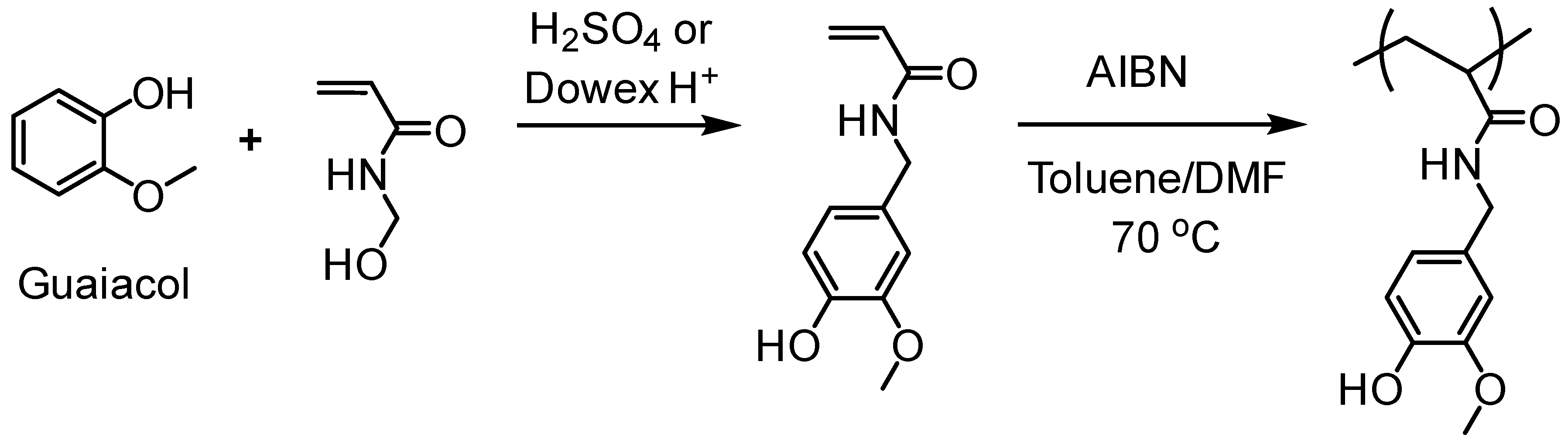
- Liu, H.; Lepoittevin, B.; Roddier, C.; Guerineau, V.; Bech, L.; Herry, J.-M.; Bellon-Fontaine, M.-N.; Roger, P. Facile synthesis and promising antibacterial properties of a new guaiacol-based polymer. Polymer 2011, 52, 1908–1916. [Google Scholar] [CrossRef]
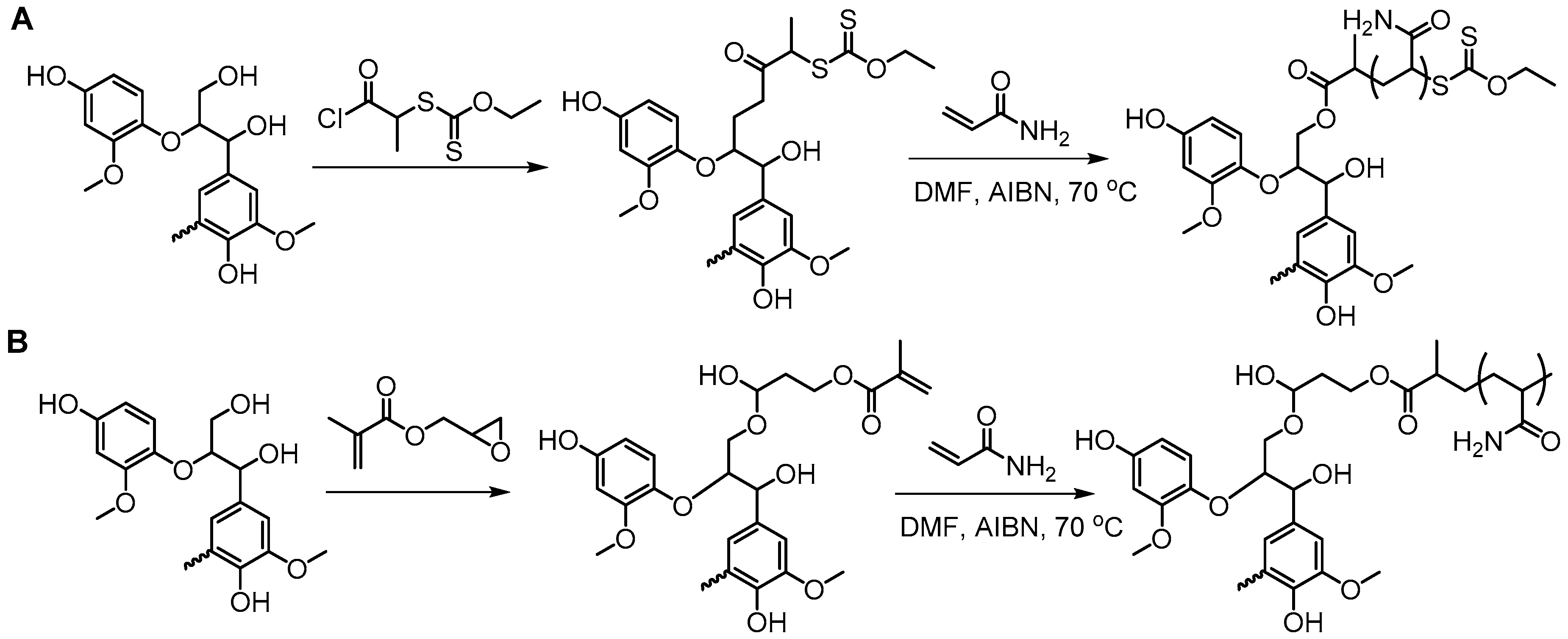
- Holmberg, A.L.; Stanzione, J.F., III; Wool, R.P.; Epps, T.H., III. A facile method for generating designer block copolymers from functionalized lignin model compounds. ACS Sustain. Chem. Eng. 2014, 2, 569–573. [Google Scholar] [CrossRef]
- Holmberg, A.L.; Karavolias, M.G.; Epps, T.H. RAFT polymerization and associated reactivity ratios of methacrylate-functionalized mixed bio-oil constituents. Polym. Chem. 2015, 6, 5728–5739. [Google Scholar] [CrossRef]
- Holmberg, A.L.; Nguyen, N.A.; Karavolias, M.G.; Reno, K.H.; Wool, R.P.; Epps, T.H., III. Softwood lignin-based methacrylate polymers with tunable thermal and viscoelastic properties. Macromolecules 2016, 49, 1286–1295. [Google Scholar] [CrossRef]
- Holmberg, A.L.; Reno, K.H.; Nguyen, N.A.; Wool, R.P.; Epps, T.H., III. Syringyl methacrylate, a hardwood lignin-based monomer for high-T g polymeric materials. ACS Macro Lett. 2016, 5, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Emerson, J.A.; Garabedian, N.T.; Burris, D.L.; Furst, E.M.; Epps, T.H., III. Exploiting feedstock diversity to tune the chemical and tribological properties of lignin-inspired polymer coatings. ACS Sustain. Chem. Eng. 2018, 6, 6856–6866. [Google Scholar] [CrossRef]
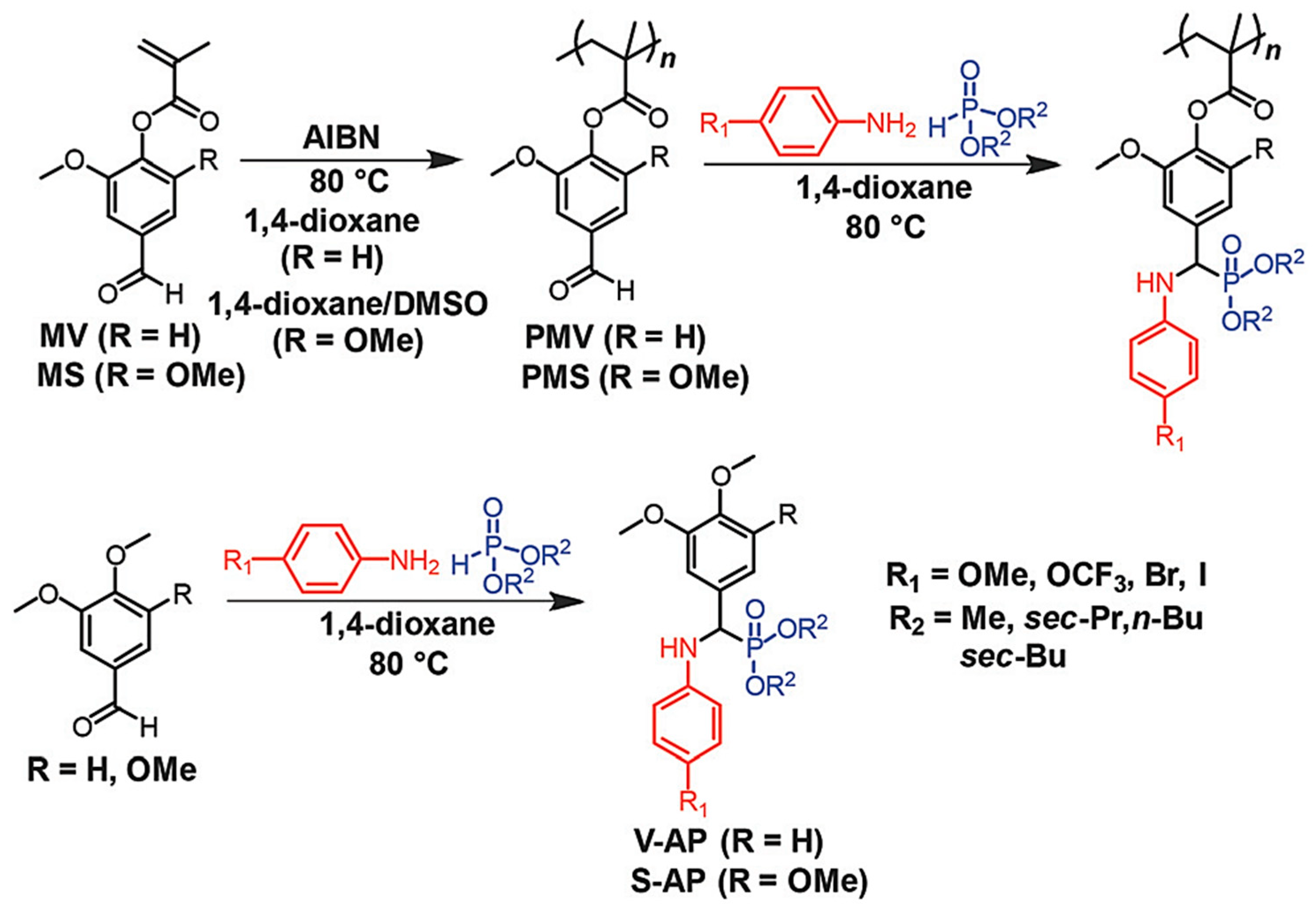
- Wang, S.; Shuai, L.; Saha, B.; Vlachos, D.G.; Epps, T.H., III. From tree to tape: Direct synthesis of pressure sensitive adhesives from depolymerized raw lignocellulosic biomass. ACS Cent. Sci. 2018, 4, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Kakuchi, R.; Yoshida, S.; Sasaki, T.; Kanoh, S.; Maeda, K. Multi-component post-polymerization modification reactions of polymers featuring lignin-model compounds. Polym. Chem. 2018, 9, 2109–2115. [Google Scholar] [CrossRef]
- Firdaus, M.; Meier, M.A. Renewable co-polymers derived from vanillin and fatty acid derivatives. Eur. Polym. J. 2013, 49, 156–166. [Google Scholar] [CrossRef]
- Barbara, I.; Flourat, A.L.; Allais, F. Renewable polymers derived from ferulic acid and biobased diols via ADMET. Eur. Polym. J. 2015, 62, 236–243. [Google Scholar] [CrossRef]
- Hollande, L.; Jaufurally, A.S.; Ducrot, P.-H.; Allais, F. ADMET polymerization of biobased monomers deriving from syringaresinol. RSC Adv. 2016, 6, 44297–44304. [Google Scholar] [CrossRef]
- Llevot, A.; Grau, E.; Carlotti, S.; Grelier, S.; Cramail, H. ADMET polymerization of bio-based biphenyl compounds. Polym. Chem. 2015, 6, 7693–7700. [Google Scholar] [CrossRef]
- Vlaminck, L.; Lingier, S.; Hufendiek, A.; Du Prez, F.E. Lignin inspired phenolic polyethers synthesized via ADMET: Systematic structure-property investigation. Eur. Polym. J. 2017, 95, 503–513. [Google Scholar] [CrossRef]
- Liu, H.; Chung, H. Lignin-based polymers via graft copolymerization. J. Polym. Sci. A 2017, 55, 3515–3528. [Google Scholar] [CrossRef]
- Feng, C.; Li, Y.; Yang, D.; Hu, J.; Zhang, X.; Huang, X. Well-defined graft copolymers: From controlled synthesis to multipurpose applications. Chem. Soc. Rev. 2011, 40, 1282–1295. [Google Scholar] [CrossRef]
- Koshijima, T.; Muraki, E. Radical grafting on lignin. Part I. Radiation-induced grafting of styrene onto hydrochloric acid lignin. J. Polym. Sci. A 1968, 6, 1431–1440. [Google Scholar] [CrossRef]
- Lu, F.J.; Chu, L.H.; Gau, R.J. Free radical-scavenging properties of lignin. Nutr. Cancer 1998, 30, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Mai, C.; Milstein, O.; Hüttermann, A. Fungal laccase grafts acrylamide onto lignin in presence of peroxides. Appl. Microbiol. Biotechnol. 1999, 51, 527–531. [Google Scholar] [CrossRef]
- Mai, C.; Milstein, O.; Hüttermann, A. Chemoenzymatical grafting of acrylamide onto lignin. J. Biotechnol. 2000, 79, 173–183. [Google Scholar] [CrossRef]
- Ye, D.Z.; Jiang, L.; Ma, C.; Zhang, M.H.; Zhang, X. The graft polymers from different species of lignin and acrylic acid: Synthesis and mechanism study. Int. J. Biol. Macromol. 2014, 63, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Zong, E.; Liu, X.; Liu, L.; Wang, J.; Song, P.; Ma, Z.; Ding, J.; Fu, S. Graft Polymerization of Acrylic Monomers onto Lignin with CaCl2-H2O2 as Initiator: Preparation, Mechanism, Characterization, and Application in Poly (lactic acid). ACS Sustain. Chem. Eng. 2017, 6, 337–348. [Google Scholar] [CrossRef]
- Wang, S.; Kong, F.; Gao, W.; Fatehi, P. Novel process for generating cationic lignin based flocculant. Ind. Eng. Chem. Res. 2018, 57, 6595–6608. [Google Scholar] [CrossRef]
- Price, J.T.; Gao, W.; Fatehi, P. Lignin–g–poly (acrylamide)–g–poly (diallyldimethyl-ammonium chloride): Synthesis, Characterization and Applications. ChemistryOpen 2018, 7, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Du, L.; Wu, W.; Xu, H.; Zhang, G.; Li, S.; Wang, C.; Lu, Z.; Deng, Y. Novel lignin-derived water-soluble binder for micro silicon anode in lithium-ion batteries. ACS Sustain. Chem. Eng. 2018, 6, 12621–12629. [Google Scholar] [CrossRef]
- Hu, L.-Q.; Dai, L.; Liu, R.; Si, C.-L. Lignin–graft–poly (acrylic acid) for enhancement of heavy metal ion biosorption. J. Mater. Sci. 2017, 52, 13689–13699. [Google Scholar] [CrossRef]
- Jeyaraj, M.; Praphakar, R.A.; Rajendran, C.; Ponnamma, D.; Sadasivuni, K.K.; Munusamy, M.A.; Rajan, M. Surface functionalization of natural lignin isolated from Aloe barbadensis Miller biomass by atom transfer radical polymerization for enhanced anticancer efficacy. RSC Adv. 2016, 6, 51310–51319. [Google Scholar] [CrossRef]
- Liu, X.; Wang, J.; Li, S.; Zhuang, X.; Xu, Y.; Wang, C.; Chu, F. Preparation and properties of UV-absorbent lignin graft copolymer films from lignocellulosic butanol residue. Ind. Crops Prod. 2014, 52, 633–641. [Google Scholar] [CrossRef]
- Ren, Y.; Luo, Y.; Zhang, K.; Zhu, G.; Tan, X. Lignin terpolymer for corrosion inhibition of mild steel in 10% hydrochloric acid medium. Corros. Sci. 2008, 50, 3147–3153. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Tsarevsky, N.V. Nanostructured functional materials prepared by atom transfer radical polymerization. Nat. Chem. 2009, 1, 276. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Kadla, J.F. Preparation of a thermoresponsive lignin-based biomaterial through atom transfer radical polymerization. Biomacromolecules 2010, 11, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yao, K.; Korich, A.L.; Li, S.; Ma, S.; Ploehn, H.J.; Iovine, P.M.; Wang, C.; Chu, F.; Tang, C. Combining renewable gum rosin and lignin: Towards hydrophobic polymer composites by controlled polymerization. J. Polym. Sci. A 2011, 49, 3728–3738. [Google Scholar] [CrossRef]
- Yu, J.; Wang, J.; Wang, C.; Liu, Y.; Xu, Y.; Tang, C.; Chu, F. UV-Absorbent Lignin-Based Multi-Arm Star Thermoplastic Elastomers. Macromol. Rapid Commun. 2015, 36, 398–404. [Google Scholar] [CrossRef]
- Gao, G.; Dallmeyer, J.I.; Kadla, J.F. Synthesis of lignin nanofibers with ionic-responsive shells: Water-expandable lignin-based nanofibrous mats. Biomacromolecules 2012, 13, 3602–3610. [Google Scholar] [CrossRef]
- Hilburg, S.L.; Elder, A.N.; Chung, H.; Ferebee, R.L.; Bockstaller, M.R.; Washburn, N.R. A universal route towards thermoplastic lignin composites with improved mechanical properties. Polymers 2014, 55, 995–1003. [Google Scholar] [CrossRef]
- Shah, T.; Gupta, C.; Ferebee, R.L.; Bockstaller, M.R.; Washburn, N.R. Extraordinary toughening and strengthening effect in polymer nanocomposites using lignin-based fillers synthesized by ATRP. Polymer 2015, 72, 406–412. [Google Scholar] [CrossRef]
- Diao, B.; Zhang, Z.; Zhu, J.; Li, J. Biomass-based thermogelling copolymers consisting of lignin and grafted poly (N-isopropylacrylamide), poly (ethylene glycol), and poly (propylene glycol). RSC Adv. 2014, 4, 42996–43003. [Google Scholar] [CrossRef]
- Kai, D.; Low, Z.W.; Liow, S.S.; Abdul Karim, A.; Ye, H.; Jin, G.; Li, K.; Loh, X.J. Development of lignin supramolecular hydrogels with mechanically responsive and self-healing properties. ACS Sustain. Chem. Eng. 2015, 3, 2160–2169. [Google Scholar] [CrossRef]
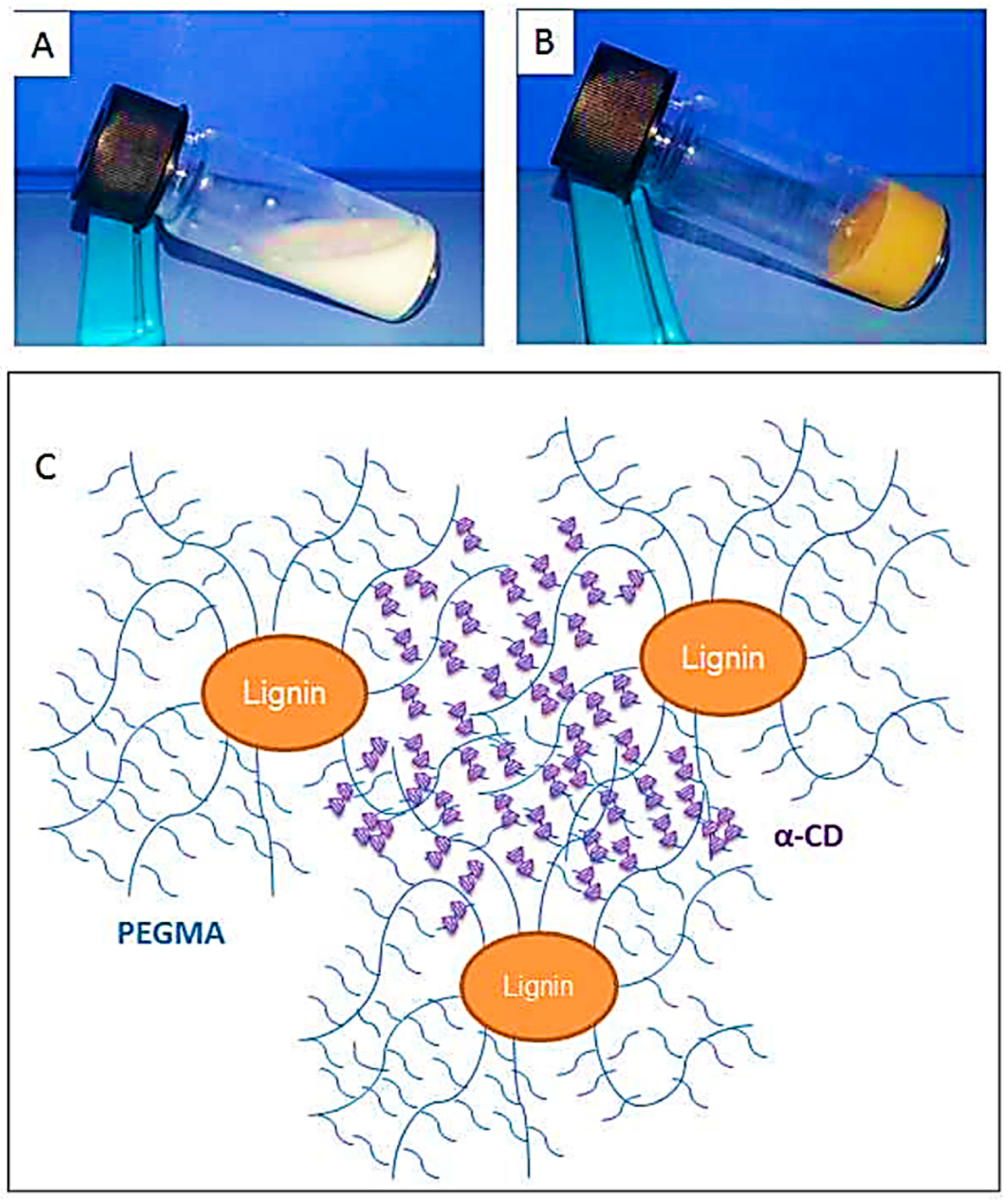
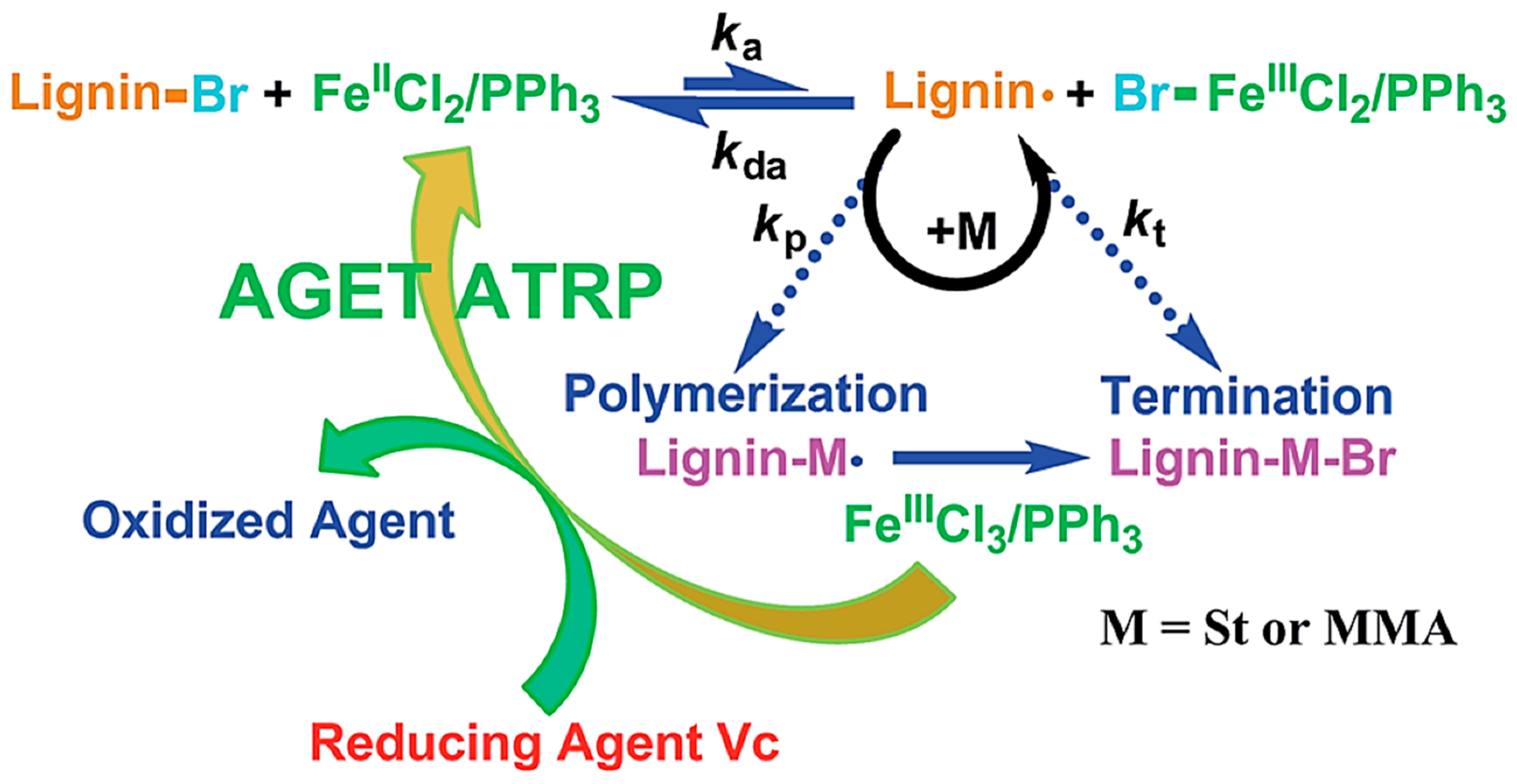
- Li, H.; Pang, Z.; Gao, P.; Wang, L. Fe (III)-catalyzed grafting copolymerization of lignin with styrene and methyl methacrylate through AGET ATRP using triphenyl phosphine as a ligand. RSC Adv. 2015, 5, 54387–54394. [Google Scholar] [CrossRef]
- Liu, X.; Yin, H.; Zhang, Z.; Diao, B.; Li, J. Functionalization of lignin through ATRP grafting of poly (2-dimethylaminoethyl methacrylate) for gene delivery. Colloids Surf. B. Biointerfaces 2015, 125, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Zhang, Q.; Qiu, X.; Zhu, S. CO2-responsive diethylaminoethyl-modified lignin nanoparticles and their application as surfactants for CO2/N2-switchable Pickering emulsions. Green Chem. 2014, 16, 4963–4968. [Google Scholar] [CrossRef]
- Jakubowski, W.; Matyjaszewski, K. activator generated by electron transfer for atom transfer radical polymerization. Macromolecules 2005, 38, 4139–4146. [Google Scholar] [CrossRef]
- Min, K.; Gao, H.; Matyjaszewski, K. Preparation of homopolymers and block copolymers in miniemulsion by ATRP using activators generated by electron transfer (AGET). J. Am. Chem. Soc. 2005, 127, 3825–3830. [Google Scholar] [CrossRef]
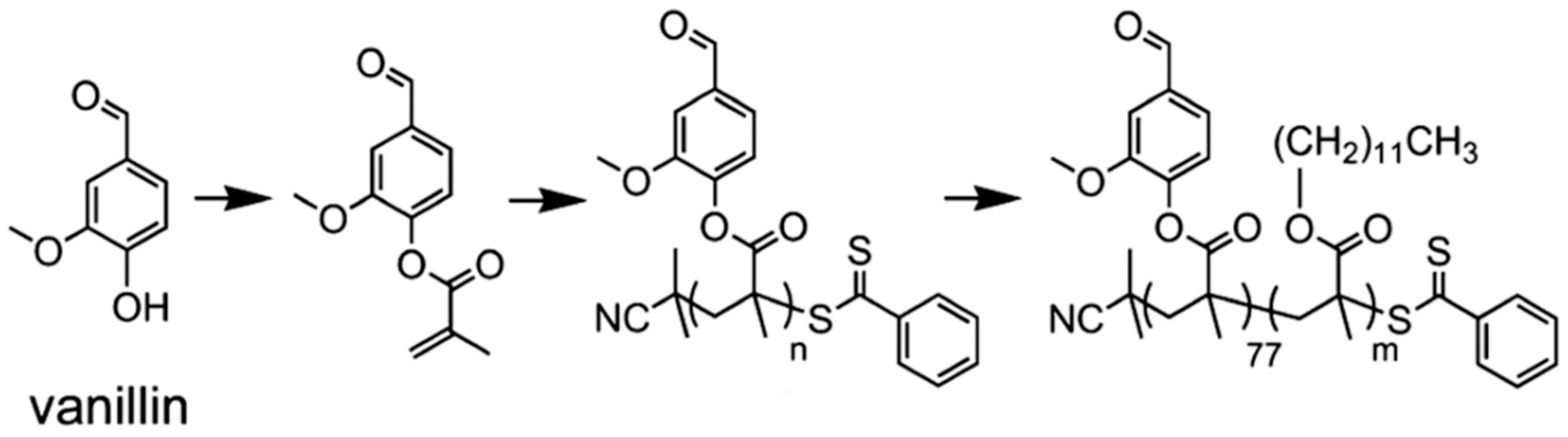
- Gupta, C.; Washburn, N.R. Polymer-grafted lignin surfactants prepared via reversible addition–fragmentation chain-transfer polymerization. Langmuir 2014, 30, 9303–9312. [Google Scholar] [CrossRef] [PubMed]
- Silmore, K.S.; Gupta, C.; Washburn, N.R. Tunable Pickering emulsions with polymer-grafted lignin nanoparticles (PGLNs). J. Colloid Interface Sci. 2016, 466, 91–100. [Google Scholar] [CrossRef]
- Gupta, C.; Nadelman, E.; Washburn, N.R.; Kurtis, K.E. Lignopolymer Superplasticizers for Low-CO2 Cements. ACS Sustain. Chem. Eng. 2017, 5, 4041–4049. [Google Scholar] [CrossRef]
- Gupta, C.; Sverdlove, M.J.; Washburn, N.R. Molecular architecture requirements for polymer-grafted lignin superplasticizers. Soft Matter 2015, 11, 2691–2699. [Google Scholar] [CrossRef]
- Xu, Y.; Yuan, L.; Wang, Z.; Wilbon, P.A.; Wang, C.; Chu, F.; Tang, C. Lignin and soy oil-derived polymeric biocomposites by “grafting from” RAFT polymerization. Green Chem. 2016, 18, 4974–4981. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, X.; Xie, J.; Feng, B.; Han, Q. Synthesis of a novel tunable lignin-based star copolymer and its flocculation performance in the treatment of kaolin suspension. Sep. Purif. Technol. 2019, 210, 355–363. [Google Scholar] [CrossRef]
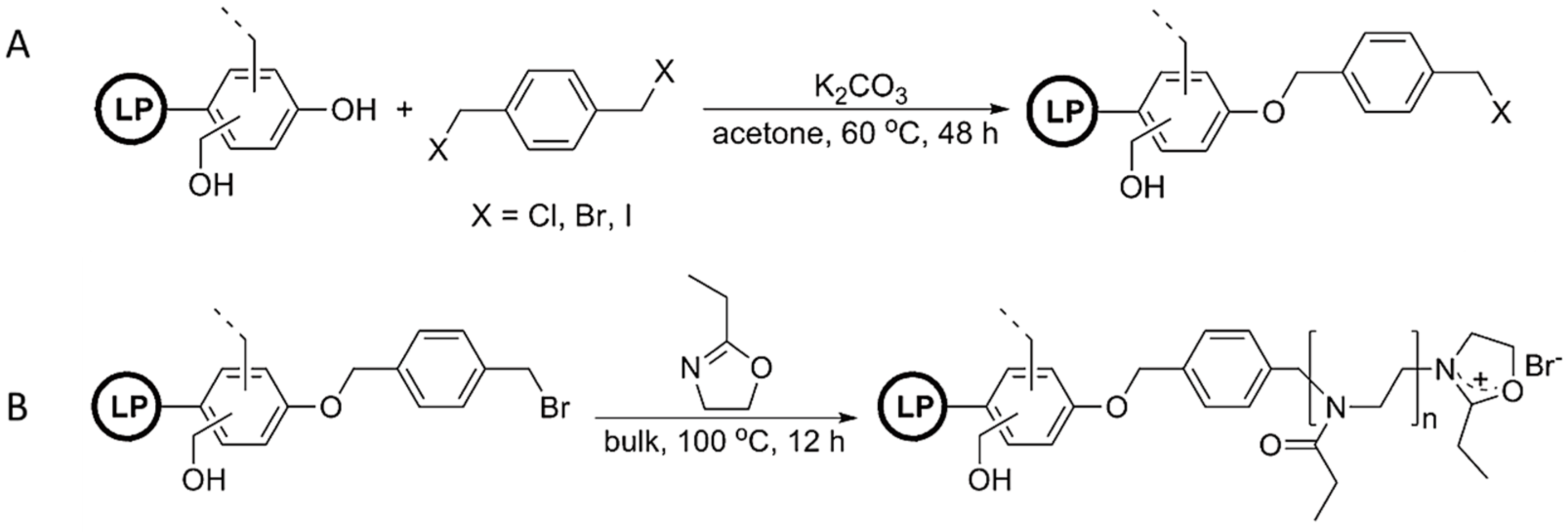
- Nemoto, T.; Konishi, G.I.; Tojo, Y.; An, Y.C.; Funaoka, M. Functionalization of lignin: Synthesis of lignophenol–graft–poly (2-ethyl-2-oxazoline) and its application to polymer blends with commodity polymers. J. Appl. Polym. Sci. 2012, 123, 2636–2642. [Google Scholar] [CrossRef]
- Mahata, D.; Jana, M.; Jana, A.; Mukherjee, A.; Mondal, N.; Saha, T.; Sen, S.; Nando, G.B.; Mukhopadhyay, C.K.; Chakraborty, R. Lignin-graft-polyoxazoline conjugated triazole a novel anti-infective ointment to control persistent inflammation. Sci. Rep. 2017, 7, 46412. [Google Scholar] [CrossRef]
- Chung, Y.-L.; Olsson, J.V.; Li, R.J.; Frank, C.W.; Waymouth, R.M.; Billington, S.L.; Sattely, E.S. A Renewable lignin-lactide copolymer and application in biobased composites. ACS Sustain. Chem. Eng. 2013, 1, 1231–1238. [Google Scholar] [CrossRef]
- Liu, X.; Zong, E.; Jiang, J.; Fu, S.; Wang, J.; Xu, B.; Li, W.; Lin, X.; Xu, Y.; Wang, C. Preparation and characterization of Lignin–graft–poly (ε-caprolactone) copolymers based on lignocellulosic butanol residue. Int. J. Biol. Macromol. 2015, 81, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yang, L.; Lu, X.; He, C. Biodegradable and renewable poly (lactide)-lignin composites: Synthesis, interface and toughening mechanism. J. Mater. Chem. A 2015, 3, 3699–3709. [Google Scholar] [CrossRef]
- Kai, D.; Zhang, K.; Jiang, L.; Wong, H.Z.; Li, Z.; Zhang, Z.; Loh, X.J. Sustainable and antioxidant lignin–polyester copolymers and nanofibers for potential healthcare applications. ACS Sustain. Chem. Eng. 2017, 5, 6016–6025. [Google Scholar] [CrossRef]
- Kai, D.; Zhang, K.; Liow, S.S.; Loh, X.J. New dual functional phb-grafted lignin copolymer: Synthesis, mechanical properties, and biocompatibility studies. ACS Appl. Bio Mater. 2018, 2, 127–134. [Google Scholar] [CrossRef]
- Schmidt, B.V.; Molinari, V.; Esposito, D.; Tauer, K.; Antonietti, M. Lignin-based polymeric surfactants for emulsion polymerization. Polymer 2017, 112, 418–426. [Google Scholar] [CrossRef]
- Pérez–Camargo, R.A.; Saenz, G.; Laurichesse, S.; Casas, M.T.; Puiggalí, J.; Avérous, L.; Müller, A.J. Nucleation, crystallization, and thermal fractionation of poly (ε-caprolactone)-grafted-lignin: Effects of grafted chains length and lignin content. J. Polym. Sci. Part B: Polym. Phys. 2015, 53, 1736–1750. [Google Scholar]
- Washburn, N.R.; Chung, H. Lignin-Containing Polymers and Compositions Including Lignin-Containing Polymers. Patent No. US9914870B2, 2018. [Google Scholar]
- Pribyl, J.; Benicewicz, B.; Bell, M.; Wagener, K.; Ning, X.; Schadler, L.; Jimenez, A.; Kumar, S. Polyethylene grafted silica nanoparticles prepared via surface-initiated ROMP. ACS Macro Lett. 2019, 8, 228–232. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Du Prez, F.E.; Espeel, P.; Hawker, C.J.; Junkers, T.; Schlaad, H.; Van Camp, W. “Clicking” polymers or just efficient linking: What is the difference? Angew. Chem. Int. Ed. 2011, 50, 60–62. [Google Scholar] [CrossRef] [PubMed]
- Golas, P.L.; Matyjaszewski, K. Marrying click chemistry with polymerization: Expanding the scope of polymeric materials. Chem. Soc. Rev. 2010, 39, 1338–1354. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Yuan, L.; Li, G.; Huang, L.; Qin, T.; Chu, F.; Tang, C. Renewable polymers from lignin via copper-free thermal click chemistry. Polymer 2016, 83, 92–100. [Google Scholar] [CrossRef]
- Yuan, L.; Zhang, Y.; Wang, Z.; Han, Y.; Tang, C. Plant oil and lignin-derived elastomers via thermal azide-alkyne cycloaddition click chemistry. ACS Sustain. Chem. Eng. 2018. [Google Scholar] [CrossRef]
- Liu, H.; Chung, H. Self-healing properties of lignin-containing nanocomposite: Synthesis of lignin-graft-poly(5-acetylaminopentyl acrylate) via RAFT and click chemistry. Macromolecules 2016, 49, 7246–7256. [Google Scholar] [CrossRef]
- Panovic, I.; Montgomery, J.R.; Lancefield, C.S.; Puri, D.; Lebl, T.; Westwood, N.J. Grafting of Technical Lignins through Regioselective Triazole Formation on β-O-4 Linkages. ACS Sustain. Chem. Eng. 2017, 5, 10640–10648. [Google Scholar] [CrossRef]
- Lowe, A.B. Thiol-ene “click” reactions and recent applications in polymer and materials synthesis. Polym. Chem. 2010, 1, 17–36. [Google Scholar] [CrossRef]
- Nair, D.P.; Podgórski, M.; Chatani, S.; Gong, T.; Xi, W.; Fenoli, C.R.; Bowman, C.N. The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry. Chem. Mater. 2014, 26, 724–744. [Google Scholar] [CrossRef]
- Jin, C.; Zhang, X.; Xin, J.; Liu, G.; Chen, J.; Wu, G.; Liu, T.; Zhang, J.; Kong, Z. Thiol-ene synthesis of cysteine-functionalized lignin for the enhanced adsorption of Cu (II) and Pb (II). Ind. Eng. Chem. Res. 2018, 57, 7872–7880. [Google Scholar] [CrossRef]
- Jawerth, M.; Johansson, M.; Lundmark, S.; Gioia, C.; Lawoko, M. Renewable thiol–ene thermosets based on refined and selectively allylated industrial lignin. ACS Sustain. Chem. Eng. 2017, 5, 10918–10925. [Google Scholar] [CrossRef]
- Liu, H.; Chung, H. Visible-light induced thiol–ene reaction on natural lignin. ACS Sustain. Chem. Eng. 2017, 5, 9160–9168. [Google Scholar] [CrossRef]
- Buono, P.; Duval, A.; Averous, L.; Habibi, Y. Lignin-based materials through thiol-maleimide “click” polymerization. ChemSusChem 2017, 10, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Gandini, A. The furan/maleimide Diels–Alder reaction: A versatile click–unclick tool in macromolecular synthesis. Prog. Polym. Sci. 2013, 38, 1–29. [Google Scholar] [CrossRef]
- Gandini, A.; Carvalho, A.J.; Trovatti, E.; Kramer, R.K.; Lacerda, T.M. Macromolecular materials based on the application of the Diels–Alder reaction to natural polymers and plant oils. Eur. J. Lipid Sci. Technol. 2018, 120, 1700091. [Google Scholar] [CrossRef]
- Buono, P.; Duval, A.; Averous, L.; Habibi, Y. Thermally healable and remendable lignin-based materials through Diels–Alder click polymerization. Polymer 2017, 133, 78–88. [Google Scholar] [CrossRef]
- Xie, S.; Natansohn, A.; Rochon, P. Recent developments in aromatic azo polymers research. Chem. Mater. 1993, 5, 403–411. [Google Scholar] [CrossRef]
- Che, P.; He, Y.; Wang, X. Hyperbranched azo-polymers synthesized by azo-coupling reaction of an AB2 monomer and postpolymerization modification. Macromolecules 2005, 38, 8657–8663. [Google Scholar] [CrossRef]
- Deng, Y.; Zhao, H.; Qian, Y.; Lü, L.; Wang, B.; Qiu, X. Hollow lignin azo colloids encapsulated avermectin with high anti-photolysis and controlled release performance. Ind. Crops Prod. 2016, 87, 191–197. [Google Scholar] [CrossRef]
- Wang, J.; Li, S.; Liang, R.; Wu, B.; He, Y. Synthesis and characterization of water-soluble PEGylated lignin-based polymers by macromolecular azo coupling reaction. Chin. Chem. Lett. 2018, 29, 143–146. [Google Scholar] [CrossRef]
- Perkins, K.M.; Gupta, C.; Charleson, E.N.; Washburn, N.R. Surfactant properties of PEGylated lignins: Anomalous interfacial activities at low grafting density. Colloids Surf. Physicochem. Eng. Aspects 2017, 530, 200–208. [Google Scholar] [CrossRef]
- Lin, X.; Zhou, M.; Wang, S.; Lou, H.; Yang, D.; Qiu, X. Synthesis, Structure, and Dispersion Property of a Novel Lignin-Based Polyoxyethylene Ether from Kraft Lignin and Poly(ethylene glycol). ACS Sustain. Chem. Eng. 2014, 2, 1902–1909. [Google Scholar] [CrossRef]
- Liu, K.; Zheng, D.; Lei, H.; Liu, J.; Lei, J.; Wang, L.; Ma, X. Development of novel lignin-based targeted polymeric nanoparticle platform for efficient delivery of anticancer drugs. ACS Biomater. Sci. Eng. 2018, 4, 1730–1737. [Google Scholar] [CrossRef]
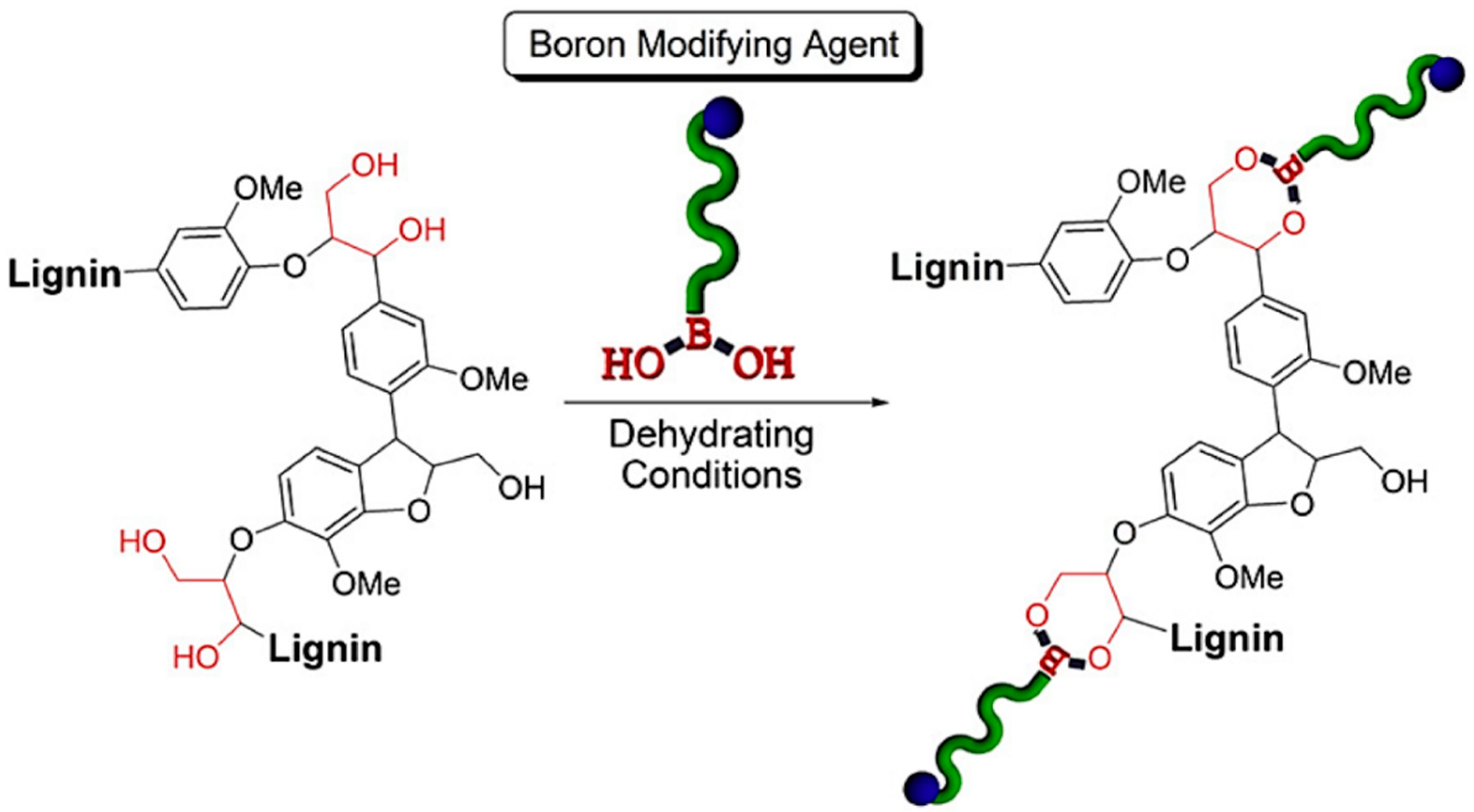
- Brooks, W.L.; Sumerlin, B.S. Synthesis and applications of boronic acid-containing polymers: From materials to medicine. Chem. Rev. 2015, 116, 1375–1397. [Google Scholar] [CrossRef] [PubMed]
- Korich, A.L.; Clarke, K.M.; Wallace, D.; Iovine, P.M. Chemical Modification of a Lignin Model Polymer via Arylboronate Ester Formation under Mild Reaction Conditions. Macromolecules 2009, 42, 5906–5908. [Google Scholar] [CrossRef]
- Korich, A.L.; Fleming, A.B.; Walker, A.R.; Wang, J.; Tang, C.; Iovine, P.M. Chemical modification of organosolv lignin using boronic acid-containing reagents. Polymers 2012, 53, 87–93. [Google Scholar] [CrossRef]
- Khan, T.A.; Lee, J.-H.; Kim, H.-J. Chapter 9-Lignin-Based Adhesives and Coatings. In Lignocellulose for Future Bioeconomy; Ariffin, H., Sapuan, S.M., Hassan, M.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 153–206. [Google Scholar]
- Gandini, A.; Belgacem, M.N.; Guo, Z.-X.; Montanari, S. Lignins as macromonomers for polyesters and polyurethanes. In Chemical Modification, Properties, and Usage of Lignin; Springer: Berlin, Germany, 2002; pp. 57–80. [Google Scholar]
- Zhang, Y.; Liao, J.; Fang, X.; Bai, F.; Qiao, K.; Wang, L. Renewable high-performance polyurethane bioplastics derived from lignin-poly (ε-caprolactone). ACS Sustain. Chem. Eng. 2017, 5, 4276–4284. [Google Scholar] [CrossRef]
- Kalami, S.; Arefmanesh, M.; Master, E.; Nejad, M. Replacing 100% of phenol in phenolic adhesive formulations with lignin. J. Appl. Polym. Sci. 2017, 134, 45124. [Google Scholar] [CrossRef]
- Mahmood, N.; Yuan, Z.; Schmidt, J.; Xu, C. Depolymerization of lignins and their applications for the preparation of polyols and rigid polyurethane foams: A review. Renew. Sustain. Energ. Rev. 2016, 60, 317–329. [Google Scholar] [CrossRef]
- Nguyen, N.A.; Bowland, C.C.; Naskar, A.K. A general method to improve 3D-printability and inter-layer adhesion in lignin-based composites. Appl. Mater. Today 2018, 12, 138–152. [Google Scholar] [CrossRef]
- Nguyen, H.T.H.; Qi, P.; Rostagno, M.; Feteha, A.; Miller, S.A. The quest for high glass transition temperature bioplastics. J. Mater. Chem. A 2018, 6, 9298–9331. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, H.; Deng, J.; Wu, Y. High glass-transition temperature acrylate polymers derived from biomasses, syringaldehyde, and vanillin. Macromol. Chem. Phys. 2016, 217, 2402–2408. [Google Scholar] [CrossRef]
- Vinardell, M.; Mitjans, M. Lignins and their derivatives with beneficial effects on human health. Int. J. Mol. Sci. 2017, 18, 1219. [Google Scholar] [CrossRef] [PubMed]
- Puziy, A.M.; Poddubnaya, O.I.; Sevastyanova, O. Carbon materials from technical lignins: Recent advances. Top. Curr. Chem. 2018, 376, 33. [Google Scholar] [CrossRef] [PubMed]
- Graichen, F.H.; Grigsby, W.J.; Hill, S.J.; Raymond, L.G.; Sanglard, M.; Smith, D.A.; Thorlby, G.J.; Torr, K.M.; Warnes, J.M. Yes, we can make money out of lignin and other bio-based resources. Ind. Crops Prod. 2017, 106, 74–85. [Google Scholar] [CrossRef]
- Fortunati, E.; Yang, W.; Luzi, F.; Kenny, J.; Torre, L.; Puglia, D. Lignocellulosic nanostructures as reinforcement in extruded and solvent casted polymeric nanocomposites: An overview. Eur. Polym. J. 2016, 80, 295–316. [Google Scholar] [CrossRef]








































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extraction Method | Extraction Conditions | Lignin Properties | Remarks |
|---|---|---|---|
| Kraft Pulping Process | Wood chips are digested in aqueous NaOH and Na2S at 150–170 °C for 2 h. This breaks down lignin and solubilizes it. After the cellulose fibers are recovered, lignin is precipitated by lowering the pH of the soap-free black liquor. | Soluble in alkali media and some organic solvents (DMSO, DMF, pyridine) Molecular weight 1000–15,000 g/mol, Đ 2.5–3.5, sulfur 1–3%, ash 0.5–3%, Tg 140–160 °C. | The globally dominant method for isolating lignin from paper pulping waste. It is estimated that more than 20 million tons of kraft lignin are produced in the United States [21]. Mostly sugar-free lignin with some condensed and –SH group attached structures are obtained by this process. All types of wood and non-wood species like bamboo can be used as the substrate for kraft pulping process. Reactive sites are present for sulfonation or other chemistries. However, large volumes of kraft lignin are used as boiler fuel in paper mills. Westvaco (now Ingevity Corporation) developed the initial patented kraft lignin recovery process [22]. More recently, Lignoboost [23] and LingoForce [24] processes are developed that enable integrated lignin isolation. |
| Sulfite Process | 140–170 °C, H2O, metal sulfites (e.g., Na2SO3, NaHSO3, (NH4)2SO3, MgSO3, CaSO3) and sulfur dioxide, 1–5 h | Soluble in water, molecular weight 1000–50,000 g/mol, Đ 6–8, sulfur 4–8%, ash 4–8%, Tg ~ 130 °C. | Lignosulfonates are obtained with highly condensed structures and –SO3 groups. An estimated 1.5 million tons of sulfite lignin is annually produced. Higher in sugar content and impurities. Mostly used as a cement additive. Less control is available over the location of sulfonate groups or the degree of sulfonation. |
| Soda Lignin | 120–170 °C, H2O, NaOH, anthraquinone as a catalyst | Soluble in alkali media, molecular weight 1000–3000 g/mol, Đ 2.5–3.5, sulfur-free, ash 0.7–2.3%, Tg ~ 140 °C. | Soda lignin is sulfur free and has less condensed structures. An estimated |
| Organosolv Lignin | Organic solvents such as alcohol or alcohol/water mixtures, formic acid, and acetic acid. Treated at 170–190 °C. | Soluble in alkali media, molecular weight 500–5000 g/mol, Đ 1.5–2.5, sulfur-free, ash 1.7%, Tg ~ 100 °C. | Organosolv lignin is obtained sulfur-free with relatively high purity. This is a mild process that results in less structural modifications. |
| Lignin Source | Monomers (Representative) | Catalyst system and Conditions | Application | Ref. |
|---|---|---|---|---|
| Kraft lignin from Westvaco Corp (Ingevity Corp) (Charleston, SC) |  | ATRP: CuBr/PMDTA in water/DMF at 50 °C | Thermoresponsive materials | [72] |
| Organosolv lignin, Lignol Corporation |  | ATRP: CuBr/Me6TREN in THF at 65 °C | Hydrophobic polymer composites | [73] |
| Kraft lignin from Westvaco Corp (Ingevity Corp) (Charleston, SC) |  | ATRP: CuBr/PMDTA in water/DMF at 80 °C | Thermoplastic elastomers | [74] |
| Softwood Kraft lignin, Ingevity Corp. (Charleston, SC) |  | SI-ATRP: CuCl/HMTETA in water at room temperature | Ionic-responsive nanofibrous mats | [75] |
| Kraft lignin (alkali), Tokyo Chemical Industry, Co. |  | ATRP: CuCl/bpy or CuBr/PMDETA in DMF at 100 °C | Thermoplastic lignin composites | [76,77] |
| Kraft lignin (alkali), Sigma-Aldrich |  | ATRP: CuBr/HMTETA in 1,4-dioxane at 60–70 °C | Thermogelling copolymers | [78] |
| Kraft lignin (alkali), Shuntai Technology Corp. (Huaihua, Hunan, China) |  | ATRP: CuBr/PMDTA in DMF at 70 °C | CO2 responsive nanoparticles for Pickering emulsions | |
| Kraft lignin (alkali), Sigma-Aldrich |  | ATRP: CuBr/HMTETA in acetone at room temperature | Supramolecular hydrogels/self-healing materials | [79] |
| Lignin, Tokyo Kasei Kogyo Co., Ltd. |  | AGET ATRP: FeCl3.6H2O/PPh3/ascorbic acid in DMF at 110 °C for St and 90 °C for MMA | Novel polymerization method | [80] |
| Kraft lignin (alkali), Sigma-Aldrich |  | CuBr/HMTETA in 1,4-dioxane at 65 °C | Gene delivery | [81] |
| Lignin Source | Monomers (Representative) | RAFT CTA and Conditions | Application | Ref. |
|---|---|---|---|---|
| Kraft lignin (alkali), Tokyo Chemical Industry, Co. |  |  AIBN/DMF at 70 °C | Surfactants, Pickering emulsions, Cement superplasticizers | [85,86,87] |
| Kraft lignin (alkali), Tokyo Chemical Industry, Co. |  |  AIBN/DMF at 70 °C | High-performance superplasticizers | [88] |
| Organosolv lignin, Lignol Corporation |  |  AIBN/toluene at 70 °C | Biocomposites | [89] |
| Kraft lignin, Guangxi Nanning Phoenix Pulp & Paper Co., Ltd., China |  |  AMBN in DMF/water at 70 °C | Cationic flocculant | [90] |
| Lignin Source | Monomers (Representative) | Initiator and Conditions | Application | Ref. |
|---|---|---|---|---|
| Lignophenol from Japanese cedar |  | Benzyl bromide moieties, bulk, 100 °C, 12 h | Composites | [91] |
| Sulfonated lignin from Sigma Aldrich, USA |  | Tosylated lignin, DMSO, 100 °C, 10 h | Anti-infective ointment | [92] |
| Indulin AT from Ingevity Corp. (Charleston, SC) |  | Lignin hydroxyls, triazabicyclodecene (TBD), bulk, 130 °C for 3.5 h | Composites | [93] |
| Biobutanol lignin from Songyuan bairui bio-polyols Co. Ltd. |  | BBL initiator, triazabicyclodecene (TBD), bulk, 135 °C | Composites, coatings | [94] |
| Alkali lignin or organosolv lignin from Sigma Aldrich, |  | Sn(Oct)2, 120–130 °C, 24–45 h, solvent free or with solvents such as toluene | Composites, nanofibers for healthcare applications | [95,96] |
| Alkali lignin |  | solvent-free ROP, tin(II) 2-ethylhexanoate, 130 °C for 24 h | Nanofibers for biomedical applications | [97] |
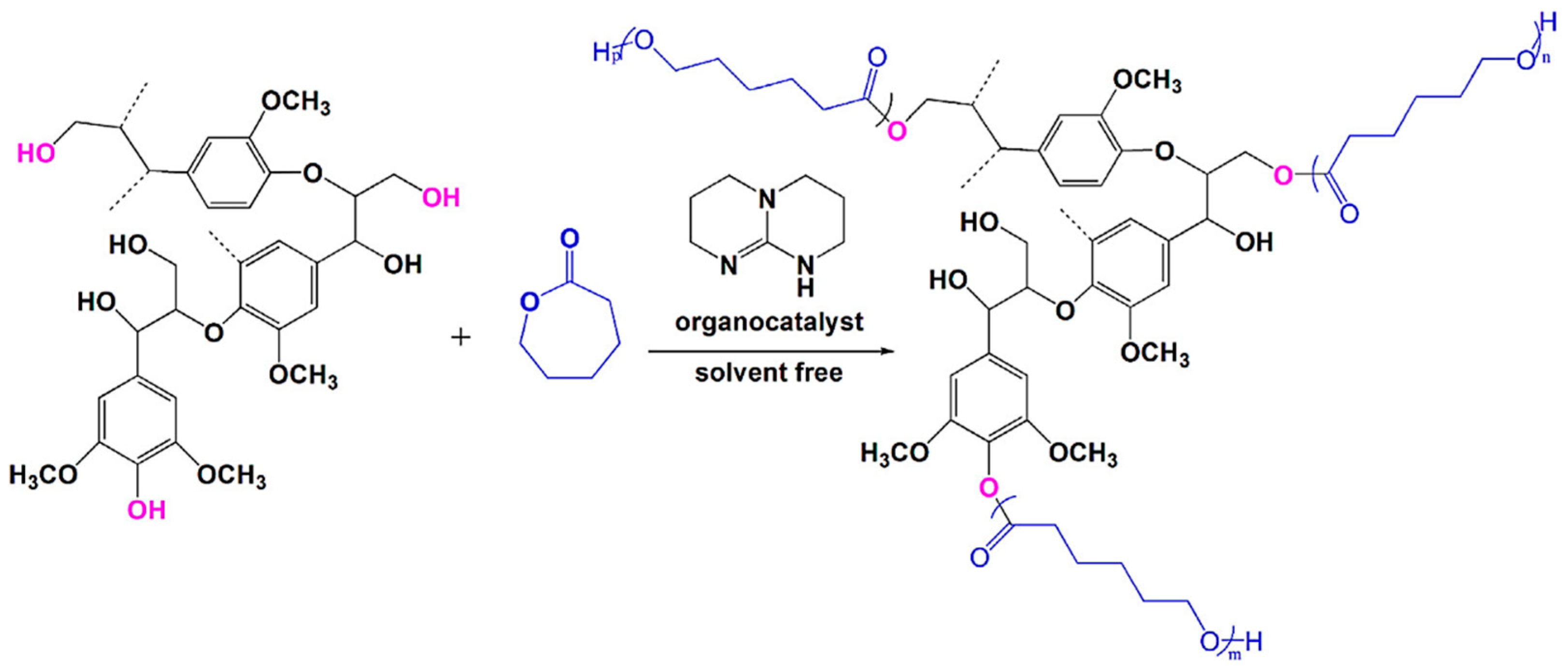
| Softwood Kraft lignin from UPM BioPiva |  | Hydrogenolyzed lignin hydroxyls, Phosphazene base P4-t-Bu, THF, at 50 °C for 2 days | Non-ionic surfactants | [98] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganewatta, M.S.; Lokupitiya, H.N.; Tang, C. Lignin Biopolymers in the Age of Controlled Polymerization. Polymers 2019, 11, 1176. https://doi.org/10.3390/polym11071176
Ganewatta MS, Lokupitiya HN, Tang C. Lignin Biopolymers in the Age of Controlled Polymerization. Polymers. 2019; 11(7):1176. https://doi.org/10.3390/polym11071176
Chicago/Turabian StyleGanewatta, Mitra S., Hasala N. Lokupitiya, and Chuanbing Tang. 2019. "Lignin Biopolymers in the Age of Controlled Polymerization" Polymers 11, no. 7: 1176. https://doi.org/10.3390/polym11071176
APA StyleGanewatta, M. S., Lokupitiya, H. N., & Tang, C. (2019). Lignin Biopolymers in the Age of Controlled Polymerization. Polymers, 11(7), 1176. https://doi.org/10.3390/polym11071176