Dual Responsive Polymersomes for Gold Nanorod and Doxorubicin Encapsulation: Nanomaterials with Potential Use as Smart Drug Delivery Systems

 ,
,
Abstract
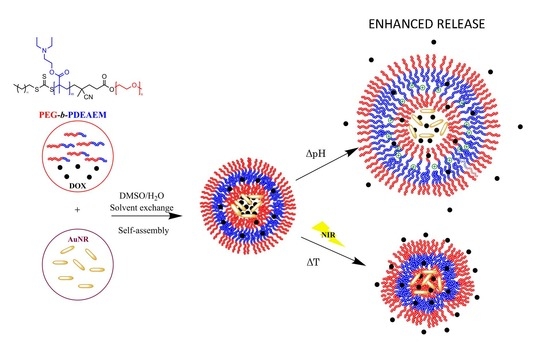
1. Introduction
2. Materials and Methods
2.1. Materials
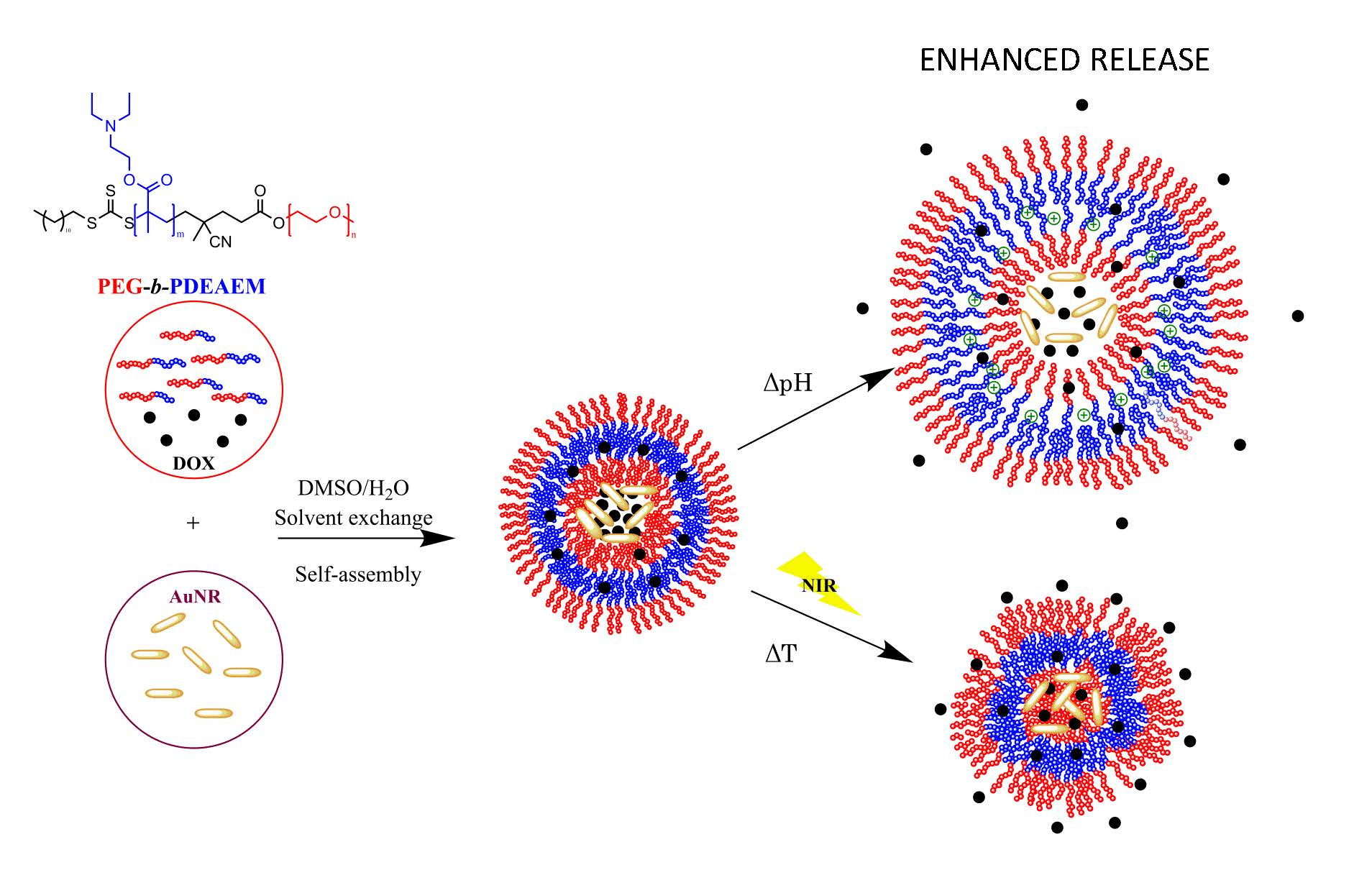
2.1.1. Macro Chain Transfer Agent (macroCTA) Synthesis
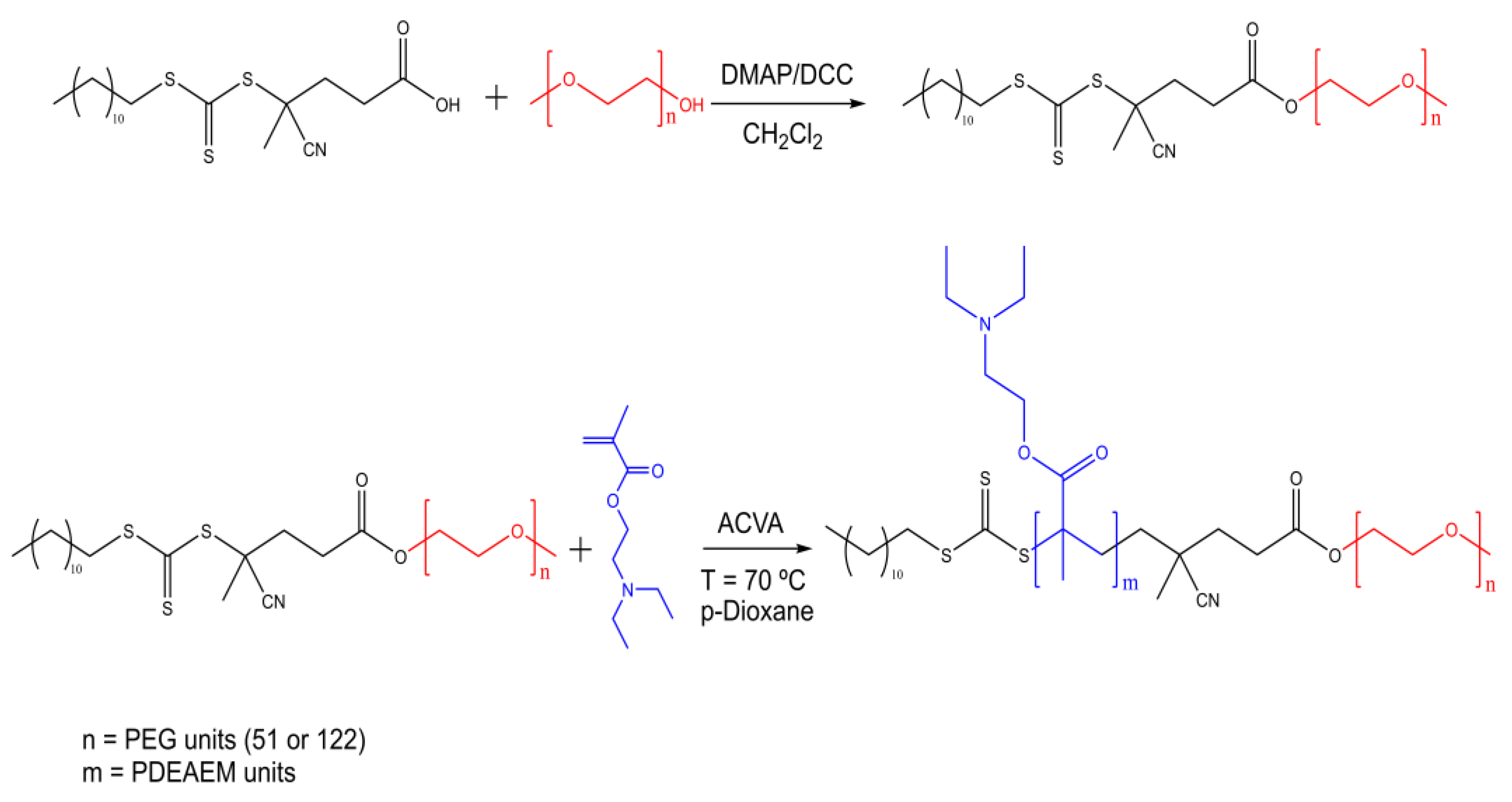
2.1.2. Synthesis of Block Copolymers PEG-b-PDEAEM
2.1.3. Preparation of Nanometric Polymer Aggregates
2.1.4. Gold Nanorod Synthesis
2.1.5. Gold Nanorod Encapsulation
2.2. Measurements
2.2.1. NIR Induced Heating Studies
2.2.2. Loading of Doxorubicin
2.2.3. In vitro release studies
3. Results and Discussion
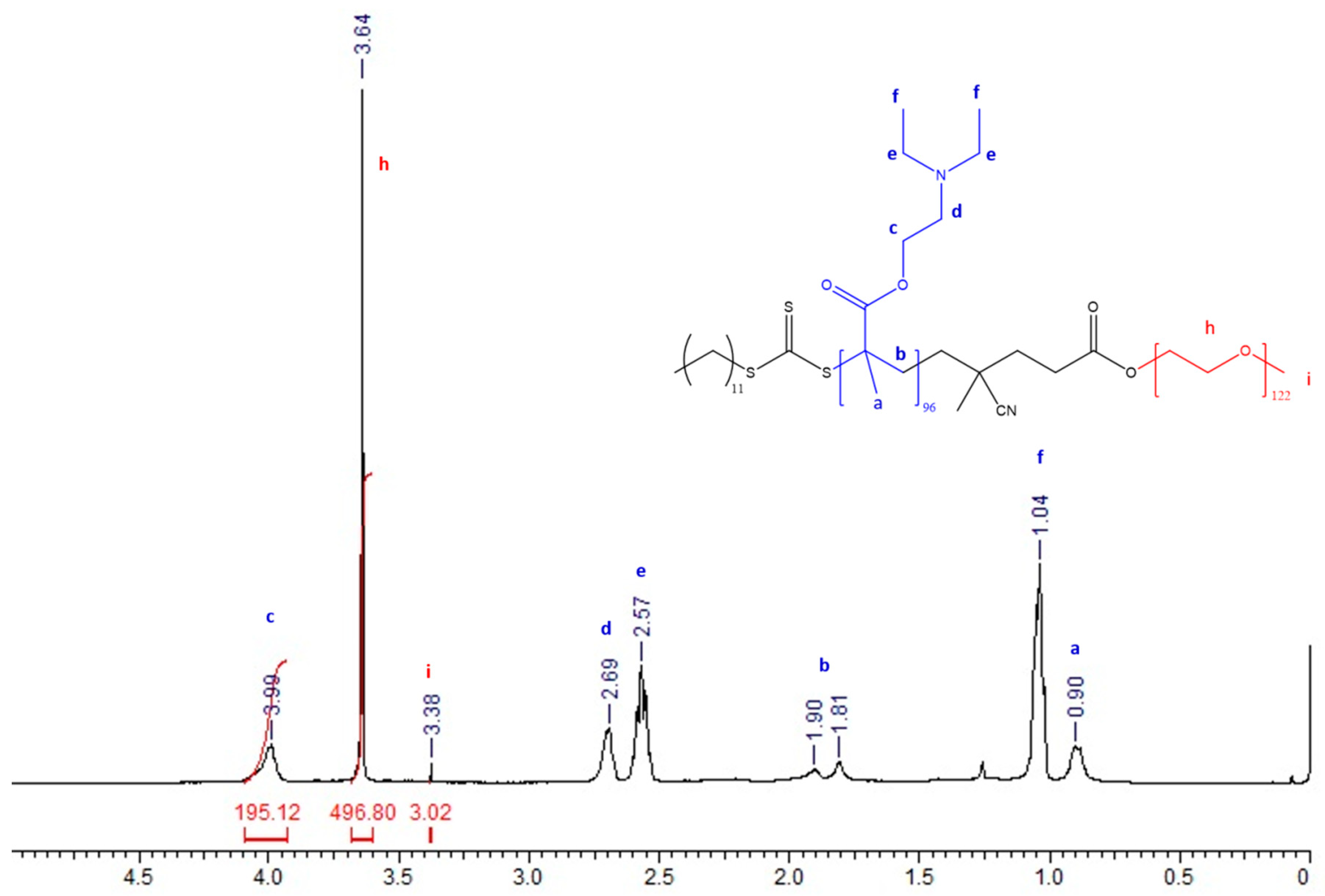
3.1. Synthesis and Characterization of PEG-b-PDEAEM Block Copolymers
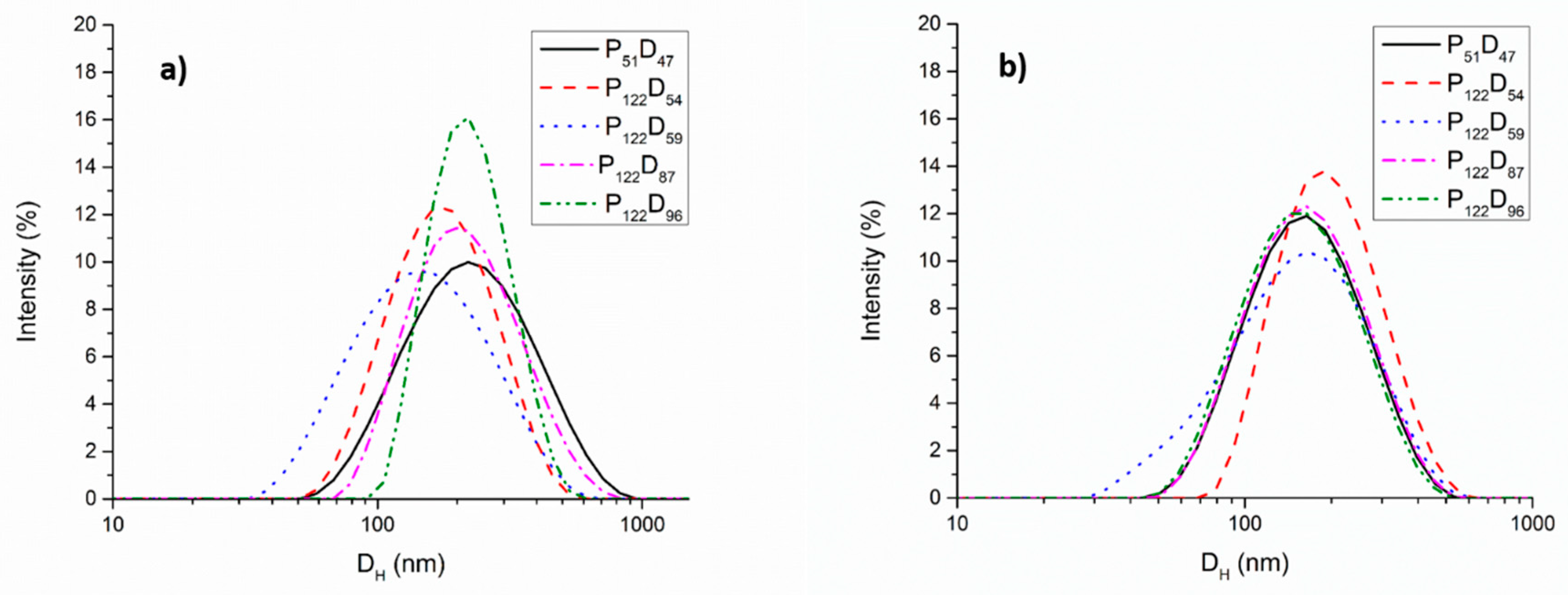
3.2. Self-assembly Studies of the PEG-b-PDEAEM Copolymers
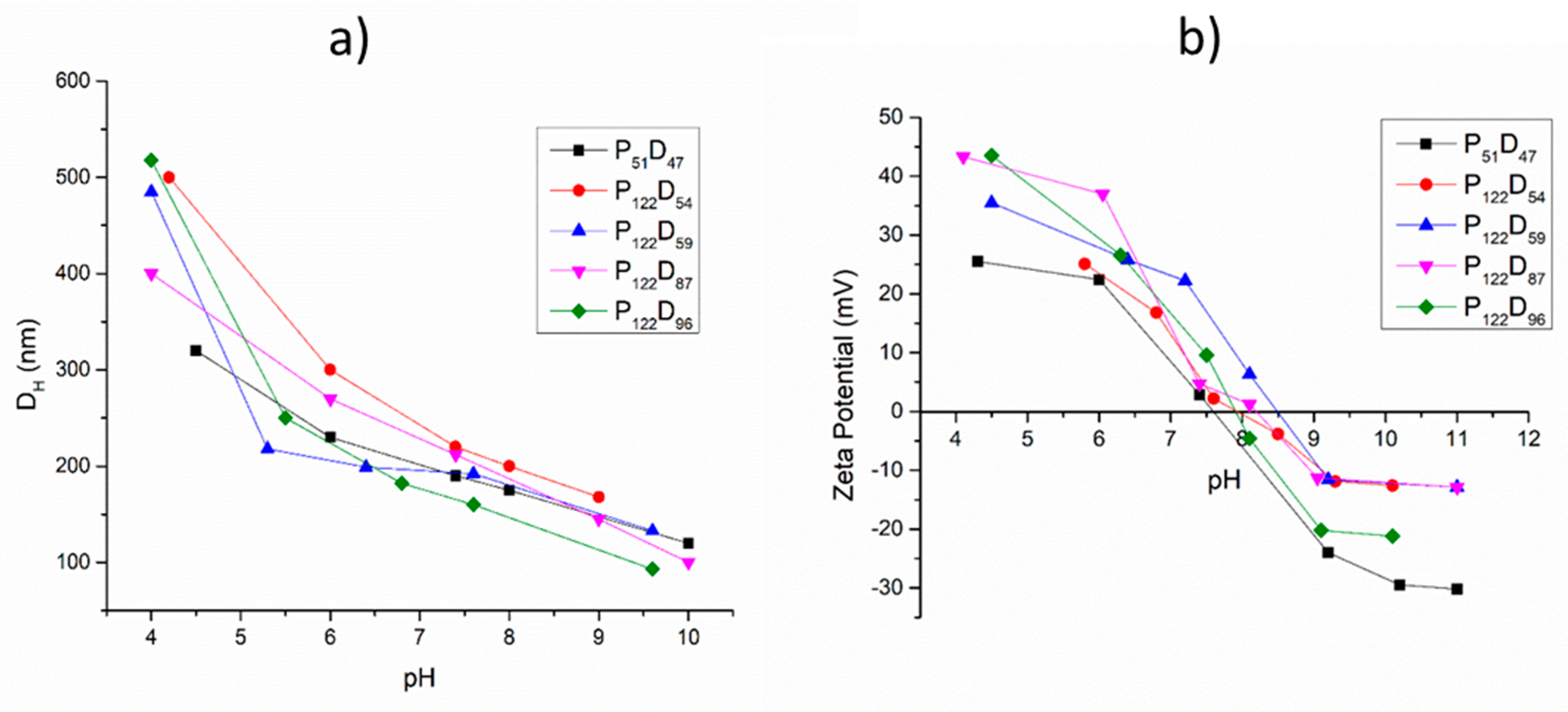
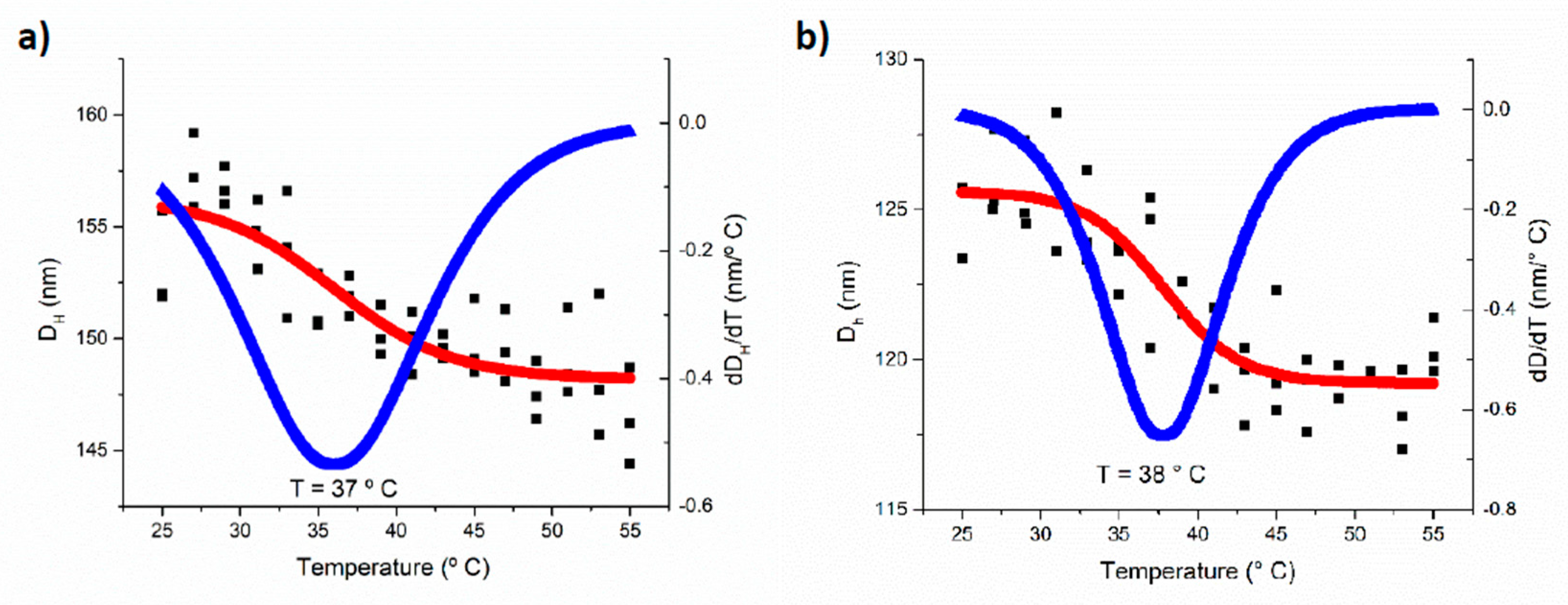
3.3. pH and Temperature Sensitive Behavior of the PEG-b-PDEAEM Aggregates
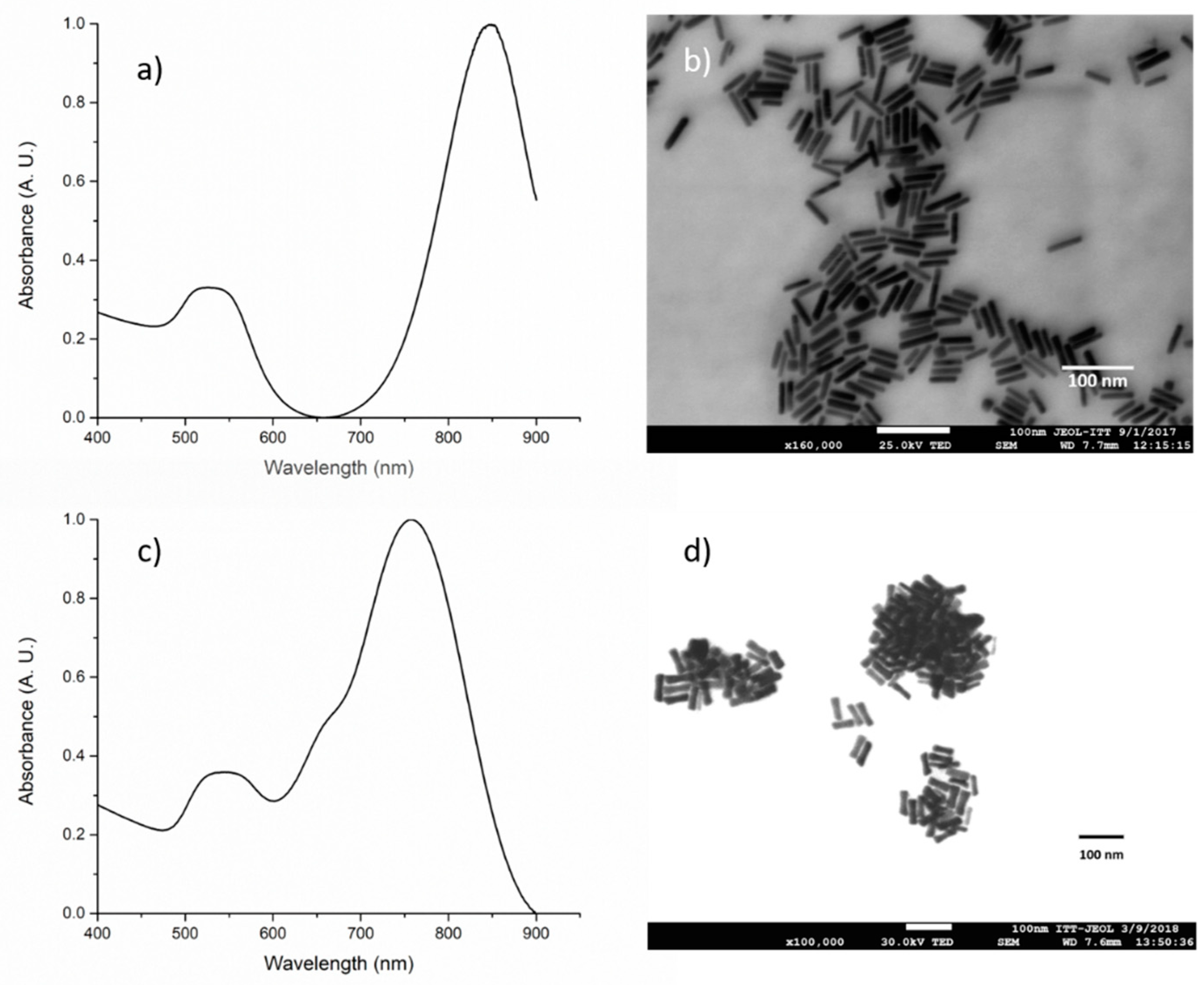
3.4. Gold Nanorod Synthesis
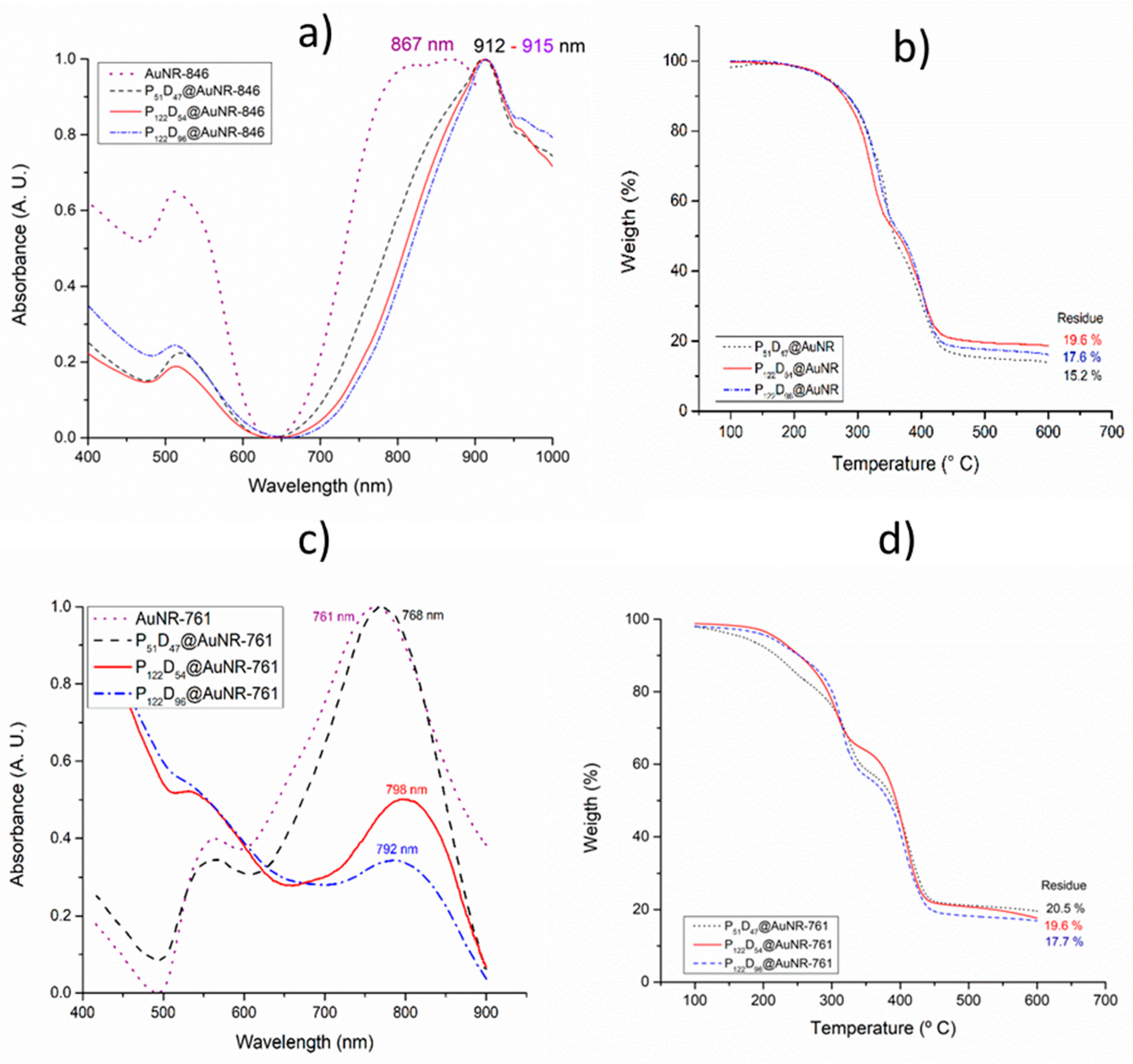
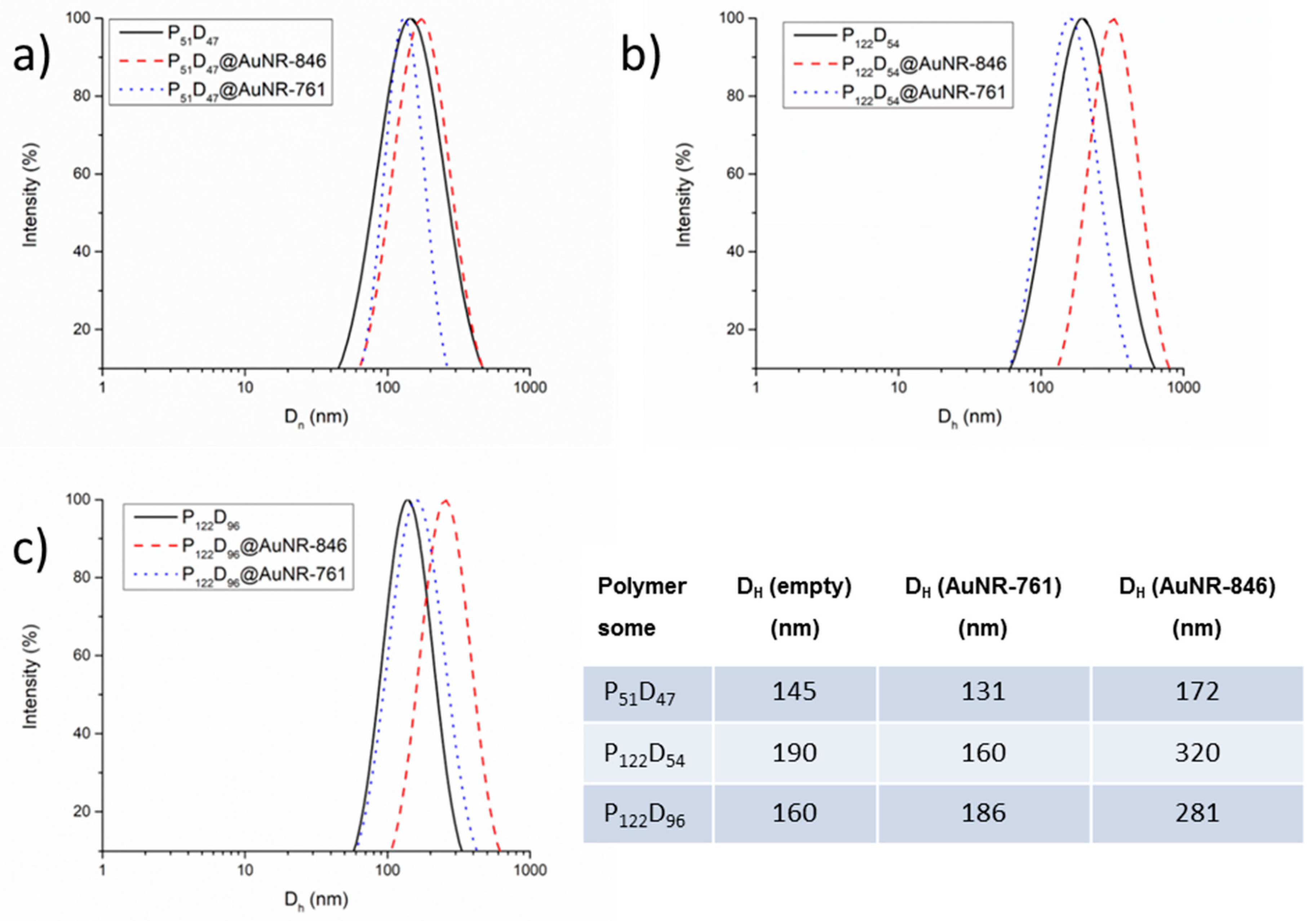
3.5. Gold Nanorod’s Encapsulation
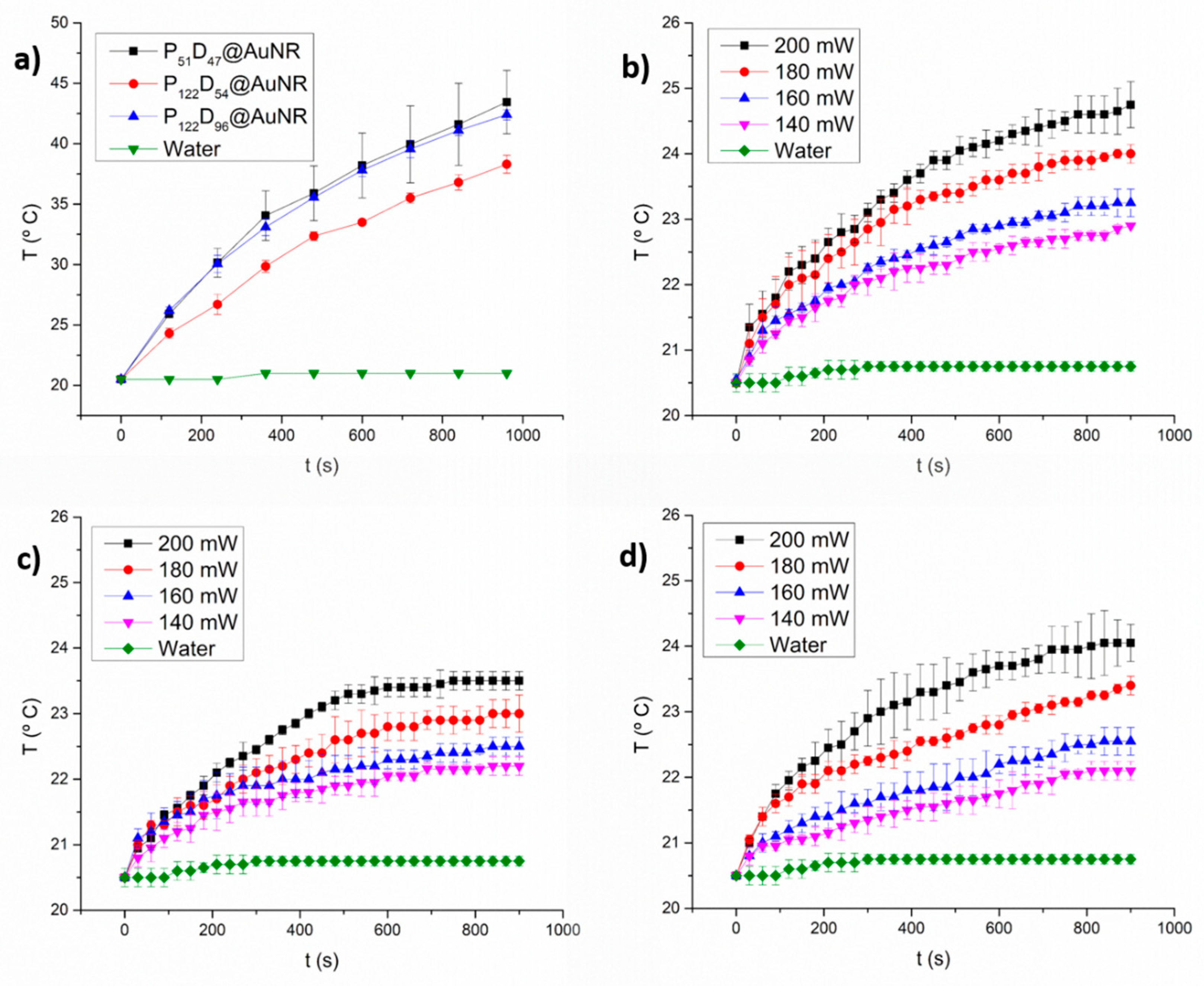
3.6. Photothermal Heating under NIR laser Irradiation
3.7. Load and Controlled Release of DOX from PEG-b-PDEAEM Polymersomes
3.7.1. DOX Loading
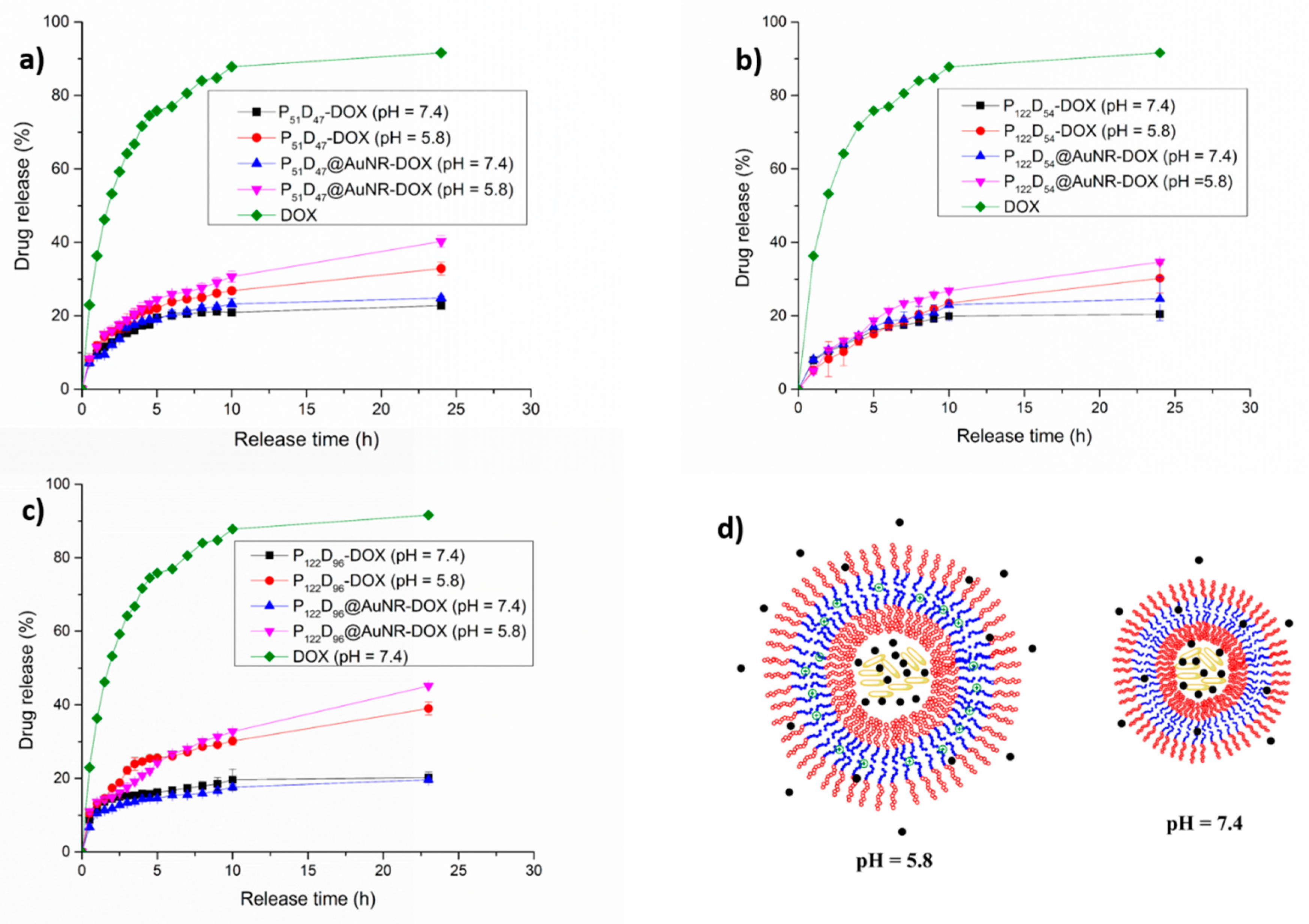
3.7.2. pH Triggered DOX Release
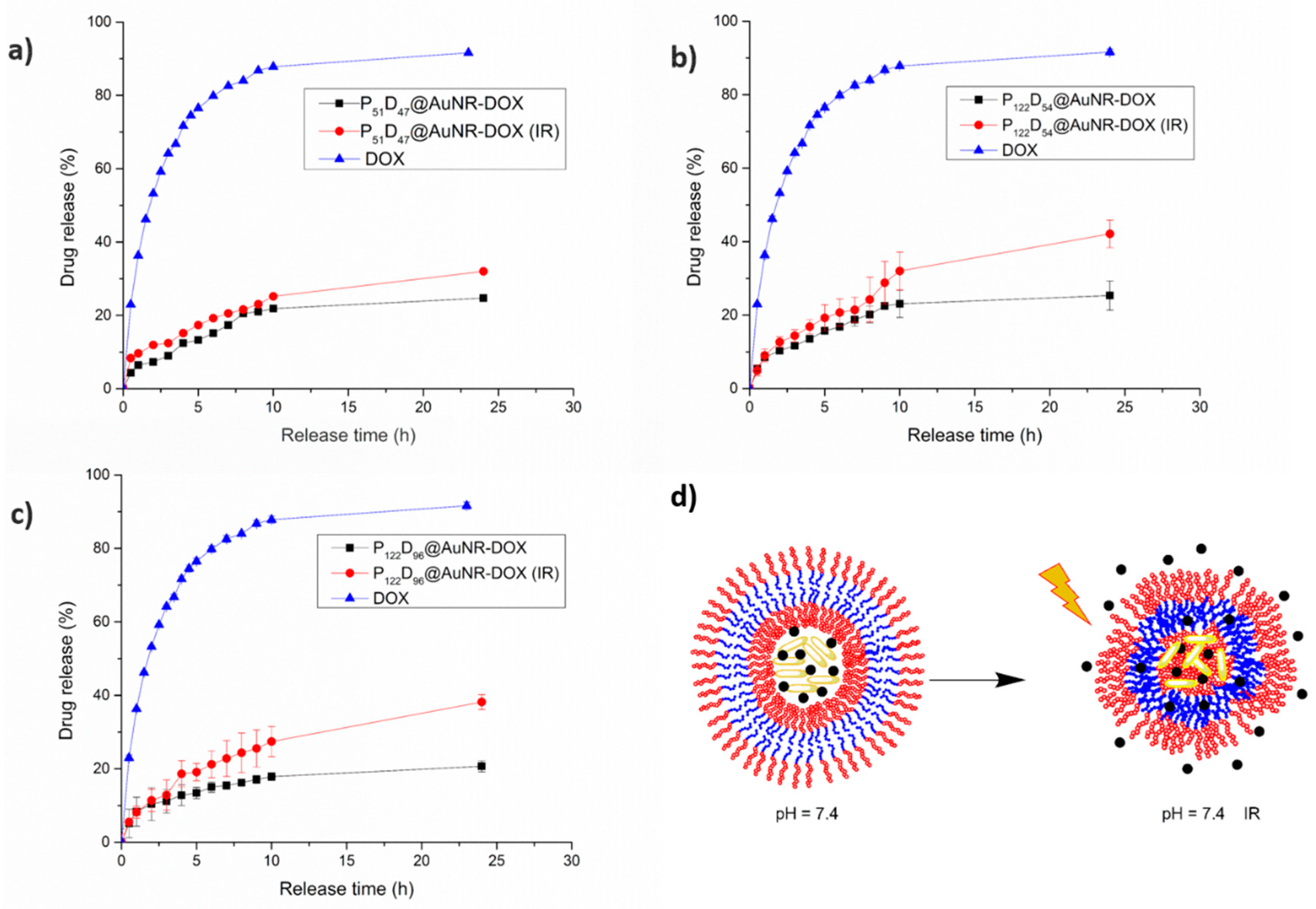
3.7.3. NIR-irradiation Triggered DOX Release
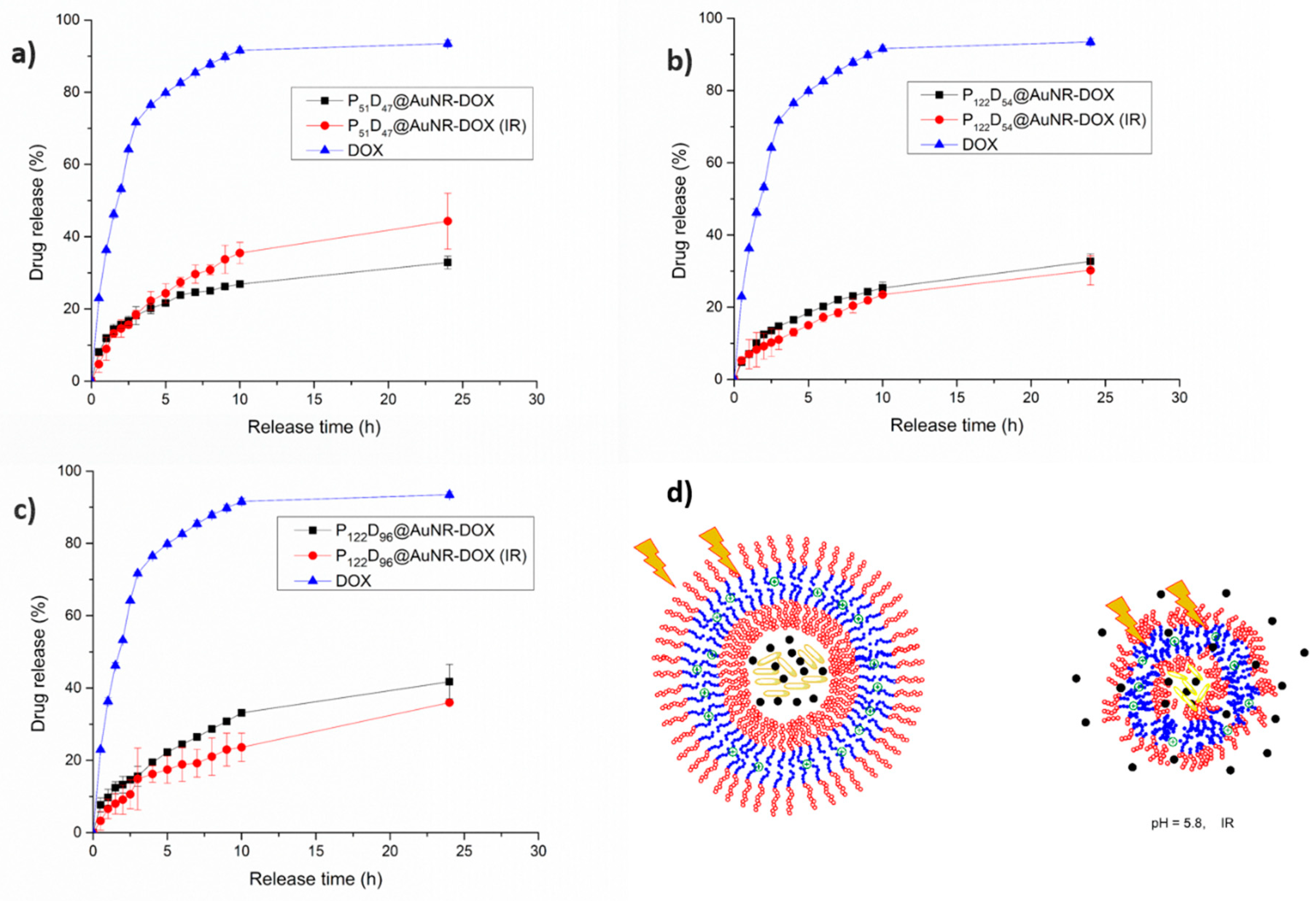
3.7.4. Combined pH and NIR-irradiation Triggered Release
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Allen, C.; Maysinger, D.; Eisenberg, A. Nano-engineering block copolymer aggregates for drug delivery. Colloids Surf. B. 1999, 16, 3–27. [Google Scholar] [CrossRef]
- Jones, M.C.; Leroux, J.C. Polymeric micelles—a new generation of colloidal drug carriers. Eur. J. Pharm. Biopharm. 1999, 48, 101–111. [Google Scholar] [CrossRef]
- Uchegbu, I.F.; Vyas, S.P. Non-ionic surfactant based vesicles (niosomes) in drug delivery. Int.J. Pharm. 1998, 172, 33–70. [Google Scholar] [CrossRef]
- Discher, D.E.; Eisenberg, A. Polymer vesicles. Science 2002, 297, 967. [Google Scholar] [CrossRef] [PubMed]
- Ayen, W.Y.; Garkhal, K.; Kumar, N. Doxorubicin-loaded (PEG)3-PLA nano-polymersomes: Effect of solvents and process parameters on formulation development and in vitro study. Mol. Pharmaceutics 2011, 8, 466–478. [Google Scholar] [CrossRef]
- Ayen, W.Y.; Garkhal, K.; Kumar, N.A. Systematic study on lyophilization process of polymersomes for long-term storage using doxorubicin-loaded (PEG)3–PLA nanopolymersomes. Eur. J. Pharm. Sci. 2012, 46, 405–414. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, Y.; Xie, Z.; Jing, X.; Belloti, A.; Gu, Z. Stimuli-responsive polymersomes for biomedical applications. Biomacromolecules 2017, 18, 649–673. [Google Scholar] [CrossRef]
- Yildirim, T.; Traeger, A.; Sungur, P.; Hoeppener, S.; Kellner, C.; Yildirim, I.; Pretzel, D.; Schubert, S.; Schubert, U.S. Polymersomes with endosomal pH-induced vesicle-to-micelle morphology transition and a potential application for controlled Doxorubicin delivery. Biomacromolecules 2017, 18, 3280–3290. [Google Scholar] [CrossRef]
- Gatish, J.; Huang, X.; Voit, B. Engineering functional polymer capsules toward smart nanoreactors. Chem. Rev. 2016, 116, 1053–1093. [Google Scholar]
- Warren, N.J.; Armes, S.P.; Mykhaylyk, O.O.; Mahmood, D.; Ryan, A.J. RAFT aqueous dispersion polymerization yields poly(ethylene glycol)-based diblock copolymer nano-objects with predictable single phase morphologies. J. Am. Chem. Soc. 2014, 136, 10174–10185. [Google Scholar] [CrossRef] [PubMed]
- Touve, M.A.; Figg, C.A.; Wright, D.B.; Park, C.; Cantlon, J.; Sumerlin, B.S.; Gianneschi, N.C. Polymerization-induced self-assembly of micelles observed by liquid cell transmission electron microscopy. Macromolecules 2017, 50, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Sanson, C.; Schatz, C.; Le Meins, J.F.; Brulet, A.; Soum, A.; Lecommandoux, S. Biocompatible and biodegradable poly(trimethylene carbonate)-b-poly (L-glutamic acid) polymersomes: Size control and stability. Langmuir 2010, 26, 2751–2760. [Google Scholar] [CrossRef]
- Schmalz, A.; Hanisch, M.; Schmalz, H.; Müller, A.H. Double stimuli-responsive behavior of linear and star-shaped poly(N,N-diethylaminoethyl methacrylate) in aqueous solution. Polymer 2010, 51, 1213–1217. [Google Scholar] [CrossRef]
- Tan, J.F.; Ravi, P.; Too, H.P.; Hatton, T.A.; Tam, K.C. Association behavior of biotinylated and non-biotinylated poly(ethylene oxide)-b-poly(2 (diethylamino)ethyl methacrylate). Biomacromolecules 2005, 6, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, C.; Huang, J. Synthesis, characterization, and stimuli-sensitive properties of triblock copolymer poly(ethylene oxide)-b-poly(2-(diethylamino)ethyl methacrylate)-b-poly(N-isopropylacrylamide). J. Appl. Polym. Sci. 2008, 108, 2180–2188. [Google Scholar] [CrossRef]
- Faraha, S.; Aviva, O.; Laoutb, N.; Ratnerb, S.; Beythc, N.; Domba, A.J. Quaternary ammonium poly(diethylaminoethyl methacrylate) possessing antimicrobial activity. Colloids Surf. B. 2015, 128, 608–613. [Google Scholar] [CrossRef]
- Cortez-Lemus, N.A.; Garcia-Soria, S.V.; Paraguay-Delgado, F.; Licea-Claverie, A. Synthesis of gold nanoparticles using poly (ethyleneglycol)-b-poly (N,N-diethylaminoethylmethacrylate) as nanoreactors. Polym. Bull. 2017, 74, 3527–3544. [Google Scholar] [CrossRef]
- Muddineti, O.S.; Ghosh, B.; Biswas, S. Current trends in using polymer coated gold nanoparticles for cancer therapy. Int. J. Pharm. 2015, 484, 252–267. [Google Scholar] [CrossRef]
- Sheng, W.; He, S.; Seare, W.J.; Almutairi, A. Review of the progress toward achieving heat confinement—the holy grail of photothermal therapy. J. Biomed. Opt. 2017, 22, 080901. [Google Scholar] [CrossRef] [PubMed]
- Haine, A.T.; Niidome, T. Gold nanorods as nanodevices for bioimaging, photothermal therapeutics, and drug delivery. Chem. Pharm. Bull. (Tokyo) 2017, 65, 625–628. [Google Scholar] [CrossRef]
- Sun, Q.; He, F.; Bi, H.; Wang, Z.; Sun, C.; Li, C.; Xu, J.; Yang, D.; Wang, X.; Gai, S.; Yang, P. An intelligent nanoplatform for simultaneously controlled chemo-, photothermal, and photodynamic therapies mediated by a single NIR light. Chem. Eng. J. 2019, 362, 679–691. [Google Scholar] [CrossRef]
- Chen, W.H.; Xu, X.D.; Jia, H.Z.; Lei, Q.; Luo, G.F.; Cheng, S.X.; Zhuo, R.X.; Zhang, X.Z. Therapeutic nanomedicine based on dual-intelligent functionalized gold nanoparticles for cancer imaging and therapy in vivo. Biomaterials 2013, 34, 8798–8807. [Google Scholar] [CrossRef]
- Liao, J.; Li, W.; Peng, J.; Yang, Q.; Wei, Y.; Zhang, X.; Qian, Z. Combined cancer photothermal-chemotherapy based on Doxorubicin/gold nanorod-loaded polymersomes. Theranostics 2015, 5, 345–356. [Google Scholar] [CrossRef]
- Liu, J.; Detrembleur, C.; De Pauw-Gillet, M.C.; Mornet, S.; Duget, E.; Jérôme, C. Gold nanorods coated with a thermo-responsive poly(ethylene glycol)-b-poly(N-vinylcaprolactam) corona as drug delivery systems for remotely near infrared-triggered release. Polym. Chem. 2014, 5, 799–813. [Google Scholar] [CrossRef]
- Zhong, Y.; Wang, C.; Cheng, L.; Meng, F.; Zhong, Z.; Liu, Z. Gold nanorod-cored biodegradable micelles as a robust and remotely controllable Doxorubicin release system for potent inhibition of drug-sensitive and -resistant cancer cells. Biomacromolecules 2013, 14, 2411–2419. [Google Scholar] [CrossRef]
- Gole, A.; Murphy, C. Seed-mediated synthesis of gold nanorods: Role of the size and nature of the seed. Chem. Mater. 2004, 16, 3633–3640. [Google Scholar] [CrossRef]
- Knights, O.B.; McLaughlan, J.R. Gold nanorods for light-based lung cancer theranostics. Int. J. Mol. Sci. 2018, 19, 3318. [Google Scholar] [CrossRef]
- Xiao, Y.; Hong, H.; Matson, V.Z.; Javadi, A.; Xu, W.; Yang, Y.; Zhang, Y.; Engle, J.W.; Nickles, R.J.; Cai, W.; Steeber, D.A.; Gong, S. Gold nanorods conjugated with doxorubicin and cRGD for combined anticancer drug delivery and PET imaging. Theranostics 2012, 2, 757–768. [Google Scholar] [CrossRef]
- Mirza, A.Z. A novel drug delivery system of gold nanorods with doxorubicin and study of drug release by single molecule spectroscopy. J. Drug Targeting 2015, 23, 52–58. [Google Scholar] [CrossRef]
- Gole, A.; Murphy, C. Polyelectrolyte-coated gold nanorods: Synthesis, characterization and immobilization. Chem. Mater. 2005, 17, 1325–1330. [Google Scholar] [CrossRef]
- Sason, C.; Schatz, C.; Le Meins, J.; Soum, A.; Thévenot, J.; Garanger, E.; Lecommanadoux, S. A simple method to achieve high doxorubicin loading in biodegradable polymersomes. J. Control. Release 2010, 147, 428–435. [Google Scholar] [CrossRef]
- Navarro-Vega, P.; Zizumbo-Lopez, A.; Licea-Claverie, A.; Vega-Rios, A.; Paraguay-Delgado, F. Equilibrium and nonequilibrium nanoscale ordering of polystyrene-b-poly(N,N-diethylaminoethyl methacrylate), a block copolymer carrying tertiary amine functional groups. J. Nanomater. 2014, 2014, 725356. [Google Scholar] [CrossRef]
- Zhang, G.; Ge, Z.; Liu, S.; Hu, J. Stimuli-responsive tertiary amine methacrylate-based blockcopolymers: Synthesis, supramolecular self-assembly and functional applications. Prog. Polym. Sci. 2014, 39, 1096–1143. [Google Scholar]
- Lee, H.; Venable, R.M.; MacKerell, A.D., Jr.; Pastor, R.W. Molecular dynamics studies of polyethylene oxide and polyethylene glycol: Hydrodynamic radius and shape anisotropy. Biophys J. 2008, 95, 1590–1599. [Google Scholar] [CrossRef]
- Zhang, Q.; Weber, C.; Schubert, U.S.; Hoogenboom, R. Thermoresponsive polymers with lower critical solution temperature: From fundamental aspects and measuring to recommended turbidimetry conditions. Mater. Horiz. 2017, 4, 109–116. [Google Scholar] [CrossRef]
- Salgado-Rodríguez, R.; Licea-Claverie, A.; Arndt, K.F. Random copolymers of N-Isopropylacrylamide and methacrylic acid monomers with hydrophobic spacers: pH-tunable temperature sensitive materials. Eur. Polym. J. 2004, 40, 1066–7857. [Google Scholar]
- Pikabea, A.; Ramos, J.; Forcada, J. Production of cationic nanogels with potential use in controlled drug delivery. Part. Part. Syst. Charact. 2014, 31, 101–109. [Google Scholar] [CrossRef]
- Burrows, N.D.; Lin, W.; Hinman, J.G.; Dennison, J.M.; Vartanian, A.M.; Abadeer, N.S.; Grzincic, E.M.; Jacob, L.M.; Li, J.; Murphy, C.J. Surface Chemistry of Gold Nanorods. Langmuir 2016, 32, 9905–9921. [Google Scholar] [CrossRef] [PubMed]
- Huff, T.B.; Tong, L.; Zhao, Y.; Hansen, M.N.; Cheng, J.; Wei, A. Hyperthermic effects of gold nanorods on tumor cells. Nanomedicine 2007, 2, 125–132. [Google Scholar] [CrossRef]
- Dickerson, E.B.; Dreaden, E.C.; Huang, X.; El-Sayed, I.H.; Chu, H.; Pushpaketh, S.; McDonald, J.F.; El-Sayed, M.A. Gold nanorods assisted near-infrared plasmonic photothermal therapy (PPTT) of squamous cell carcinoma in mice. Cancer Lett. 2008, 269, 57–66. [Google Scholar] [CrossRef]
- Sanchis, A.; Salvador, J.P.; Marco, M.P. Light-induced mechanisms for nanocarriers cargo release. Colloids Surf. B 2019, 173, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Han, J.; Zhu, C.; Xi, J.; Fan, L.; Guo, R. GNRs/PPy/m-SiO2 Core/Shell hybrids as drug nanocarriers for efficient chemo-photothermal therapy. Langmuir 2018, 34, 14661–14669. [Google Scholar] [CrossRef]
- Chen, J.; Li, X.; Zhao, X.; Wu, Q.; Zhu, H.; Mao, Z.; Gao, C. Doxorubicin-conjugated pH-responsive gold nanorods for combined photothermal therapy and chemotherapy of cancer. Bioact. Mater. 2018, 3, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Messager, L.; Gaitzsch, J.; Chierico, L.; Battalia, G. Novel aspects of encapsulation and delivery using polymersomes. Curr. Opin. Pharmacol. 2014, 18, 104–108. [Google Scholar] [CrossRef]
- Gallois, L.; Fiallo, M.; Garnier-Suillerot, A. Comparison of interaction of doxorubicin, daunorubicin, idarubicin and idarubicinol with large unilamellar vesicles circular dischroism study. Biochim. Biophys. Acta 1998, 1370, 31–40. [Google Scholar] [CrossRef]
- Chen, W.; Du, J.Z. Ultrasound and pH dually responsive polymer vesicles for anticancer drug delivery. Sci. Rep. 2013, 3, 2162. [Google Scholar] [CrossRef]
- Kumar-Kankala, R.; Liu, C.-G.; Chen, A.-Z.; Wang, S.-B.; Xu, P.-Y.; Kumar-Mende, L.; Liu, C.-L.; Lee, C.-H.; Hu, Y.-F. Overcoming Multidrug Resistance through the Synergistic Effects of Hierarchical pH-Sensitive, ROS-Generating Nanoreactors. ACS Biomater. Sci. Eng. 2017, 3, 2431–2442. [Google Scholar] [CrossRef]
- Kumar-Kankala, R.; Tsai, P.-Y.; Kuthati, Y.; Wei, P.-R.; Liu, C.-L.; Lee, C.-H. Overcoming multidrug resistance through co-delivery of ROS-generating nano-machinery in cancer therapeutics. J. Mater. Chem. B 2017, 5, 1507–1517. [Google Scholar] [CrossRef]
- Chen, Y.; Cheng, Y.; Zhao, P.; Zhang, S.; Li, M.; He, C.; Zhang, X.; Yang, T.; Yan, R.; Ye, P.; Ma, X.; Xiang, G. Co-delivery of doxorubicin and imatinib by pH sensitive cleavable PEGylated nanoliposomes with folate-mediated targeting to overcome multidrug resistance. Int. J. Pharm. 2018, 542, 266–279. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample a | M:macroCTA:I | Yield b (%) | Mn (CALC) c (g mol−1) | Mn (GPC) d (g mol−1) | Ð d | Mn (NMR) e (g mol−1) | PEG:PDEAEM e (Molar ratio) |
|---|---|---|---|---|---|---|---|
| P51D47 | 432:8:1 | 51.7 | 12408 | 21020 | 1.11 | 11283 | 52: 48 |
| P122D54 | 432:6:1 f | 38.9 | 19038 | 19619 | 1.35 | 15807 | 69: 31 |
| P122D59 | 432:4:1 f | 39.1 | 25708 | 24431 | 1.14 | 16734 | 67: 33 |
| P122D87 | 432:6:1 | 40.0 | 19038 | 23187 | 1.03 | 22047 | 58: 42 |
| P122D96 | 432:4:1 | 43.7 | 25708 | 28082 | 1.22 | 23715 | 56: 44 |
| Sample a | PEG:PDEAEM a | DH (direct dispersion) b | DH (solvent exchange) b | D (AFM) | D c (TEM) | nPDEAEM a | L (nm) d |
|---|---|---|---|---|---|---|---|
| P51D47 | 52:48 | 208 | 100 | 109.3 ± 13.8 | 119.5 ± 7.4 | 46 | 7.63–16.14 |
| P122D54 | 69:31 | 197 | 150 | 206.7 ± 26.5 | 105 ± 3.7 | 54 | 14.65–24.59 |
| P122D59 | 67:33 | 140 | 180 | 163.3 ± 42.7 | - | 59 | 14.95–25.87 |
| P122D87 | 58:42 | 217 | 120 | 122.0 ± 18.6 | - | 87 | 16.88–32.98 |
| P122D96 | 56:44 | 238 | 115 | 173.4 ± 44.0 | 137.6 ± 9.6 | 96 | 17.50–35.26 |
| Polymersome | DLEDOX (%) | LEAuNR a (%) | DLCDOX (%) | LCAuNR b |
|---|---|---|---|---|
| P51D47 | 52.0 | - | 12.0 | - |
| P51D47@AuNR-761 | 47.4 | 44.7 | 10.9 | 16.1 |
| P122D54 | 46.7 | - | 10.8 | - |
| P122D54@AuNR-761 | 44.3 | 42.5 | 10.2 | 15.3 |
| P122D96 | 48.3 | - | 11.1 | - |
| P122D96@AuNR-761 | 44.7 | 33.8 | 10.3 | 12.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
DiazDuarte-Rodriguez, M.; Cortez-Lemus, N.A.; Licea-Claverie, A.; Licea-Rodriguez, J.; Méndez, E.R. Dual Responsive Polymersomes for Gold Nanorod and Doxorubicin Encapsulation: Nanomaterials with Potential Use as Smart Drug Delivery Systems. Polymers 2019, 11, 939. https://doi.org/10.3390/polym11060939
DiazDuarte-Rodriguez M, Cortez-Lemus NA, Licea-Claverie A, Licea-Rodriguez J, Méndez ER. Dual Responsive Polymersomes for Gold Nanorod and Doxorubicin Encapsulation: Nanomaterials with Potential Use as Smart Drug Delivery Systems. Polymers. 2019; 11(6):939. https://doi.org/10.3390/polym11060939
Chicago/Turabian StyleDiazDuarte-Rodriguez, Melissa, Norma A. Cortez-Lemus, Angel Licea-Claverie, Jacob Licea-Rodriguez, and Eugenio R. Méndez. 2019. "Dual Responsive Polymersomes for Gold Nanorod and Doxorubicin Encapsulation: Nanomaterials with Potential Use as Smart Drug Delivery Systems" Polymers 11, no. 6: 939. https://doi.org/10.3390/polym11060939
APA StyleDiazDuarte-Rodriguez, M., Cortez-Lemus, N. A., Licea-Claverie, A., Licea-Rodriguez, J., & Méndez, E. R. (2019). Dual Responsive Polymersomes for Gold Nanorod and Doxorubicin Encapsulation: Nanomaterials with Potential Use as Smart Drug Delivery Systems. Polymers, 11(6), 939. https://doi.org/10.3390/polym11060939