Polylactide Composite Pins Reinforced with Bioresorbable Continuous Glass Fibers Demonstrating Bone-like Apatite Formation and Spiral Delamination Degradation



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Biocomposite Pins
2.2. Soaking Medium
2.3. In Vitro Degradation
2.4. Characterization of Degradation Behavior
2.5. Characterization of Calcium Phosphate Formation
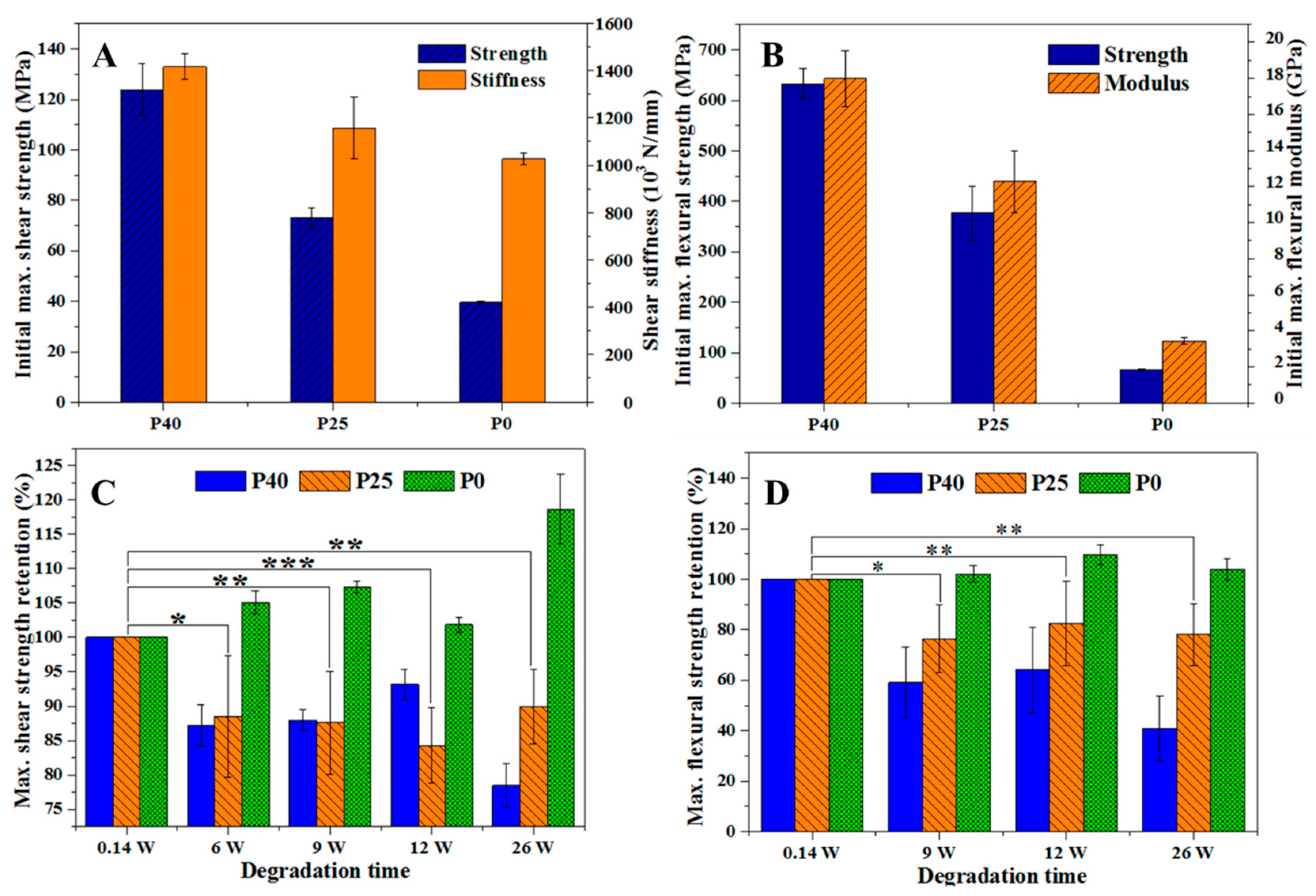
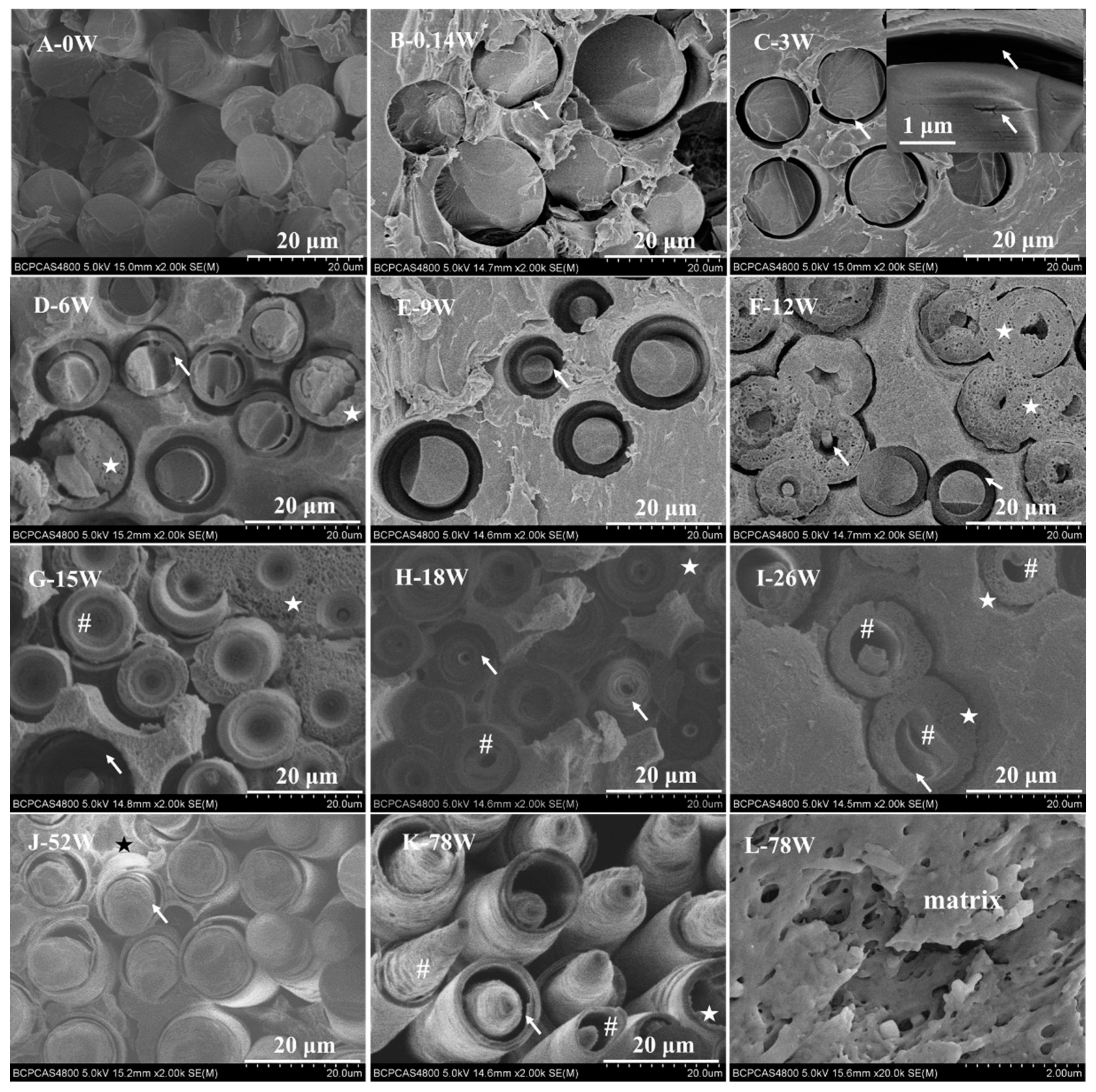
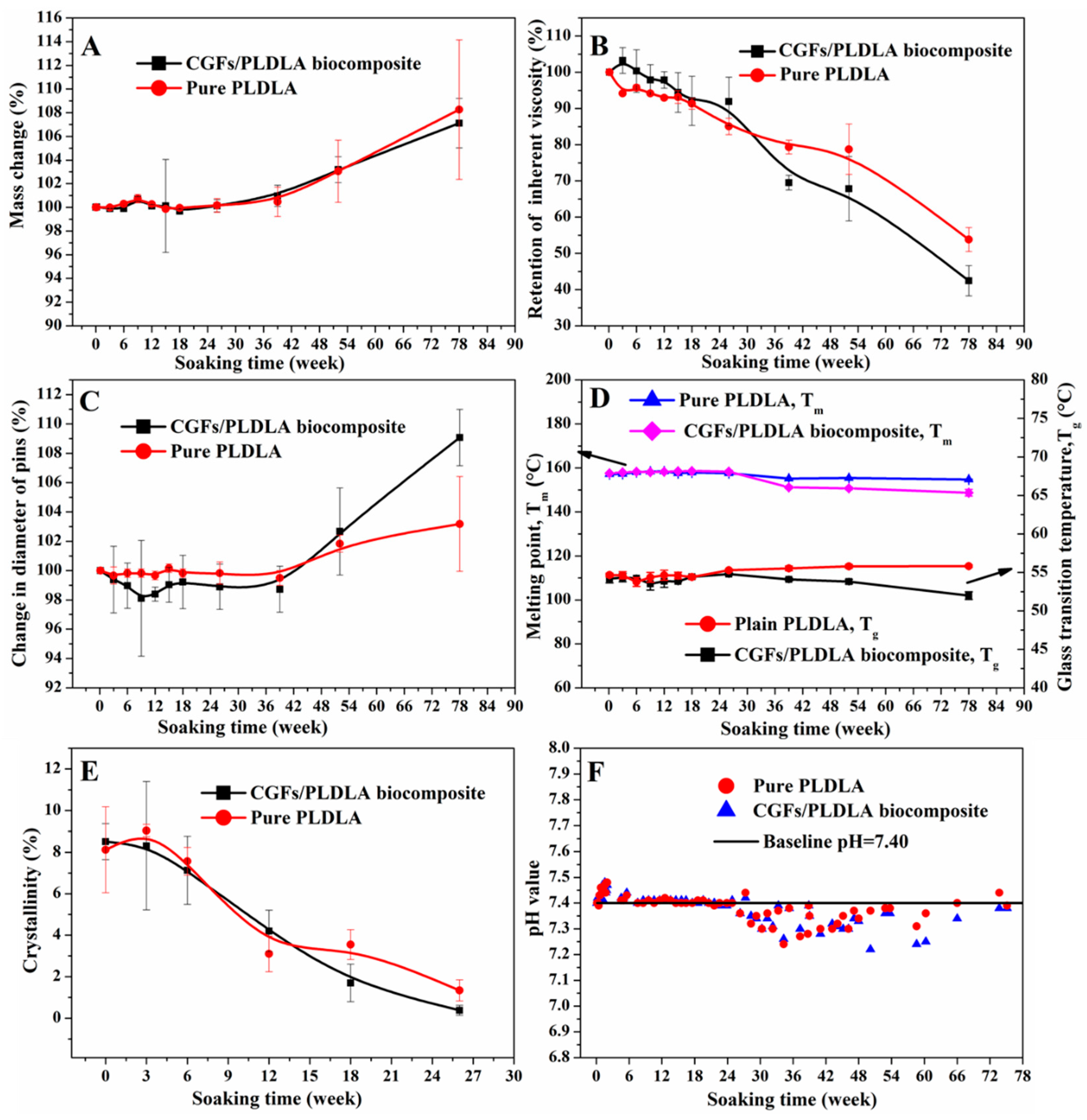
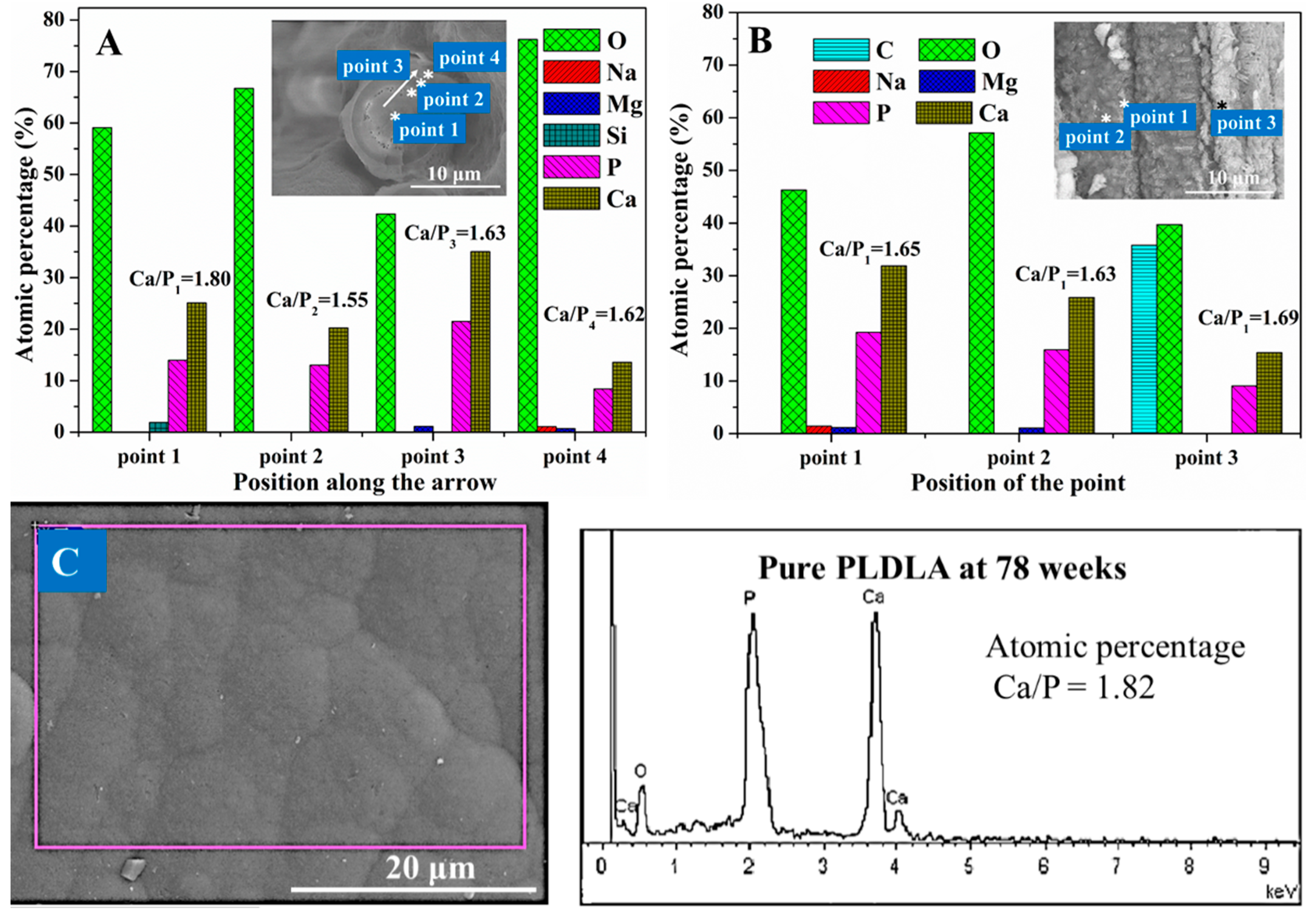
3. Results
3.1. Degradation Behavior
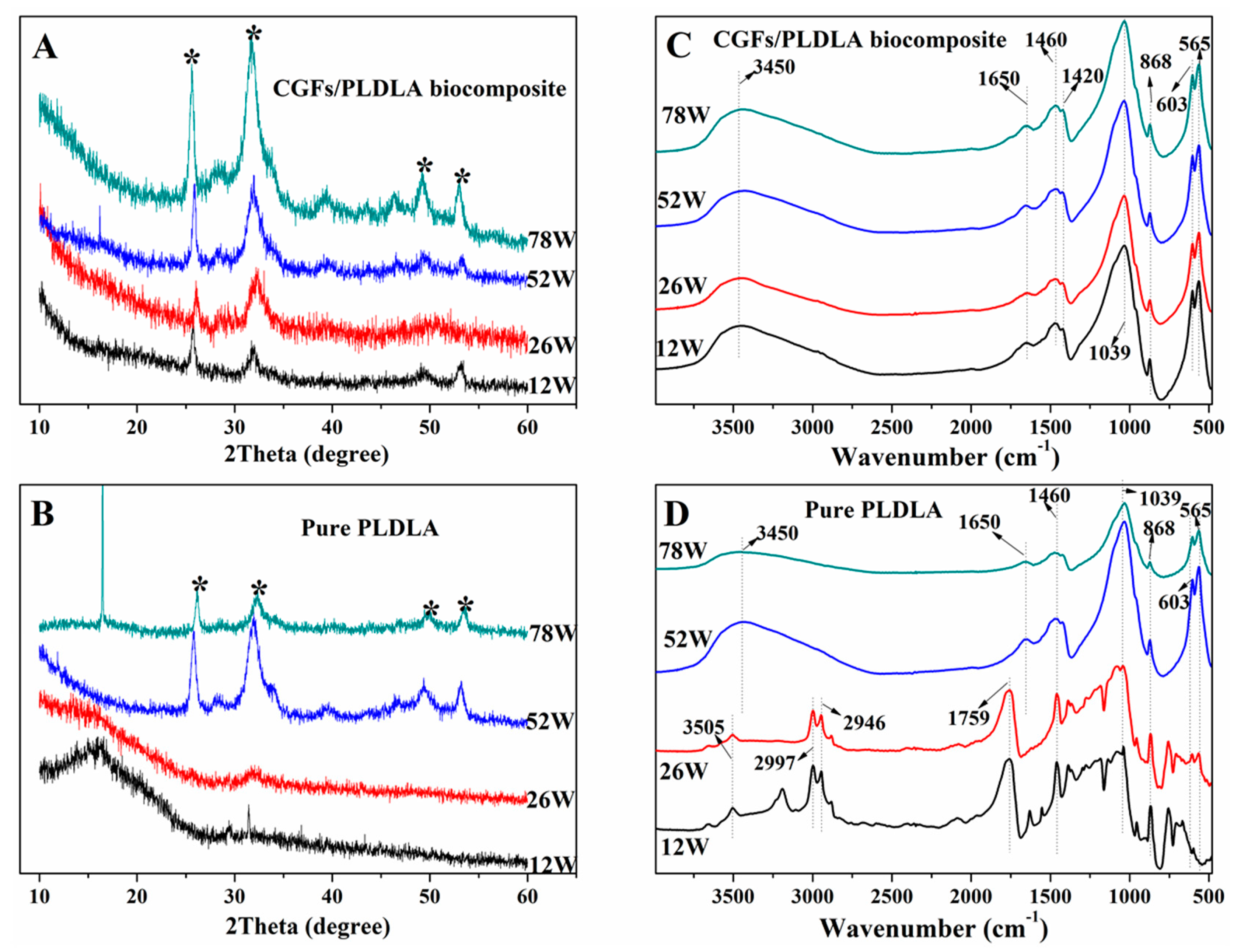
3.2. Calcium Phosphate Formation
4. Discussion
4.1. Degradation Behavior
4.2. Calcium Phosphate Formation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


References
- Lehtonen, T.J.; Tuominen, J.U.; Hiekkanen, E. Resorbable composites with bioresorbable glass fibers for load-bearing applications. In vitro degradation and degradation mechanism. Acta Biomater. 2013, 9, 4868–4877. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.C.; Wang, T.W.; Sun, J.S.; Lee, Y.H.; Shen, M.H.; Tsai, Z.R.; Chen, C.Y.; Hsu, H.C. Development and characterization of a bioinspired bone matrix with aligned nanocrystalline hydroxyapatite on collagen nanofibers. Materials 2016, 9, 198. [Google Scholar] [CrossRef]
- Zagho, M.M.; Hussein, E.A.; Elzatahry, A.A. Recent overviews in functional polymer composites for biomedical applications. Polymers 2018, 10, 739. [Google Scholar] [CrossRef]
- Daculsi, G.; Goyenvalle, E.; Cognet, R.; Aguado, E.; Suokas, E.O. Osteoconductive properties of poly(96l/4d-lactide)/beta-tricalcium phosphate in long term animal model. Biomaterials 2011, 32, 3166–3177. [Google Scholar] [CrossRef]
- Varila, L.; Fagerlund, S.; Lehtonen, T.; Tuominen, J.; Hupa, L. Surface reactions of bioactive glasses in buffered solutions. J. Eur. Ceram. Soc. 2012, 32, 2757–2763. [Google Scholar] [CrossRef]
- Varila, L.; Lehtonen, T.; Tuominen, J.; Hupa, M.; Hupa, L. In vitro behaviour of three biocompatible glasses in composite implants. J. Mater. Sci. Mater. Med. 2012, 23, 2425–2435. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, D.; de Bruijn, J.D.; Luo, X.; Fare, S.; Grijpma, D.W.; Yuan, H. Controlling dynamic mechanical properties and degradation of composites for bone regeneration by means of filler content. J. Mech. Behav. Biomed. Mater. 2013, 20, 162–172. [Google Scholar] [CrossRef]
- Vallittu, P.K.; Narhi, T.O.; Hupa, L. Fiber glass-bioactive glass composite for bone replacing and bone anchoring implants. Dent. Mater. 2015, 31, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Abert, J.; Amella, A.; Weigelt, S.; Fischer, H. Degradation and swelling issues of poly-(d,l-lactide)/beta-tricalcium phosphate/calcium carbonate composites for bone replacement. J. Mech. Behav. Biomed. Mater. 2016, 54, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, M.; Hamdi, M.; Basirun, W.J. Bioglass(r) 45s5-based composites for bone tissue engineering and functional applications. J. Biomed. Mater. Res. Part A 2017, 105, 3197–3223. [Google Scholar] [CrossRef]
- Fernandez de Grado, G.; Keller, L.; Idoux-Gillet, Y.; Wagner, Q.; Musset, A.M.; Benkirane-Jessel, N.; Bornert, F.; Offner, D. Bone substitutes: A review of their characteristics, clinical use, and perspectives for large bone defects management. J. Tissue Eng. 2018, 9, 2041731418776819. [Google Scholar] [CrossRef]
- Winkler, T.; Sass, A.; Duda, G.; Schmidt-Bleek, K. A review of biomaterials in bone defect healing, remaining shortcomings and future opportunities for bone tissue engineering: The unsolved challenge. Bone & Joint Res. 2018, 7, 232–243. [Google Scholar]
- Kulkova, J.; Moritz, N.; Suokas, E.O.; Strandberg, N.; Leino, K.A.; Laitio, T.T.; Aro, H.T. Osteointegration of plga implants with nanostructured or microsized beta-tcp particles in a minipig model. J. Mech. Behav. Biomed. Mater. 2014, 40, 190–200. [Google Scholar] [CrossRef]
- Jones, J.R. Review of bioactive glass: From hench to hybrids. Acta Biomater. 2015, 9, 4457–4486. [Google Scholar] [CrossRef]
- Heimbach, B.; Tonyali, B.; Zhang, D.; Wei, M. High performance resorbable composites for load-bearing bone fixation devices. J. Mech. Behav. Biomed. Mater. 2018, 81, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, T.J.; Tuominen, J.U.; Hiekkanen, E. Dissolution behavior of high strength bioresorbable glass fibers manufactured by continuous fiber drawing. J. Mech. Behav. Biomed. Mater. 2013, 20, 376–386. [Google Scholar] [CrossRef]
- Felfel, R.M.; Ahmed, I.; Parsons, A.J.; Palmer, G.; Sottile, V.; Rudd, C.D. Cytocompatibility, degradation, mechanical property retention and ion release profiles for phosphate glass fibre reinforced composite rods. Mater. Sci. Eng. C Mater. Biol. Appl. 2013, 33, 1914–1924. [Google Scholar] [CrossRef] [PubMed]
- Sharmin, N.; Hasan, M.S.; Parsons, A.J.; Rudd, C.D.; Ahmed, I. Cytocompatibility, mechanical and dissolution properties of high strength boron and iron oxide phosphate glass fibre reinforced bioresorbable composites. J. Mech. Behav. Biomed. Mater. 2016, 59, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Sang, L.; Zhao, M.; Liang, Q.; Wei, Z. Silane-treated basalt fiber–reinforced poly(butylene succinate) biocomposites: Interfacial crystallization and tensile properties. Polymers 2017, 9, 351. [Google Scholar] [CrossRef]
- Cingolani, A.; Casalini, T.; Caimi, S.; Klaue, A.; Sponchioni, M.; Rossi, F.; Perale, G. A methodologic approach for the selection of bio-resorbable polymers in the development of medical devices: The case of poly(l-lactide-co-epsilon-caprolactone). Polymers 2018, 10, 851. [Google Scholar] [CrossRef]
- Ginjupalli, K.; Shavi, G.V.; Averineni, R.K.; Bhat, M.; Udupa, N.; Nagaraja Upadhya, P. Poly(α-hydroxy acid) based polymers: A review on material and degradation aspects. Polym. Degrad. Stab. 2017, 144, 520–535. [Google Scholar] [CrossRef]
- Felfel, R.M.; Ahmed, I.; Parsons, A.J.; Walker, G.S.; Rudd, C.D. In vitro degradation, flexural, compressive and shear properties of fully bioresorbable composite rods. J. Mech. Behav. Biomed. Mater. 2011, 4, 1462–1472. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Wang, Y.; Xue, H.; Dou, Y. Biomimetic growth of hydroxyapatite on electrospun ca/pvp core(-)shell nanofiber membranes. Polymers 2018, 10, 1032. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Y.; Niu, W.; Chen, M.; Xue, Y.; Ge, J.; Ma, P.X.; Lei, B. Biodegradable multifunctional bioactive glass-based nanocomposite elastomers with controlled biomineralization activity, real-time bioimaging tracking, and decreased inflammatory response. ACS Appl. Mater. Interfaces 2018, 10, 17722–17731. [Google Scholar] [CrossRef] [PubMed]
- Haugen, H.J.; Lyngstadaas, S.P.; Rossi, F.; Perale, G. Bone grafts: Which is the ideal biomaterial? J. Clin. Periodontol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Niemelä, T. Effect of β-tricalcium phosphate addition on the in vitro degradation of self-reinforced poly-l,d-lactide. Polym. Degrad. Stab. 2005, 89, 492–500. [Google Scholar] [CrossRef]
- Lafon, J.P.; Champion, E.; Bernache-Assollant, D.; Gibert, R.; Danna, A.M. Termal decomposition of carbonated calcium phosphate apatites. J. Therm. Anal. Calorim. 2003, 72, 1127–1134. [Google Scholar] [CrossRef]
- Rehman, I.; Bonfield, W. Characterization of hydroxyapatite and carbonated apatite by photo acoustic ftir spectroscopy. J. Mater. Sci. Mater. Med. 1997, 8, 1–4. [Google Scholar] [CrossRef]
- Shaltout, A.A.; Allam, M.A.; Moharram, M.A. Ftir spectroscopic, thermal and xrd characterization of hydroxyapatite from new natural sources. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 83, 56–60. [Google Scholar] [CrossRef]
- Meaurio, E.; Zuza, E.; López-Rodríguez, N.; Sarasua, J.R. Conformational behavior of poly(l-lactide) studied by infrared spectroscopy. J. Phys. Chem. B 2006, 110, 5790–5800. [Google Scholar] [CrossRef]
- Kokubo, T. Bioactive glass ceramics: Properties and applications. Biomaterials 1991, 12, 155–163. [Google Scholar] [CrossRef]
- Xue, W.; Liu, X.; Zheng, X.; Ding, C. In vivo evaluation of plasma-sprayed wollastonite coating. Biomaterials 2005, 26, 3455–3460. [Google Scholar] [CrossRef] [PubMed]
- Hench, L.L. The story of bioglass®. J. Mater. Sci. Mater. Med. 2006, 17, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Lin, K.; Wang, Z.; Chang, J.; Wang, L.; Lu, J.; Ning, C. Reconstruction of calvarial defect of rabbits using porous calcium silicate bioactive ceramics. Biomaterials 2008, 29, 2588–2596. [Google Scholar] [CrossRef] [PubMed]
- Esmaeilzadeh, J.; Hesaraki, S.; Hadavi, S.M.; Ebrahimzadeh, M.H.; Esfandeh, M. Poly(D/L) lactide/polycaprolactone/bioactive glasss nanocomposites materials for anterior cruciate ligament reconstruction screws: The effect of glass surface functionalization on mechanical properties and cell behaviors. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 77, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Weiler, A.; Hoffmann, R.F.G.; Stähelin, A.C.; Helling, H.-J.; Südkamp, N.P. Biodegradable implants in sports medicine: The biological base. Arthrosc. J. Arthrosc. Relat. Surg. 2000, 16, 305–321. [Google Scholar] [CrossRef]
- Abou Neel, E.A.; Young, A.M.; Nazhat, S.N.; Knowles, J.C. A facile synthesis route to prepare microtubes from phosphate glass fibres. Adv. Mater. 2007, 19, 2856–2862. [Google Scholar] [CrossRef]
- Choueka, J.; Charvet, J.L.; Alexander, H.; Oh, Y.O.; Joseph, G.; Blumenthal, N.C.; Lacourse, W.C. Effect of annealing temperature on the degradation of reinforcing fibers for absorbable implants. J. Biomed. Mater. Res. 1995, 29, 1309–1315. [Google Scholar] [CrossRef]
- Delahaye, F.; Montagne, L.; Palavit, G.; Touray, J.C.; Baillif, P. Acid dissolution of sodium–calcium metaphosphate glasses. J. Non-Cryst. Solids 1998, 242, 25–32. [Google Scholar] [CrossRef]
- Cle´Ment, J.; Planell, J.A.; Avila, G.; Marti’Nez, S. Analysis of the structural changes of a phosphate glass during its dissolution in simulated body fluid. J. Mater. Sci. Mater. Med. 1999, 10, 729–732. [Google Scholar] [CrossRef]
- Gao, H.; Tan, T.; Wang, D. Dissolution mechanism and release kinetics of phosphate controlled release glasses in aqueous medium. J. Control. Release 2004, 96, 29–36. [Google Scholar] [CrossRef]
- Kokubo, T.; Takadama, H. How useful is sbf in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef]
- Yang, M.; Zhou, G.; Shuai, Y.; Wang, J.; Zhu, L.; Mao, C. Ca2+-induced self-assembly of bombyx mori silk sericin into a nanofibrous network-like protein matrix for directing controlled nucleation of hydroxylapatite nano-needles. J. Mater. Chem. B 2015, 3, 2455–2462. [Google Scholar] [CrossRef]
- Liu, K.L.; Choo, E.S.G.; Wong, S.Y.; Li, X.; He, C.B.; Wang, J.; Li, J. Designing poly[(r)-3-hydroxybutyrate]-based polyurethane block copolymers for electrospun nanofiber scaffolds with improved mechanical properties and enhanced mineralization capability. J. Phys. Chem. B 2010, 114, 7489–7498. [Google Scholar] [CrossRef]
- Pina, S.; Olhero, S.M.; Gheduzzi, S.; Miles, A.W.; Ferreira, J.M. Influence of setting liquid composition and liquid-to-powder ratio on properties of a mg-substituted calcium phosphate cement. Acta Biomater. 2009, 5, 1233–1240. [Google Scholar] [CrossRef]
- Zima, A.; Paszkiewicz, Z.; Siek, D.; Czechowska, J.; Ślósarczyk, A. Study on the new bone cement based on calcium sulfate and mg, co3 doped hydroxyapatite. Ceram. Int. 2012, 38, 4935–4942. [Google Scholar] [CrossRef]
- Mróz, W.; Bombalska, A.; Burdyńska, S.; Jedyński, M.; Prokopiuk, A.; Budner, B.; Ślósarczyk, A.; Zima, A.; Menaszek, E.; Ścisłowska-Czarnecka, A.; et al. Structural studies of magnesium doped hydroxyapatite coatings after osteoblast culture. J. Mol. Struct. 2010, 977, 145–152. [Google Scholar] [CrossRef]
- Lu, S.; Mcenery, M.A.P.; Rogers, B.R.; Wenke, J.C.; Dan, S.; Guelcher, S.A. Resorbable nanocomposites with bone-like strength and enhanced cellular activity. J. Mater. Chem. B 2017, 5, 4198–4206. [Google Scholar] [CrossRef]
- Wei, P.; Yuan, Z.; Jing, W.; Huang, Y.; Cai, Q.; Guan, B.; Liu, Z.; Zhang, X.; Mao, J.; Chen, D.; et al. Strengthening the potential of biomineralized microspheres in enhancing osteogenesis via incorporating alendronate. Chem. Eng. J. 2019, 368, 577–588. [Google Scholar] [CrossRef]
- Cao, X.-Y.; Tian, N.; Dong, X.; Cheng, C.-K. Effects of varying the content of bioresorbable continuous glass fibers on the mechanical behavior of biocomposite pins for surgical load-bearing musculoskeletal applications. J. Mech. Behave. Biomed. Mater. 2019. submitted for publication. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ion | Concentration (10−3 mol/L) | |
|---|---|---|
| SBF (pH 7.40) | Blood Plasma (pH 7.2 to 7.4) | |
| Na+ | 142.0 | 142.0 |
| K+ | 5.0 | 5.0 |
| Mg2+ | 1.5 | 1.5 |
| Ca2+ | 2.5 | 2.5 |
| Cl− | 147.8 | 103.0 |
| HCO3− | 4.2 | 27.0 |
| HPO42− | 1.0 | 1.0 |
| SO42− | 0.5 | 0.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, X.-Y.; Tian, N.; Dong, X.; Cheng, C.-K. Polylactide Composite Pins Reinforced with Bioresorbable Continuous Glass Fibers Demonstrating Bone-like Apatite Formation and Spiral Delamination Degradation. Polymers 2019, 11, 812. https://doi.org/10.3390/polym11050812
Cao X-Y, Tian N, Dong X, Cheng C-K. Polylactide Composite Pins Reinforced with Bioresorbable Continuous Glass Fibers Demonstrating Bone-like Apatite Formation and Spiral Delamination Degradation. Polymers. 2019; 11(5):812. https://doi.org/10.3390/polym11050812
Chicago/Turabian StyleCao, Xiao-Yan, Na Tian, Xiang Dong, and Cheng-Kung Cheng. 2019. "Polylactide Composite Pins Reinforced with Bioresorbable Continuous Glass Fibers Demonstrating Bone-like Apatite Formation and Spiral Delamination Degradation" Polymers 11, no. 5: 812. https://doi.org/10.3390/polym11050812
APA StyleCao, X.-Y., Tian, N., Dong, X., & Cheng, C.-K. (2019). Polylactide Composite Pins Reinforced with Bioresorbable Continuous Glass Fibers Demonstrating Bone-like Apatite Formation and Spiral Delamination Degradation. Polymers, 11(5), 812. https://doi.org/10.3390/polym11050812