Crystalline Structures and Structural Transitions of Copolyamides Derived from 1,4-Diaminobutane and Different Ratios of Glutaric and Azelaic Acids

 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Synthesis of Copolyamides Based on 1,4-Diaminobutane and Glutaric and Azelaic Acids
2.2. Measurements
3. Results and Discussion
3.1. Synthesis of Nylons 4,5+9_X
3.2. Thermal Properties of Nylons 4,5+9_X
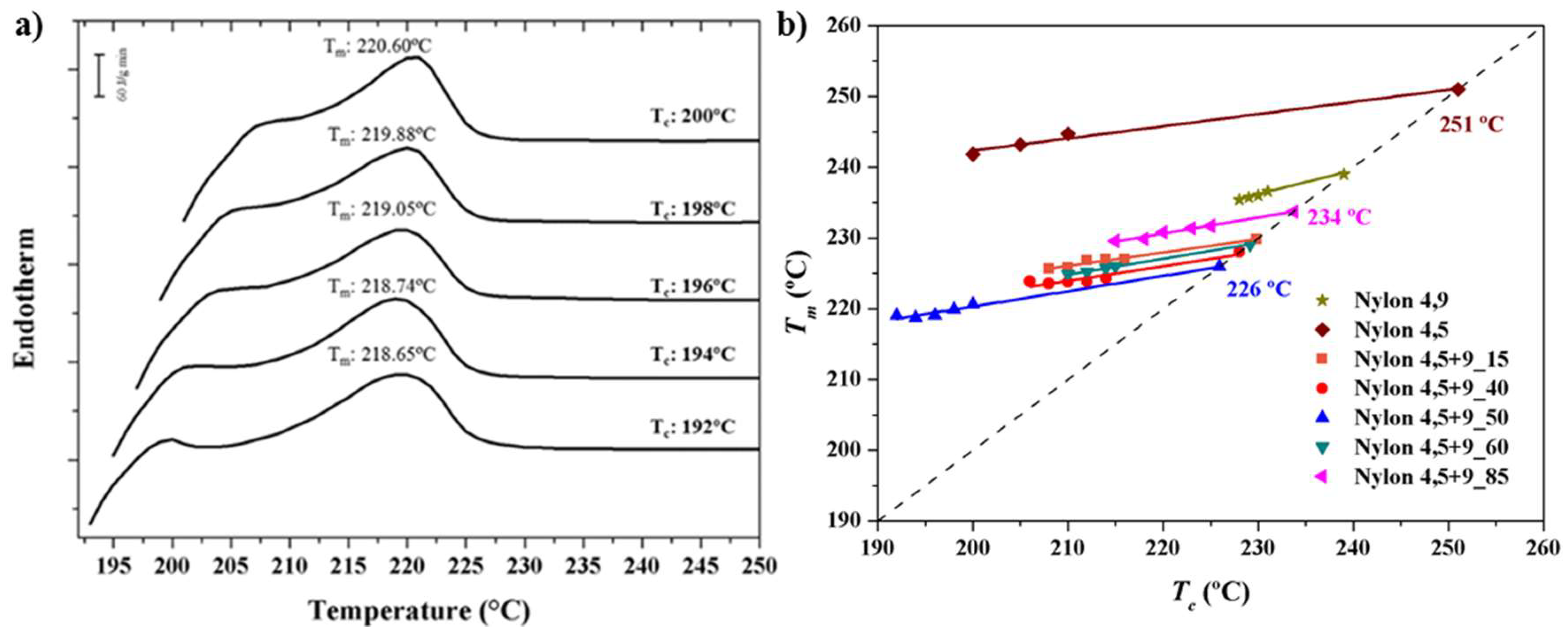
- (a)
- Melting enthalpies were clearly dependent on the specific composition and specifically decreased as the comonomer content increased (i.e., nylons 4,5+9_40 and 4,5+9_50 with fA values of 0.45 and 0.55 showed the lower melting enthalpies) for compositions with fA higher than 0.15. Thus, the enthalpies observed in the second heating scan varied from a minimum of 43.5 J/g (for fA = 0.55) and a maximum of 86.8 J/g (for fA = 1). Nevertheless, the minimum enthalpy was detected for nylon 4,5+9_15 (i.e., 31.0 J/g) probably due to the occurrence of some degradation before fusion as discussed later.
- (b)
- Melting enthalpies of solution-precipitated samples were clearly higher than those determined from melt crystallized samples and obviously than exhibited after fast cooling. Nevertheless, it is significant that the capacity to crystallize even for samples with a high comonomer content and even after the quenching process (e.g., 37.7 J/g for nylon 4,5+9_50).
- (c)
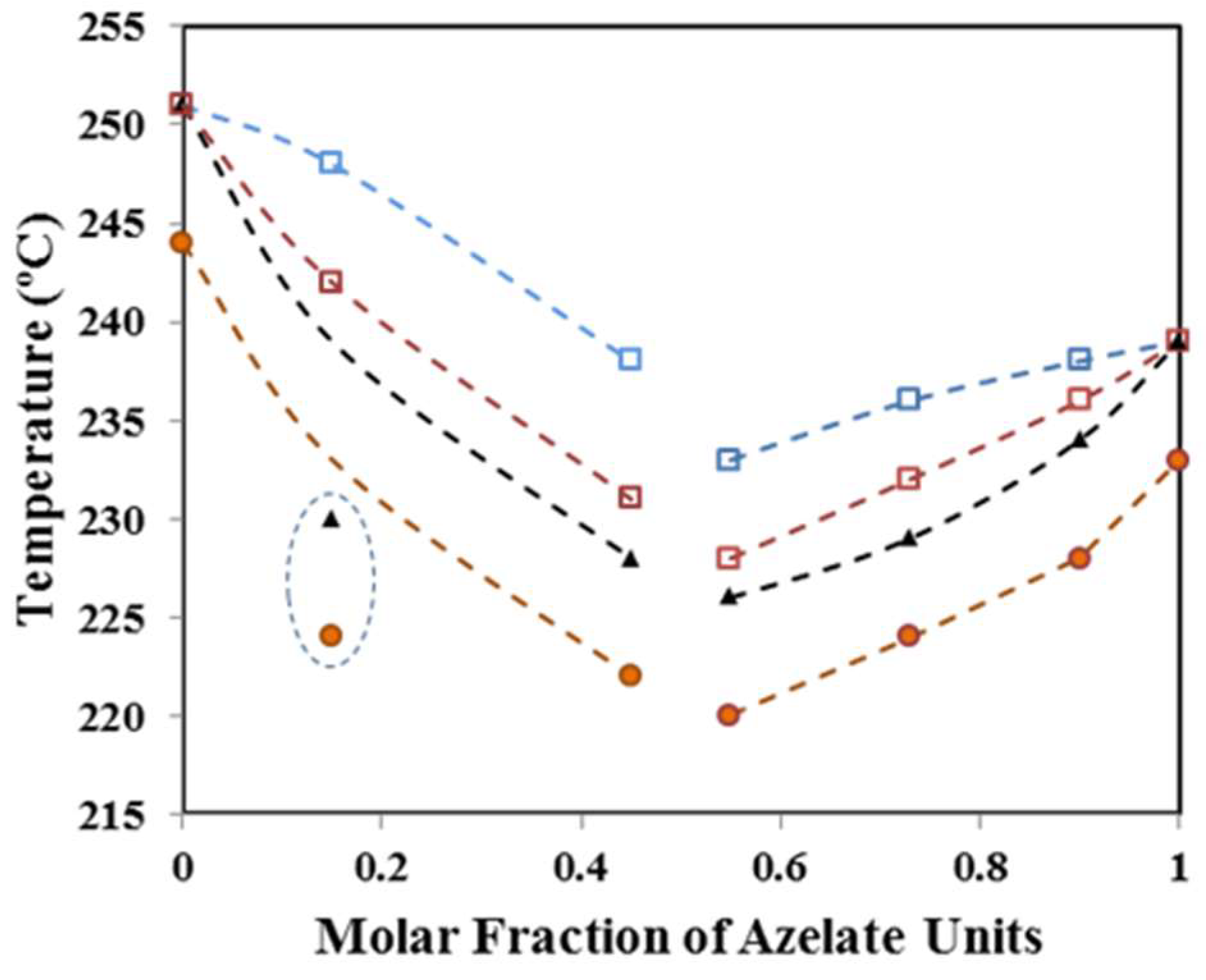
- A predominant melting peak in the 220–244 °C interval (second heating run) was always observed. This peak is associated to the fusion of lamellar crystals formed during the previous crystallization process. Peak temperature decreased with the comonomer content (e.g., 233 °C, 220 °C and 244 °C for samples with X equal to 100, 50 and 0, respectively).
- (d)
- A peak of variable intensity in the 228–244 °C interval is also usually observed. This peak can be associated with the fusion of lamellae recrystallized during the heating process and appeared at a practically constant temperature when copolymer composition was varied. Thus, the predominant initial lamellae followed a typical reorganization process that led to an increase of the thickness and obviously of the melting temperature. A hot crystallization exotherm can be observed in some cases between the two main melting peaks (e.g., see nylon 4,5+9_100 in Figure 2b and data corresponding to the third heating run in Table 2). Comparison between second and third heating traces points out that reordering was favoured when the sample was less crystalline (i.e., samples obtained by fast cooling) since probably more segments able to be incorporated into the crystalline lamellae are disposable on the lamellar surface.
- (e)
- Additional low-intensity endothermic processes can be envisaged at temperatures lower than the main melting peak temperature. These processes may correspond to the fusion of high defective crystals or the melting of crystals having a different crystalline structure.
- (f)
- Samples easily crystallized in the cooling run from the melt state, decreasing the crystallization peak temperature (i.e., from 212/218 °C to 196 °C) and enthalpy (i.e., from 69.2 to 39.3 J/g for fA > 0.15) with the comonomer content. Logically, the presence of structural foreign units hindered the crystallization process.
- (g)
- A single glass transition temperature was always detected as evidence that phase separation did not occur in the amorphous fraction. In any case the Tg varied in a narrow interval (e.g., between 50 and 71 °C) being difficult to deduce a specific trend since both composition and molecular weight played a significant influence. Logically the homopolymer derived from glutaric acid was slightly more rigid than that constituted by azelaic acid (i.e., 71 °C with respect to 50 °C).
3.3. Equilibrium Melting Point of Nylon 4,5+9_X Copolymers
3.4. Melting Point Depression
3.5. Morphology of Nylon 4,5+9 Single Crystals
3.6. Morphology of Nylon 4,5+9_X Spherulites Obtained from Melt Crystallization
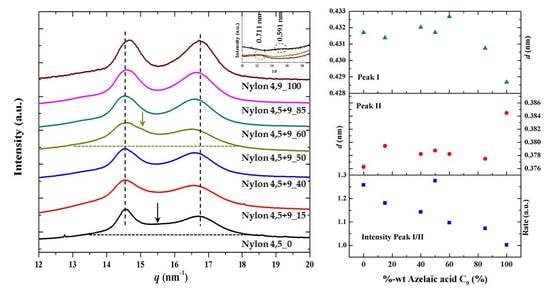
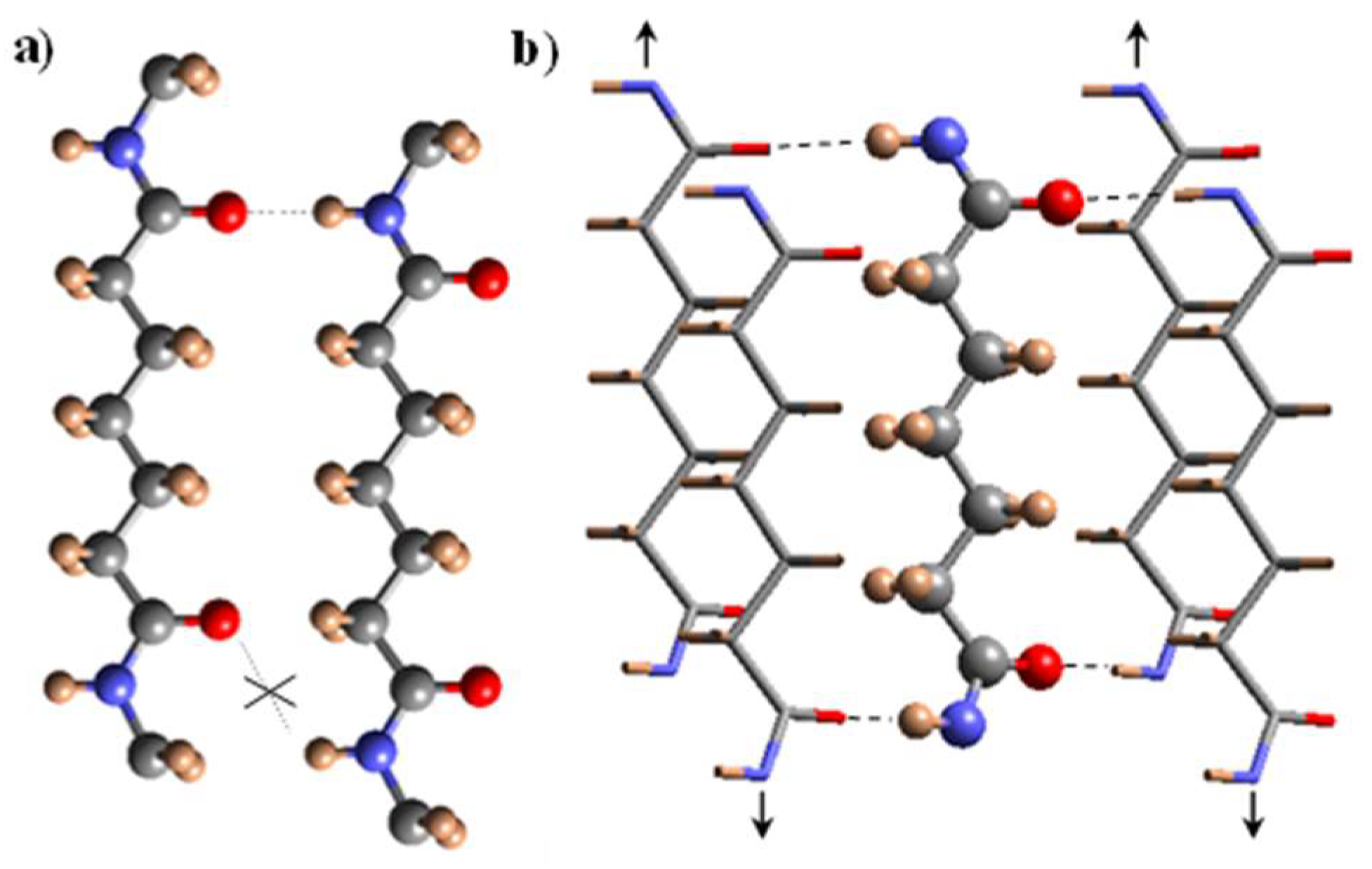
3.7. X-ray Diffraction Patterns of Solution Crystallized Samples
3.8. Structural Transitions During the Heating Process of Solution Crystallized Homopolymer Samples
3.9. Structural Transitions During the Heating Process of Solution Crystallized Copolymer Samples
3.10. Structural Transitions During the Cooling and Reheating Process of Nylon 4,5+9 Samples
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Kohan, M.I. Nylon Plastics Handbook; Distributed in the USA and in Canada; Hanser/Gardner Publications: Munich, Germany; New York, NY, USA; Cincinnati, OH, USA, 1995. [Google Scholar]
- Marchildon, K. Polyamides—Still Strong After Seventy Years. Macromol. React. Eng. 2011, 5, 22–54. [Google Scholar] [CrossRef]
- Winnacker, M. Polyamides and their functionalization: Recent concepts for their applications as biomaterials. Biomater. Sci. 2017, 5, 1230–1235. [Google Scholar] [CrossRef]
- Bunn, C.W.; Garner, E.V.; Bragg, W.L. The crystal structures of two polyamides (‘nylons‘). Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1947, 189, 39–68. [Google Scholar]
- Holmes, D.R.; Bunn, C.W.; Smith, D.J. The crystal structure of polycaproamide: Nylon 6. J. Polym. Sci. 1955, 17, 159–177. [Google Scholar] [CrossRef]
- Kinoshita, Y. An investigation of the structures of polyamide series. Die Makromol. Chem. 1959, 33, 1–20. [Google Scholar] [CrossRef]
- Villaseñor, P.; Franco, L.; Subirana, J.A.; Puiggalí, J. On the crystal structure of odd–even nylons: Polymorphism of nylon 5,10. J. Polym. Sci. Part B Polym. Phys. 1999, 37, 2383–2395. [Google Scholar] [CrossRef]
- Franco, L.; Subirana, J.A.; Puiggalí, J. Structure and Morphology of Odd Polyoxamides [Nylon 9,2]. A New Example of Hydrogen-Bonding Interactions in Two Different Directions. Macromolecules 1998, 31, 3912–3924. [Google Scholar] [CrossRef]
- Nakagawa, T.; Nozaki, K.; Maeda, S.; Yamamoto, T. Polymorphism of poly(nonamethyleneoxamide) crystal. Polymer 2014, 57, 99–104. [Google Scholar] [CrossRef]
- Cui, X.; Yan, D. Preparation, characterization and crystalline transitions of odd–even polyamides 11,12 and 11,10. Eur. Polym. J. 2005, 41, 863–870. [Google Scholar] [CrossRef]
- Samanta, S.; He, J.; Selvakumar, S.; Lattimer, J.; Ulven, C.; Sibi, M.; Bahr, J.; Chisholm, B.J. Polyamides based on the renewable monomer, 1,13-tridecane diamine II: Synthesis and characterization of nylon 13,6. Polymer 2013, 54, 1141–1149. [Google Scholar] [CrossRef]
- Morales-Gámez, L.; Soto, D.; Franco, L.; Puiggalí, J. Brill transition and melt crystallization of nylon 56: An odd–even polyamide with two hydrogen-bonding directions. Polymer 2010, 51, 5788–5798. [Google Scholar] [CrossRef]
- Navarro, E.; Subirana, J.A.; Puiggali, J. The structure of nylon 12,5 is characterized by two hydrogen bond directions as are other polyamides derived from glutaric acid. Polymer 1997, 38, 3429–3432. [Google Scholar] [CrossRef]
- Cui, X.; Yan, D.; Xiao, D. Synthesis and characterization of novel polyamides based on tridecanedioic acid: Nylons 3 13, 5 13, 6 13, 7 13, 9 13, 10 13, 11 13. e-Polymers 2004, 4. [Google Scholar] [CrossRef]
- Morales-Gámez, L.; Casas, M.T.; Franco, L.; Puiggalí, J. Structural transitions of nylon 47 and clay influence on its crystallization behavior. Eur. Polym. J. 2013, 49, 1354–1364. [Google Scholar] [CrossRef]
- Franco, L.; Cooper, S.J.; Atkins, E.D.T.; Hill, M.J.; Jones, N.A. Nylon 6 9 can crystallize with hydrogen bonding in two and in three interchain directions. J. Polym. Sci. Part B Polym. Phys. 1998, 36, 1153–1165. [Google Scholar] [CrossRef]
- Murase, S.K.; Casas, M.T.; Martínez, J.C.; Estrany, F.; Franco, L.; Puiggalí, J. Reversible changes induced by temperature in the spherulitic birefringence of nylon 6 9. Polymer 2015, 76, 34–45. [Google Scholar] [CrossRef]
- Olmo, C.; Rota, R.; Carlos Martínez, J.; Puiggalí, J.; Franco, L. Temperature-induced structural changes in even-odd nylons with long polymethylene segments. J. Polym. Sci. Part B Polym. Phys. 2016, 54, 2494–2506. [Google Scholar] [CrossRef]
- Olmo, C.; Casas, M.T.; Martínez, J.C.; Franco, L.; Puiggalí, J. Thermally Induced Structural Transitions of Nylon 4 9 as a New Example of Even–Odd Polyamides. Polymers 2018, 10, 198. [Google Scholar] [CrossRef]
- Brill, V.R. Beziehungen zwischen wasserstoffbindung und einigen eigenschaften von polyamiden. Die Makromol. Chem. Macromol. Chem. Phys. 1956, 18, 294–309. [Google Scholar] [CrossRef]
- Wendoloski, J.J.; Gardner, K.H.; Hirschinger, J.; Miura, H.; English, A.D. Molecular Dynamics in Ordered Structures: Computer Simulation and Experimental Results for Nylon 66 Crystals. Science 1990, 247, 431–436. [Google Scholar] [CrossRef]
- Feldman, A.Y.; Wachtel, E.; Vaughan, G.B.M.; Weinberg, A.; Marom, G. The Brill Transition in Transcrystalline Nylon-66. Macromolecules 2006, 39, 4455–4459. [Google Scholar]
- Nair, S.S.; Ramesh, C.; Tashiro, K. Crystalline Phases in Nylon-11: Studies Using HTWAXS and HTFTIR. Macromolecules 2006, 39, 2841–2848. [Google Scholar] [CrossRef]
- Wolanov, Y.; Feldman, A.; Harel, H.; Marom, G. Amorphous and crystalline phase interaction during the Brill transition in nylon 66. Express Polym. Lett. 2009, 3, 452–457. [Google Scholar] [CrossRef]
- Chi Wang, S.-Y.T. Hsuan-Sheng Lin Brill transition of nylon-6 in electrospun nanofibers. Colloid Polym. Sci. 2012, 290, 1799–1809. [Google Scholar] [CrossRef]
- Suzuki, H.; Ishii, S.; Sato, H.; Yamamoto, S.; Morisawa, Y.; Ozaki, Y.; Uchiyama, T.; Otani, C.; Hoshina, H. Brill transition of nylon-6 characterized by low-frequency vibration through terahertz absorption spectroscopy. Chem. Phys. Lett. 2013, 575, 36–39. [Google Scholar] [CrossRef]
- Franco, L.; Navarro, E.; Subirana, J.A.; Puiggali, J. Synthesis and Structure of Nylons 1,n. Macromolecules 1994, 27, 4284–4297. [Google Scholar] [CrossRef]
- Puiggalí, J.; Aceituno, J.E.; Navarro, E.; Campos, J.L.; Subirana, J.A. Structure of n,3 Polyamides, a Group of Nylons with Two Spatial Hydrogen-Bond Orientations. Macromolecules 1996, 29, 8170–8179. [Google Scholar] [CrossRef]
- Tereshko, V.; Navarro, E.; Puiggali, J.; Subirana, J.A. Structures of two malonamide derivatives as models of nylons n,3 and of peptidomimetic compounds. Macromolecules 1993, 26, 7024–7028. [Google Scholar] [CrossRef]
- Puiggali, J.; Muñoz-Guerra, S.; Subirana, J.A. Morphology and crystalline structure of nylon-2/6. Polymer 1987, 28, 209–212. [Google Scholar] [CrossRef]
- Bella, J.; Puiggalí, J.; Subirana, J.A. Glycine residues induce a helical structure in polyamides. Polymer 1994, 35, 1291–1297. [Google Scholar]
- Li, X.; Hong, Z.; Sun, J.; Geng, Y.; Huang, Y.; An, H.; Ma, Z.; Zhao, B.; Shao, C.; Fang, Y.; et al. Identifying the Phase Behavior of Biodegradable Poly(hexamethylene succinate-co-hexamethylene adipate) Copolymers with FTIR. J. Phys. Chem. B 2009, 113, 2695–2704. [Google Scholar]
- Li, X.; Sun, J.; Huang, Y.; Geng, Y.; Wang, X.; Ma, Z.; Shao, C.; Zhang, X.; Yang, C.; Li, L. Inducing New Crystal Structures through Random Copolymerization of Biodegradable Aliphatic Polyester. Macromolecules 2008, 41, 3162–3168. [Google Scholar]
- Liang, Z.; Pan, P.; Zhu, B.; Dong, T.; Hua, L.; Inoue, Y. Crystalline Phase of Isomorphic Poly(hexamethylene sebacate-co-hexamethylene adipate) Copolyester: Effects of Comonomer Composition and Crystallization Temperature. Macromolecules 2010, 43, 2925–2932. [Google Scholar] [CrossRef]
- Liang, Z.; Pan, P.; Zhu, B.; Inoue, Y. Isomorphic Crystallization of Poly(hexamethylene adipate-co-butylene adipate): Regulating Crystal Modification of Polymorphic Polyester from Internal Crystalline Lattice. Macromolecules 2010, 43, 6429–6437. [Google Scholar] [CrossRef]
- Liang, Z.; Pan, P.; Zhu, B.; Inoue, Y. Isomorphic crystallization of aliphatic copolyesters derived from 1,6-hexanediol: Effect of the chemical structure of comonomer units on the extent of cocrystallization. Polymer 2011, 52, 2667–2676. [Google Scholar] [CrossRef]
- Díaz, A.; Franco, L.; Puiggalí, J. Study on the crystallization of poly(butylene azelate-co-butylene succinate) copolymers. Thermochim. Acta 2014, 575, 45–54. [Google Scholar] [CrossRef]
- Weeks, J.D.H.a.J.J. Melting Process and Equilibrium Melting Temperature of Polychlorotrifluoroethylene. J. Res. Natl. Bureau Stand. Sect. Phys. Chem. 1962, 66, 13–28. [Google Scholar]
- Strobl, G. From the melt via mesomorphic and granular crystalline layers to lamellar crystallites: A major route followed in polymer crystallization? Eur. Phys. J. E 2000, 3, 165–183. [Google Scholar] [CrossRef]
- Marand, H.; Xu, J.; Srinivas, S. Determination of the Equilibrium Melting Temperature of Polymer Crystals: Linear and Nonlinear Hoffman−Weeks Extrapolations. Macromolecules 1998, 31, 8219–8229. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Lauritzen, J.I., Jr. Treatise on Solid State Chemistry; Plentum: New York, NY, USA, 1976. [Google Scholar]
- Park, J.W.; Kim, D.K.; Im, S.S. Crystallization behaviour of poly(butylene succinate) copolymers. Polymer Int. 2002, 51, 239–244. [Google Scholar] [CrossRef]
- Flory, P.J. Theory of crystallization in copolymers. Trans. Faraday Soc 1955, 51, 848–857. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Bikiaris, D.N. Crystallization and melting behavior of three biodegradable poly(alkylene succinates). A comparative study. Polymer 2005, 46, 12081–12092. [Google Scholar] [CrossRef]
- Baur, V.H. Einfluß der sequenzlängenverteilung auf das schmelz-ende von copolymeren. Die Makromol. Chem. 1966, 98, 297–301. [Google Scholar] [CrossRef]
- Krevelen, D.W.V. Properties of Polymers; Elsevier Science: Amsterdam, The Netherlands, 1997. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Yield (%) | fA | Mn (g/mol) | Mw (g/mol) | PDI |
|---|---|---|---|---|---|
| Nylon 4,5+9_0 | 50 | 0 | 16,000 | 40,000 | 2.5 |
| Nylon 4,5+9_15 | 54 | 0.15 | 18,000 | 43,000 | 2.4 |
| Nylon 4,5+9_40 | 53 | 0.45 | 17,000 | 41,000 | 2.4 |
| Nylon 4,5+9_50 | 60 | 0.55 | 22,000 | 51,000 | 2.3 |
| Nylon 4,5+9_60 | 58 | 0.73 | 20,000 | 44,000 | 2.2 |
| Nylon 4,5+9_85 | 57 | 0.90 | 21,000 | 47,000 | 2.2 |
| Nylon 4,5+9_100 | 63 | 1 | 20,000 | 46,000 | 2.3 |
| Sample | 1st Heating Scan | Cooling Scan | |||||
| Tf (°C) | ΔHf (J/g) | Tc (°C) | ΔHc (J/g) | ||||
| Nylon 4,5+9_0 | 233,249 | 56.8 | 212 | 30.1 | |||
| Nylon 4,5+9_15 | 207,225,237 | 50.3 | 203 | 29.4 | |||
| Nylon 4,5+9_40 | 205,224,239 | 57.2 | 199 | 39.3 | |||
| Nylon 4,5+9_50 | 206,224,234 | 65.0 | 196 | 38.9 | |||
| Nylon 4,5+9_60 | 207,225,236 | 72.0 | 200 | 48.6 | |||
| Nylon 4,5+9_85 | 213,229,239 | 82.2 | 209 | 56.0 | |||
| Nylon 4,5+9_100 | 215,233,244 | 104 | 218 | 69.2 | |||
| Sample | 2nd Heating Scan | 3rd Heating Scan | |||||
| Tf (°C) | ΔHf (J/g) | Tg (°C) | Tf (°C) | Thc (°C) | ΔHhc (J/g) | ΔHf (J/g) | |
| Nylon 4,5+9_0 | 221,244 | 35.9 | 71.2 | 239 | - | - | 36.6 |
| Nylon 4,5+9_15 | 205,224,228 | 31.0 | 54.9 | 188,209 | - | - | 208 |
| Nylon 4,5+9_40 | 175,201,222 | 43.5 | 50.2 | 178,218 | 197 | 2.7 | 5.5,32.3 |
| Nylon 4,5+9_50 | 178,198,220 | 45.3 | 50.6 | 178,220,228 | 199 | 0.6 | 5.6,32.1 |
| Nylon 4,5+9_60 | 177,201,224,234 | 59.9 | 52.6 | 177,222,232 | 200 | 3.1 | 5.6,45.7 |
| Nylon 4,5+9_85 | 185,212,228,237 | 64.4 | 52.6 | 183,227,237 | 207 | 3.5 | 1.9,54.4 |
| Nylon 4,5+9_100 | 195,233,244 | 86.6 | 50.0 | 195,231,243 | 236 | 0.4 | 39.9,39.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olmo, C.; Casas, M.T.; Martínez, J.C.; Franco, L.; Puiggalí, J. Crystalline Structures and Structural Transitions of Copolyamides Derived from 1,4-Diaminobutane and Different Ratios of Glutaric and Azelaic Acids. Polymers 2019, 11, 572. https://doi.org/10.3390/polym11040572
Olmo C, Casas MT, Martínez JC, Franco L, Puiggalí J. Crystalline Structures and Structural Transitions of Copolyamides Derived from 1,4-Diaminobutane and Different Ratios of Glutaric and Azelaic Acids. Polymers. 2019; 11(4):572. https://doi.org/10.3390/polym11040572
Chicago/Turabian StyleOlmo, Cristian, María T. Casas, Juan C. Martínez, Lourdes Franco, and Jordi Puiggalí. 2019. "Crystalline Structures and Structural Transitions of Copolyamides Derived from 1,4-Diaminobutane and Different Ratios of Glutaric and Azelaic Acids" Polymers 11, no. 4: 572. https://doi.org/10.3390/polym11040572
APA StyleOlmo, C., Casas, M. T., Martínez, J. C., Franco, L., & Puiggalí, J. (2019). Crystalline Structures and Structural Transitions of Copolyamides Derived from 1,4-Diaminobutane and Different Ratios of Glutaric and Azelaic Acids. Polymers, 11(4), 572. https://doi.org/10.3390/polym11040572