Dapivirine Bioadhesive Vaginal Tablets Based on Natural Polymers for the Prevention of Sexual Transmission of HIV

 ,
, 
 ,
,  ,
, 

Abstract
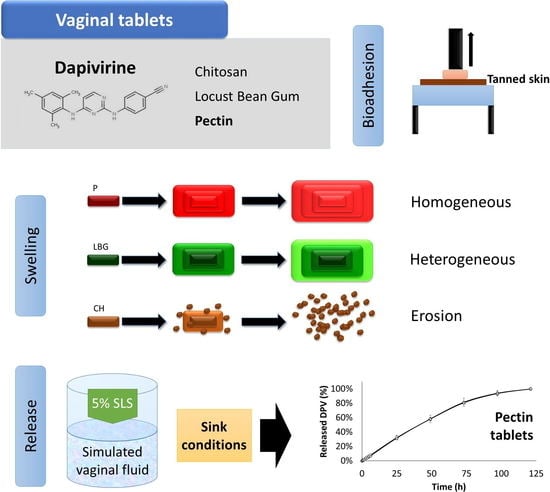
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preliminary Studies
Characterization of the Texture of Polymer Gels
Dapivirine Solubility Tests
2.2.2. Preparation of the Tablets
2.2.3. Assessment of the Tablets
Swelling Test
Drug Release Study
Bioadhesion Test
Cytotoxicity Evaluation
3. Results and Discussion
3.1. Preliminary Studies
3.1.1. Characterization of the Texture of Polymer Gels
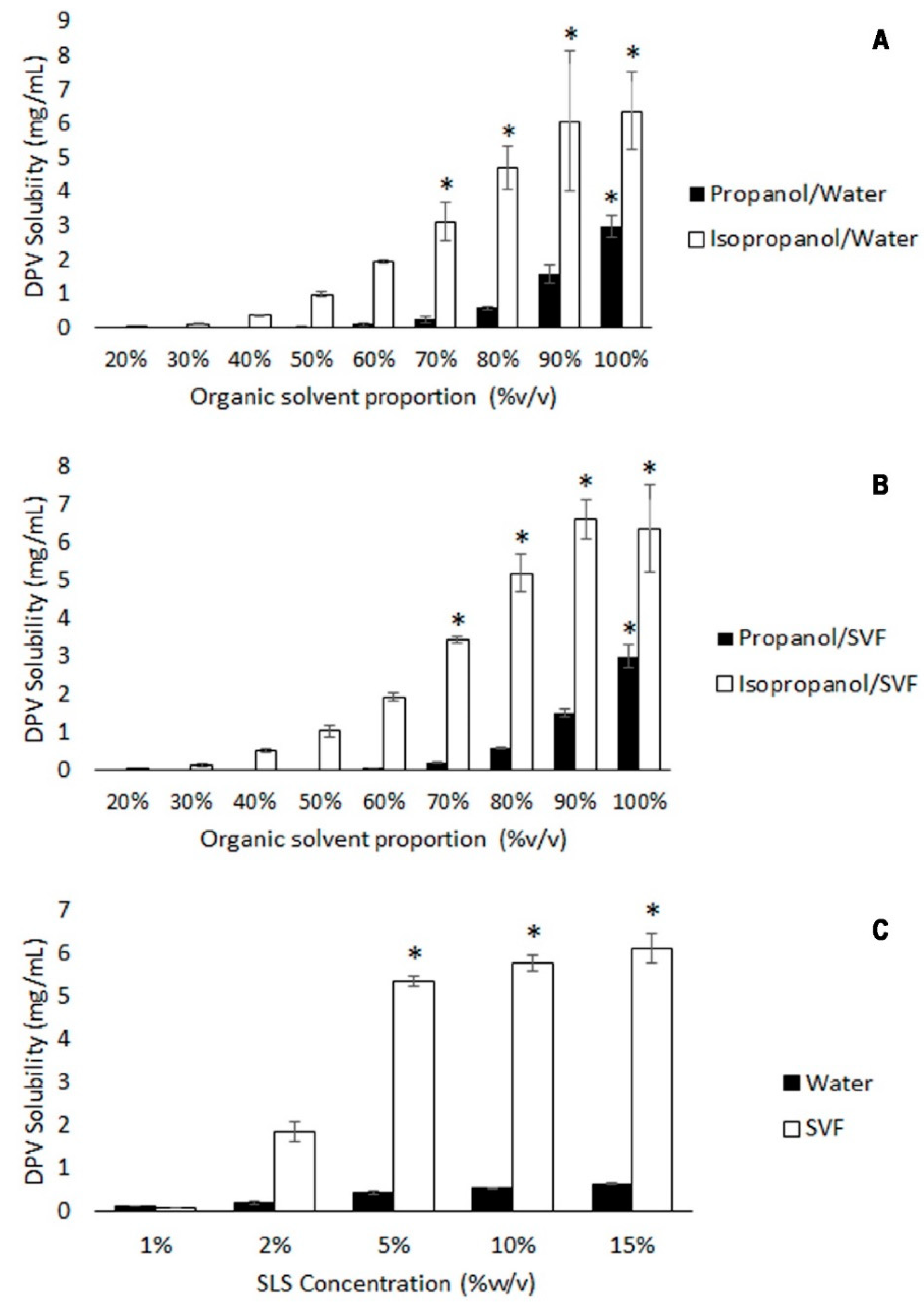
3.1.2. DPV Solubility Tests
3.2. Assessment of the Tablets
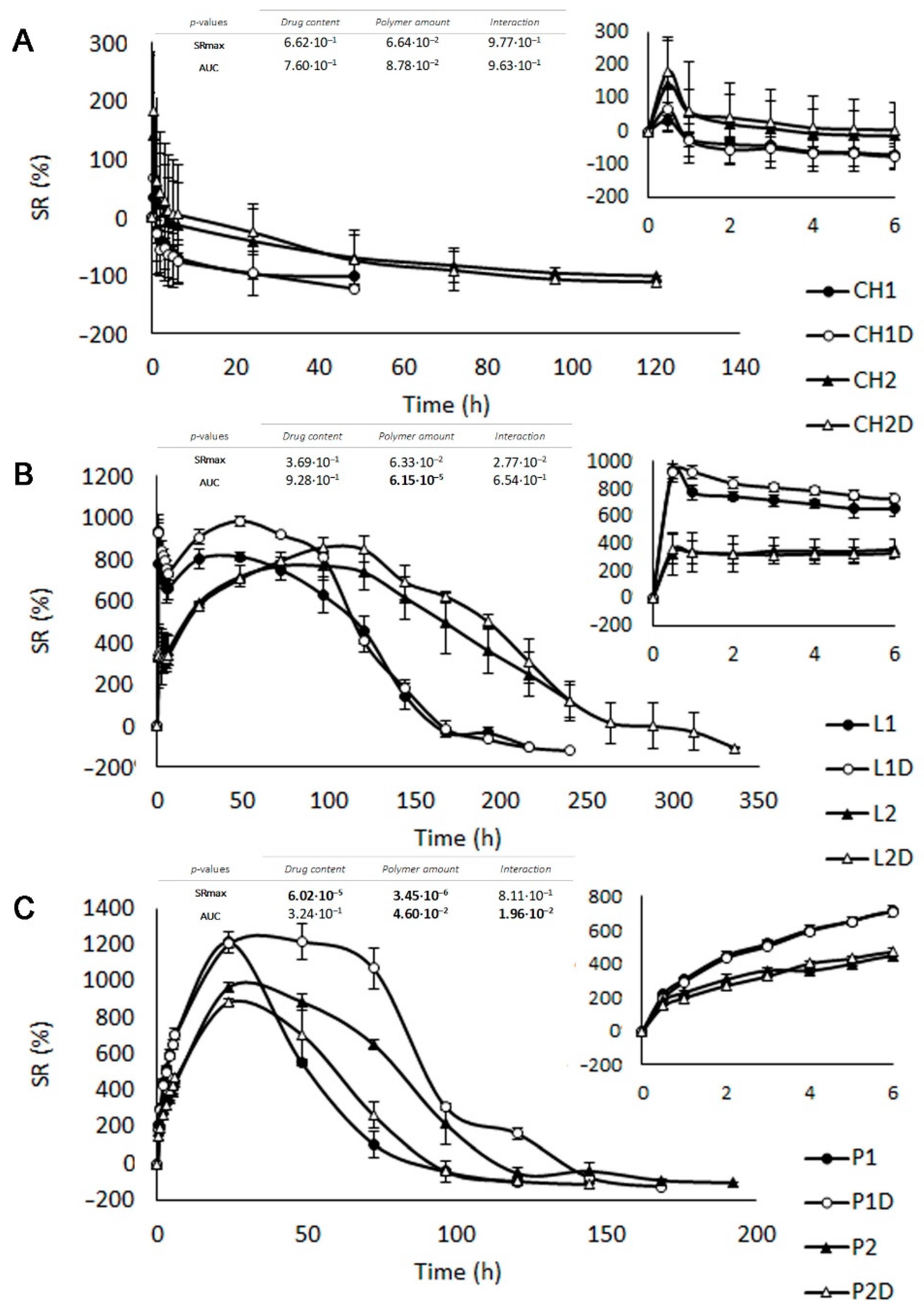
3.2.1. Swelling Test
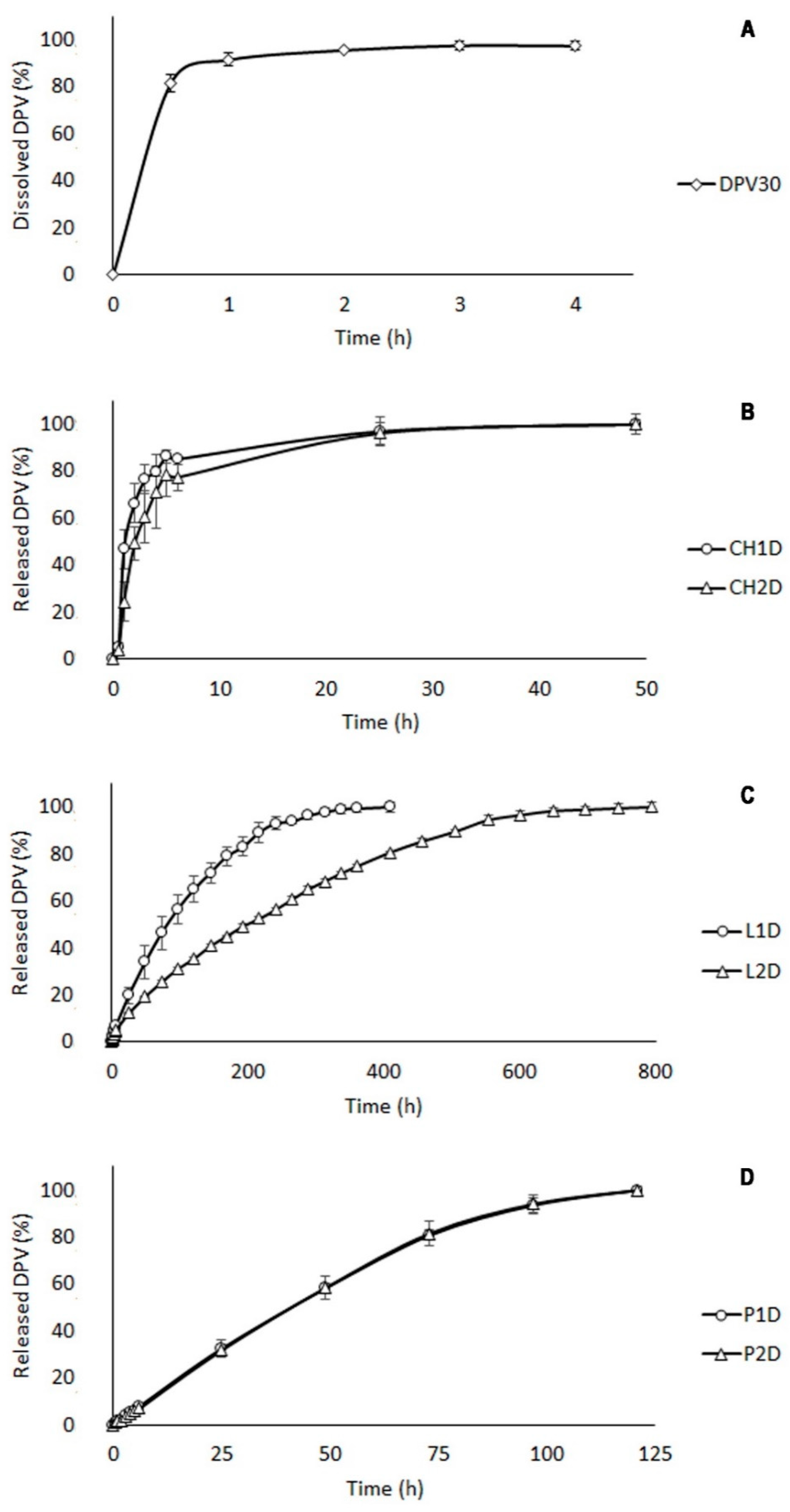
3.2.2. Drug Release Study
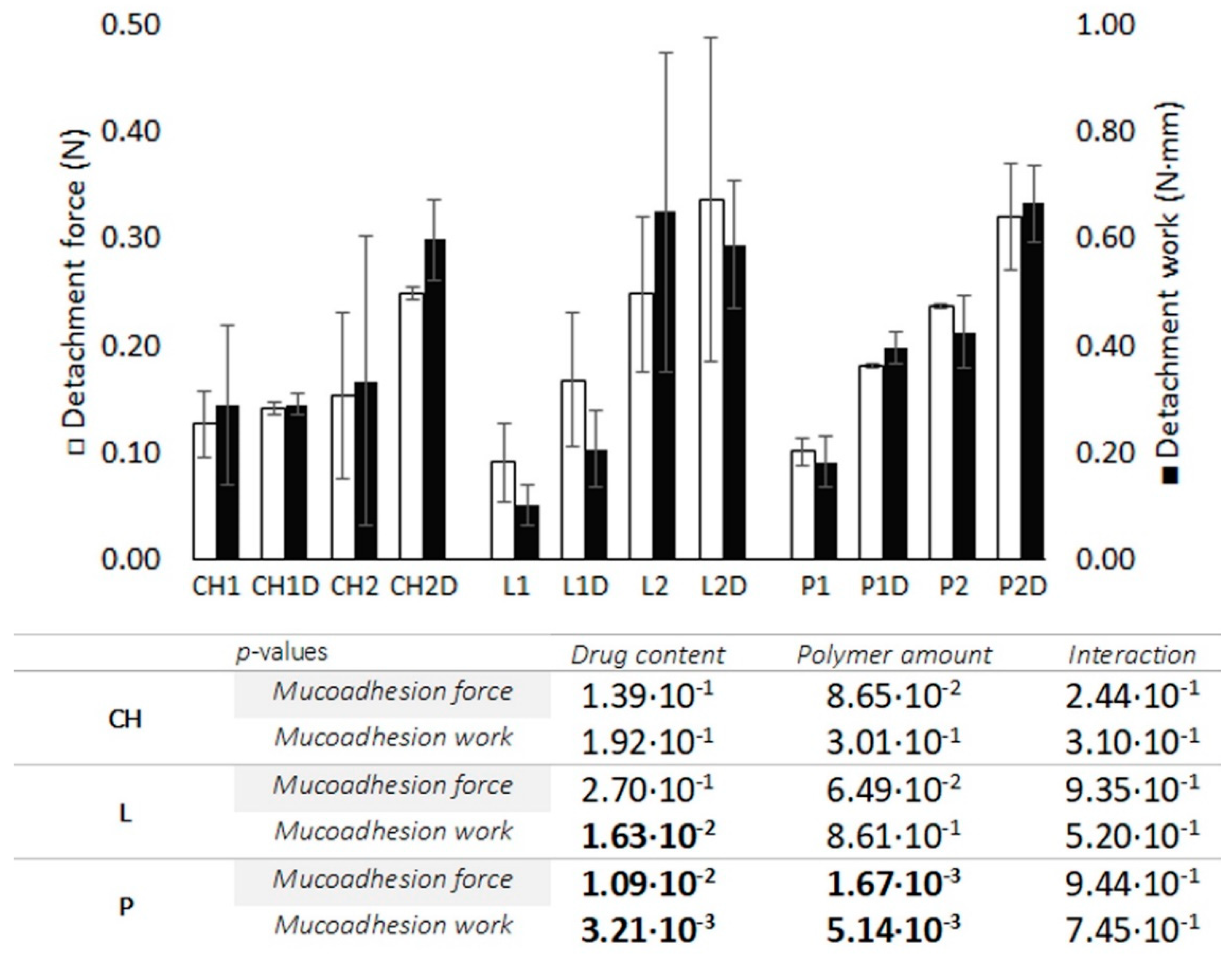
3.2.3. Bioadhesion Test
3.2.4. Cytotoxicity
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- UNAIDS. Fact Sheet—Latest Global and Regional Statistics on the Status of the AIDS Epidemic. 2017. Available online: http://www.unaids.org/sites/default/files/media_asset/unaids-data-2018_en.pdf (accessed on 15 December 2018).
- Abdool Karim, Q.; Humphries, H.; Stein, Z. Empowering women in human immunodeficiency virus prevention. Best Pract. Res. Clin. Obstet. Gynaecol. 2012, 26, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Abdool Karim, S.S.; Baxter, C. Overview of microbicides for the prevention of human immunodeficiency virus. Best Pract. Res. Clin. Obstet. Gynaecol. 2012, 26, 487–493. [Google Scholar] [CrossRef]
- Gengiah, T.N.; Abdool Karim, Q. Implementing microbicides in low-income countries. Best Pract. Res. Clin. Obstet. Gynaecol. 2012, 26, 495–501. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Das Neves, J.; Martins, J.P.; Sarmento, B. Will dapivirine redeem the promises of anti-HIV microbicides? Overview of product design and clinical testing. Adv. Drug Deliv. Rev. 2016, 103, 495–501. [Google Scholar] [CrossRef]
- Friend, D.R.; Kiser, P.F. Assessment of topical microbicides to prevent HIV-1 transmission: Concepts, testing, lessons learned. Antivir. Res. 2013, 99, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.L.; Kashuba, A.D.M. Formulation, pharmacokinetics and pharmacodynamics of topical microbicides. Best Pract. Res. Clin. Obstet. Gynaecol. 2012, 26, 451–462. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Notario-Pérez, F.; Ruiz-Caro, R.; Veiga-Ochoa, M.D. Historical development of vaginal microbicides to prevent sexual transmission of HIV in women: From past failures to future hopes. Drug Des. Devel. Ther. 2017, 11, 1767–1787. [Google Scholar] [CrossRef]
- Roberts, L.; Liebenberg, L.; Barnabas, S.; Passmore, J.A. Vaginal microbicides to prevent human immunodeficiency virus infection in women: Perspectives on the female genital tract, sexual maturity and mucosal inflammation. Best Pract. Res. Clin. Obstet. Gynaecol. 2012, 26, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.; Bekker, L.G.; Bukusi, E.; Hellström, E.; Kotze, P.; Louw, C.; Martinson, F.; Masenga, G.; Montgomery, E.; Ndaba, N.; et al. Safety, acceptability and adherence of dapivirine vaginal ring in a microbicide clinical trial conducted in multiple countries in sub-Saharan Africa. PLoS ONE 2016, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.; van Niekerk, N.; Kapiga, S.; Bekker, L.-G.; Gama, C.; Gill, K.; Kamali, A.; Kotze, P.; Louw, C.; Mabude, Z.; et al. Safety and Efficacy of a Dapivirine Vaginal Ring for HIV Prevention in Women. N. Engl. J. Med. 2016, 375, 2133–2143. [Google Scholar] [CrossRef] [PubMed]
- Halwes, M.E.; Steinbach-Rankins, J.M.; Frieboes, H.B. Pharmacokinetic modeling of a gel-delivered dapivirine microbicide in humans. Eur. J. Pharm. Sci. 2016, 93, 410–418. [Google Scholar] [CrossRef]
- Major, I.; Boyd, P.; Kilbourne-Brook, M.; Saxon, G.; Cohen, J.; Malcolm, R.K. A modified SILCS contraceptive diaphragm for long-term controlled release of the HIV microbicide dapivirine. Contraception 2013, 88, 58–66. [Google Scholar] [CrossRef]
- Akil, A.; Devlin, B.; Cost, M.; Rohan, L.C. Increased dapivirine tissue accumulation through vaginal film codelivery of dapivirine and tenofovir. Mol. Pharm. 2014, 11, 1533–1541. [Google Scholar] [CrossRef]
- McConville, C.; Major, I.; Devlin, B.; Brimer, A. Development of a multi-layered vaginal tablet containing dapivirine, levonorgestrel and acyclovir for use as a multipurpose prevention technology. Eur. J. Pharm. Biopharm. 2016, 104, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Valenta, C. The use of mucoadhesive polymers in vaginal delivery. Adv. Drug Deliv. Rev. 2005, 57, 1692–1712. [Google Scholar] [CrossRef]
- Cazorla-luna, R.; Notario-pérez, F.; Martín-illana, A.; Ruiz-caro, R. Chitosan-Based Mucoadhesive Vaginal Tablets for Controlled Release of the Anti-HIV Drug Tenofovir. Pharmaceutics 2019, 11, 20. [Google Scholar] [CrossRef]
- Berger, J.; Reist, M.; Mayer, J.M.; Felt, O.; Peppas, N.A.; Gurny, R. Structure and interactions in covalently and ionically crosslinked chitosan hydrogels for biomedical applications. Eur. J. Pharm. Biopharm. 2004, 57, 19–34. [Google Scholar] [CrossRef]
- Garcia, M.T.J.; de Paula Freitas, C.; Graciano, T.B.; Coutinho, T.S.; Cressoni, C.B.; de Lima Pereira, S.A.; Shimano, M.M. Chitosan-based mucoadhesive gel for oral mucosal toluidine blue O delivery: The influence of a non-ionic surfactant. Photodiagnosis Photodyn. Ther. 2017, 20, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Nady, N.; Kandil, S.H. Novel blend for producing porous chitosan-based films suitable for biomedical applications. Membranes 2018, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Notario-Pérez, F.; Martín-Illana, A.; Cazorla-Luna, R.; Ruiz-Caro, R.; Bedoya, L.M.; Tamayo, A.; Rubio, J.; Veiga, M.D. Influence of chitosan swelling behaviour on controlled release of tenofovir from mucoadhesive vaginal systems for prevention of sexual transmission of HIV. Mar. Drugs 2017, 15, 50. [Google Scholar] [CrossRef]
- Andersen, T.; Bleher, S.; Flaten, G.E.; Tho, I.; Mattsson, S.; Škalko-Basnet, N. Chitosan in mucoadhesive drug delivery: Focus on local vaginal therapy. Mar. Drugs 2015, 13, 222–236. [Google Scholar] [CrossRef]
- Dhanapal, J.; Balaraman Ravindrran, M. Chitosan/poly (lactic acid)-coated piceatannol nanoparticles exert an in vitro apoptosis activity on liver, lung and breast cancer cell lines. Artif. Cells Nanomed. Biotechnol. 2018, 1–9. [Google Scholar] [CrossRef]
- Sharma, A.K.; Gupta, L.; Sahu, H.; Qayum, A.; Singh, S.K.; Nakhate, K.T.; Ajazuddin; Gupta, U. Chitosan Engineered PAMAM Dendrimers as Nanoconstructs for the Enhanced Anti-Cancer Potential and Improved In vivo Brain Pharmacokinetics of Temozolomide. Pharm. Res. 2018, 35, 9. [Google Scholar] [CrossRef]
- Notario-Pérez, F.; Cazorla-Luna, R.; Martín-Illana, A.; Ruiz-Caro, R.; Tamayo, A.; Rubio, J.; Veiga, M.-D. Optimization of tenofovir release from mucoadhesive vaginal tablets by polymer combination to prevent sexual transmission of HIV. Carbohydr. Polym. 2018, 179, 305–316. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, M.P.; Martín-Illana, A.; Ruiz-Caro, R.; Bermejo, P.; Abad, M.J.; Carro, R.; Bedoya, L.M.; Tamayo, A.; Rubio, J.; Fernández-Ferreiro, A.; et al. Chitosan and Kappa-Carrageenan Vaginal Acyclovir Formulations for Prevention of Genital Herpes. In Vitro and Ex Vivo Evaluation. Mar. Drugs 2015, 13, 5976–5992. [Google Scholar] [CrossRef]
- Prajapati, V.D.; Jani, G.K.; Moradiya, N.G.; Randeria, N.P.; Nagar, B.J. Locust bean gum: A versatile biopolymer. Carbohydr. Polym. 2013, 94, 814–821. [Google Scholar] [CrossRef]
- Chakravorty, A.; Barman, G.; Mukherjee, S.; Sa, B. Effect of carboxymethylation on rheological and drug release characteristics of locust bean gum matrix tablets. Carbohydr. Polym. 2016, 144, 50–58. [Google Scholar] [CrossRef]
- Goulas, V.; Stylos, E.; Chatziathanasiadou, M.V.; Mavromoustakos, T.; Tzakos, A.G. Functional components of carob fruit: Linking the chemical and biological space. Int. J. Mol. Sci. 2016, 17, 1875. [Google Scholar] [CrossRef]
- Andersen, T.; Vanić, Ž.; Eide Flaten, G.; Mattsson, S.; Tho, I.; Škalko-Basnet, N. Pectosomes and chitosomes as delivery systems for metronidazole: The one-pot preparation method. Pharmaceutics 2013, 5, 445–456. [Google Scholar] [CrossRef]
- Baloǧlu, E.; Özyazici, M.; Yaprak Hizarcioǧlu, S.; Şenyiǧit, T.; Özyurt, D.; Pekçetin, C. Bioadhesive controlled release systems of ornidazole for vaginal delivery. Pharm. Dev. Technol. 2006, 11, 477–484. [Google Scholar] [CrossRef]
- Caswell, M.; Kane, M. Comparison of the moisturization efficacy of two vaginal moisturizers: Pectin versus polycarbophil technologies. J. Cosmet. Sci. 2002, 53, 81–87. [Google Scholar]
- Owen, D.H.; Katz, D.F. A vaginal fluid simulant. Contraception 1999, 59, 91–95. [Google Scholar] [CrossRef]
- Boyd, P.; Fetherston, S.M.; McCoy, C.F.; Major, I.; Murphy, D.J.; Kumar, S.; Holt, J.; Brimer, A.; Blanda, W.; Devlin, B.; et al. Matrix and reservoir-type multipurpose vaginal rings for controlled release of dapivirine and levonorgestrel. Int. J. Pharm. 2016, 511, 619–629. [Google Scholar] [CrossRef]
- Nakamura, S.; Ishii, N.; Nakashima, N.; Sakamoto, T.; Yuasa, H. Evaluation of Sucrose Fatty Acid Esters as Lubricants in Tablet Manufacturing. Chem. Pharm. Bull. (Tokyo) 2017, 65, 432–441. [Google Scholar] [CrossRef][Green Version]
- Rojewska, M.; Olejniczak-Rabinek, M.; Bartkowiak, A.; Snela, A.; Prochaska, K.; Lulek, J. The wettability and swelling of selected mucoadhesive polymers in simulated saliva and vaginal fluids. Colloids Surf. B Biointerfaces 2017, 156, 366–374. [Google Scholar] [CrossRef]
- Shah, V.P.; Tsong, Y.; Sathe, P.; Liu, J.P. In vitro dissolution profile comparison- Statistics and analysis of the similarity factor, f2. Pharm. Res. 1998, 15, 889–896. [Google Scholar] [CrossRef]
- Campaña-Seoane, M.; Peleteiro, A.; Laguna, R.; Otero-Espinar, F.J. Bioadhesive emulsions for control release of progesterone resistant to vaginal fluids clearance. Int. J. Pharm. 2014, 477, 495–505. [Google Scholar] [CrossRef]
- Harada, S.; Koyanagi, Y.; Yamamoto, N. Infection of HTLV-III/LAV in HTLV-I-carrying cells MT-2 and MT-4 and application in a plaque assay. Science 1985, 229, 563–566. [Google Scholar] [CrossRef]
- Rowe, R.C.; Sheskey, P.J.; Fenton, M.E. Handbook of Pharmaceutical Excipients: Pharmaceutical Excipients, 7th ed.; Pharmaceutical Press: London, UK, 2012. [Google Scholar] [CrossRef]
- Linhares, I.M.; Summers, P.R.; Larsen, B.; Giraldo, P.C.; Witkin, S.S. Contemporary perspectives on vaginal pH and lactobacilli. Am. J. Obstet. Gynecol. 2011, 204, 120.e1–120.e5. [Google Scholar] [CrossRef]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Kaur, M.; Gupta, K.M.; Poursaid, A.E.; Karra, P.; Mahalingam, A.; Aliyar, H.A.; Kiser, P.F. Engineering a degradable polyurethane intravaginal ring for sustained delivery of dapivirine. Drug Deliv. Transl. Res. 2011, 1, 223–237. [Google Scholar] [CrossRef]
- Woolfson, A.D.; Umrethia, M.L.; Kett, V.L.; Malcolm, R.K. Freeze-dried, mucoadhesive system for vaginal delivery of the HIV microbicide, dapivirine: Optimisation by an artificial neural network. Int. J. Pharm. 2010, 388, 136–143. [Google Scholar] [CrossRef]
- Thirawong, N.; Nunthanid, J.; Puttipipatkhachorn, S.; Sriamornsak, P. Mucoadhesive properties of various pectins on gastrointestinal mucosa: An in vitro evaluation using texture analyzer. Eur. J. Pharm. Biopharm. 2007, 67, 132–140. [Google Scholar] [CrossRef]
- Fletcher, P.; Harman, S.; Azijn, H.; Armanasco, N.; Manlow, P.; Perumal, D.; De Bethune, M.P.; Nuttall, J.; Romano, J.; Shattock, R. Inhibition of human immunodeficiency virus type 1 infection by the candidate microbicide dapivirine, a nonnucleoside reverse transcriptase inhibitor. Antimicrob. Agents Chemother. 2009, 53, 487–495. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Batch | DPV (mg) | CH (mg) | P (mg) | LBG (mg) | MgSt (mg) |
|---|---|---|---|---|---|
| CH1 | 145 | 3 | |||
| CH2 | 290 | 3 | |||
| CH1D | 30 | 145 | 3 | ||
| CH2D | 30 | 290 | 3 | ||
| L1 | 145 | 3 | |||
| L2 | 290 | 3 | |||
| L1D | 30 | 145 | 3 | ||
| L2D | 30 | 290 | 3 | ||
| P1 | 145 | 3 | |||
| P2 | 290 | 3 | |||
| P1D | 30 | 145 | 3 | ||
| P2D | 30 | 290 | 3 |
| Batch | Smax (%) | tmax (h) | terosion (h) | AUC (%·h) | S/a (mm2/g) |
|---|---|---|---|---|---|
| CH1 | 32.91 | 0.5 | 48 | 7.295 | 1.17 |
| CH2 | 138.57 | 0.5 | 120 | 40.224 | 1.03 |
| CH1D | 54.32 | 0.5 | 48 | 11.647 | 0.70 |
| CH2D | 162.51 | 0.5 | 120 | 46.494 | 0.68 |
| L1 | 793.64 | 48 | 216 | 1103.871 | 1.20 |
| L2 | 758.05 | 96 | 312 | 1587.312 | 1.06 |
| L1D | 799.63 | 48 | 240 | 1078.568 | 0.73 |
| L2D | 766.71 | 96 | 360 | 1626.576 | 0.69 |
| P1 | 1191.71 | 24 | 120 | 696.036 | 1.20 |
| P2 | 960.91 | 24 | 192 | 782.980 | 1.06 |
| P1D | 993.50 | 72 | 168 | 1001.369 | 0.73 |
| P2D | 796.31 | 24 | 144 | 594.549 | 0.67 |
| Batch | CH1D | CH2D | L1D | L2D | P1D | P2D |
|---|---|---|---|---|---|---|
| CH1D | 43.16 | 13.72 | 13.44 | 13.71 | 13.57 | |
| CH2D | 14.84 | 14.30 | 14.84 | 14.68 | ||
| L1D | 36.48 | 43.83 | 43.58 | |||
| L2D | 33.52 | 33.37 | ||||
| P1D | 97.61 | |||||
| P2D |
| CH1D | CH2D | L1D | L2D | P1D | P2D | ||
|---|---|---|---|---|---|---|---|
| Zero Order | K0 | 0.0084 | 0.0101 | 0.0029 | 0.0018 | 0.0090 | 0.0087 |
| R2 | 0.3361 | 0.4755 | 0.9011 | 0.9779 | 0.9723 | 0.9714 | |
| Hopfenberg | KHP | 0.0103 | 0.0113 | 0.0027 | 0.0016 | 0.0080 | 0.0081 |
| R2 | 0.6162 | 0.7402 | 0.9873 | 0.9844 | 0.9973 | 0.9978 | |
| Higuchi | KH | 0.3863 | 0.3603 | 0.0590 | 0.0380 | 0.0847 | 0.0821 |
| R2 | 0.6221 | 0.8866 | 0.9815 | 0.9908 | 0.9389 | 0.9322 | |
| Korsmeyer‒Peppas | nK | 1.8712 | 1.4864 | 0.8126 | 0.6597 | 0.9249 | 0.9511 |
| KKP | 0.2431 | 0.1478 | 0.0150 | 0.0151 | 0.0151 | 0.0134 | |
| R2 | 0.8480 | 0.9155 | 0.9895 | 0.9992 | 0.9957 | 0.9895 |
| Drug | Cell Line | CC50 |
|---|---|---|
| DPV | MT-2 | 1.9 µg/mL |
| HEC-1A | 11.2 µg/mL | |
| CH | MT-2 | >1000 µg/mL |
| HEC-1A | >1000 µg/mL | |
| LBG | MT-2 | >1000 µg/mL |
| HEC-1A | >1000 µg/mL | |
| P | MT-2 | >1000 µg/mL |
| HEC-1A | >1000 µg/mL |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cazorla-Luna, R.; Martín-Illana, A.; Notario-Pérez, F.; Bedoya, L.-M.; Bermejo, P.; Ruiz-Caro, R.; Veiga, M.-D. Dapivirine Bioadhesive Vaginal Tablets Based on Natural Polymers for the Prevention of Sexual Transmission of HIV. Polymers 2019, 11, 483. https://doi.org/10.3390/polym11030483
Cazorla-Luna R, Martín-Illana A, Notario-Pérez F, Bedoya L-M, Bermejo P, Ruiz-Caro R, Veiga M-D. Dapivirine Bioadhesive Vaginal Tablets Based on Natural Polymers for the Prevention of Sexual Transmission of HIV. Polymers. 2019; 11(3):483. https://doi.org/10.3390/polym11030483
Chicago/Turabian StyleCazorla-Luna, Raúl, Araceli Martín-Illana, Fernando Notario-Pérez, Luis-Miguel Bedoya, Paulina Bermejo, Roberto Ruiz-Caro, and María-Dolores Veiga. 2019. "Dapivirine Bioadhesive Vaginal Tablets Based on Natural Polymers for the Prevention of Sexual Transmission of HIV" Polymers 11, no. 3: 483. https://doi.org/10.3390/polym11030483
APA StyleCazorla-Luna, R., Martín-Illana, A., Notario-Pérez, F., Bedoya, L.-M., Bermejo, P., Ruiz-Caro, R., & Veiga, M.-D. (2019). Dapivirine Bioadhesive Vaginal Tablets Based on Natural Polymers for the Prevention of Sexual Transmission of HIV. Polymers, 11(3), 483. https://doi.org/10.3390/polym11030483