An Ab Initio Investigation of the 4,4′-Methlylene Diphenyl Diamine (4,4′-MDA) Formation from the Reaction of Aniline with Formaldehyde



Abstract

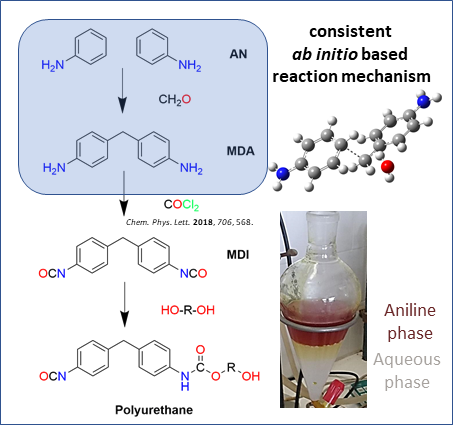
1. Introduction
2. Methods
3. Results and Discussion
3.1. Main Reaction Mechanism in Gas Phase
3.2. Gas Phase Thermodynamic Properties of Reactants, Intermediates, and Products
3.3. Solvent Effect
3.4. Proton Dissociation Constants (pKa) of Intermediates
3.5. Important Side Reactions
4. Conclusions
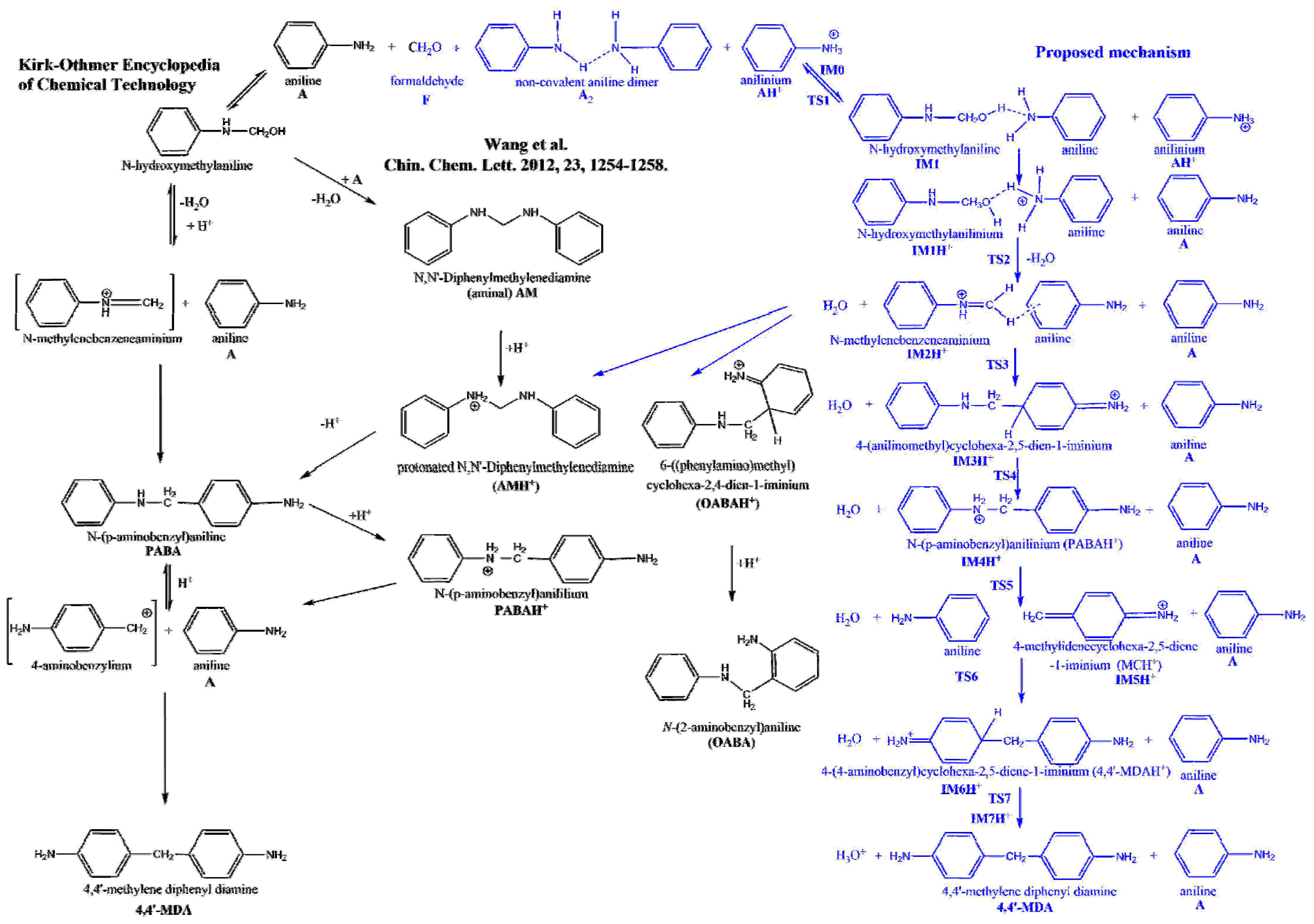
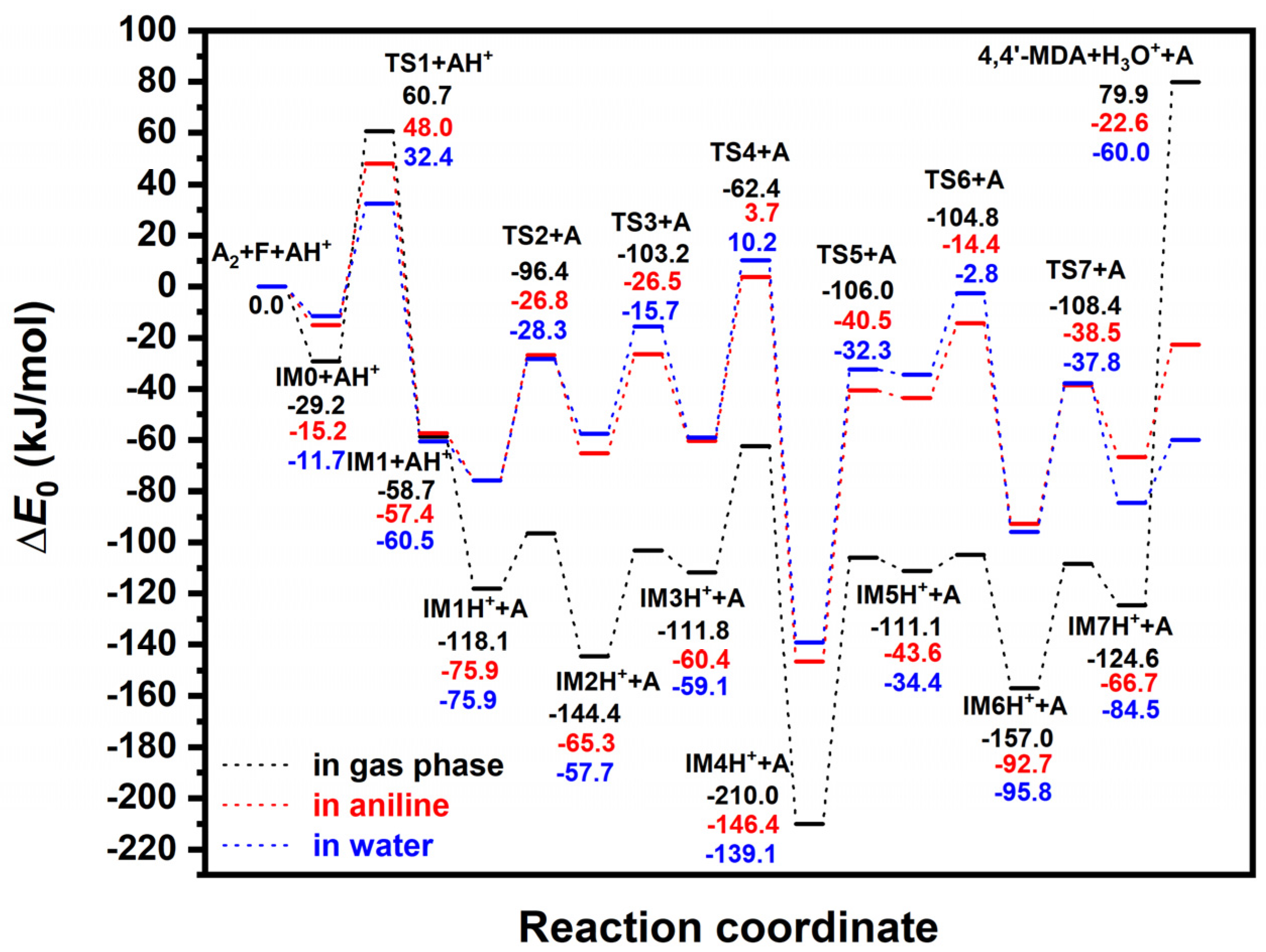
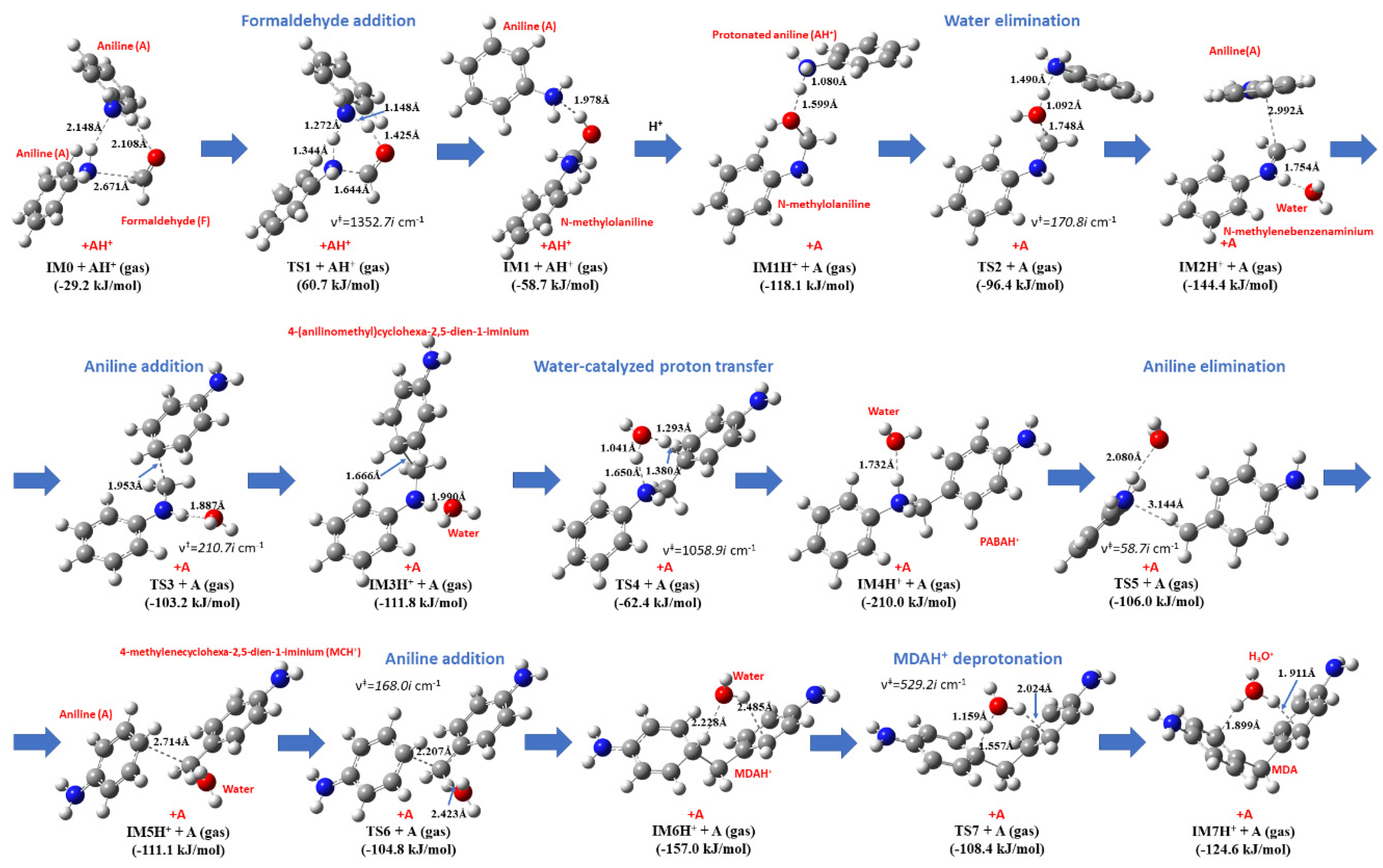
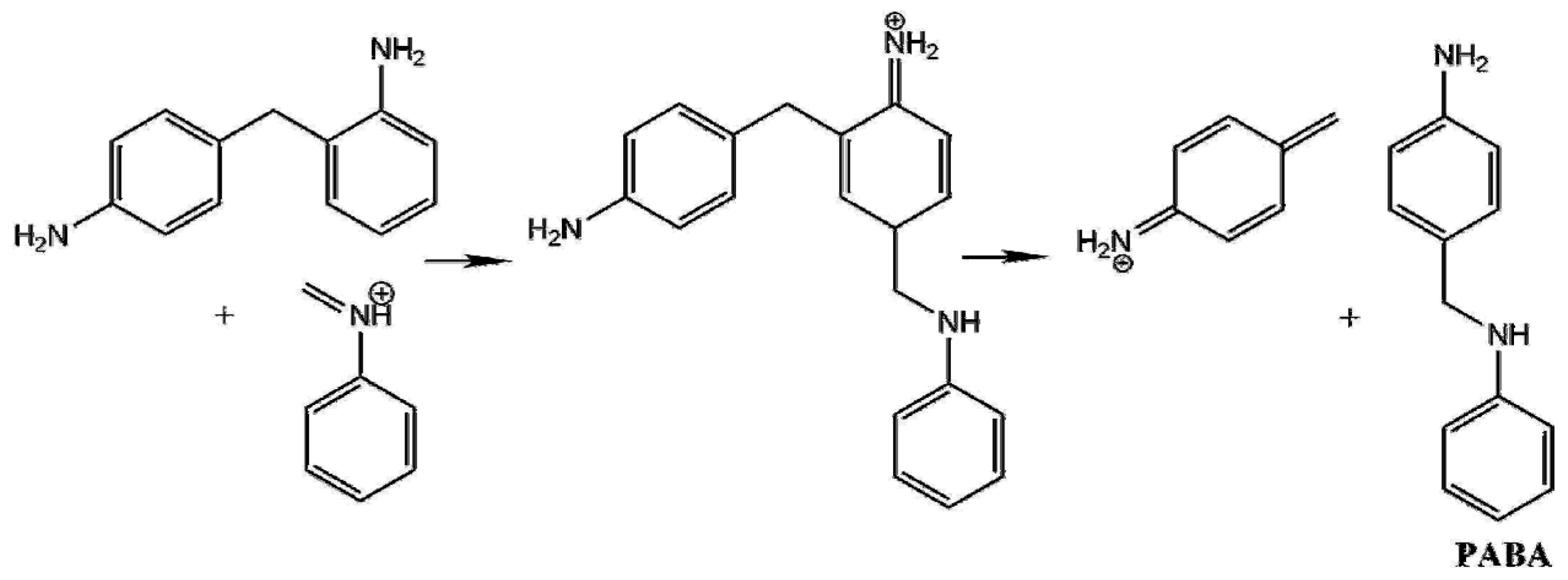
- The highest lying transition state (60.7 kJ/mol) corresponds to formaldehyde addition to aniline (TS1) leading to N-hydroxymethylaniline formation and its barrier heights significantly decreased by solvation. This step can be the main kinetic bottleneck for 4,4′-MDA production. After exothermic water elimination, aniline addition to N-methylenebenzenaminium took place through either tight transition state (resulted in 4-methylidenecyclohexa-2,5-diene-1-iminium) or loose transition state to form protonated aminal. However, aminal formation is both thermodynamically and kinetically preferred, there is no exit channel belong to it. Afterwards, proton shift can undergo in 4-methylidenecyclohexa-2,5-diene-1-iminium to produce protonated PABA, PABAH+, being the global minimum at this reactive potential energy surface, which can be also along with the line with experimental observation of PABA during MDA production. Two steps rearrangement of aniline resulted the protonated MDA isomers (MDAH+). It was found that aniline addition in ortho position is competitive with that of in the para position from both kinetic and thermodynamic points of view. The deprotonation of MDAH+ is thermodynamically more favorable in water phase.
- The species in proposed mechanism are proton activated, their possible deprotonation can lead to side reactions and appearance of deactivated intermediates in the industrial process. Therefore, the acid strength of four important intermediates such as N-methylenebenzeneanilium (4.2), PABAH+ (6.7), MCH+ (11.4), and AMH+ (5.1) was estimated using relative pKa calculation. Although, most of them found to be weak acid in aqueous solution, but they got more acidic in aniline (basic) environment which can then deactivate the intermediates.
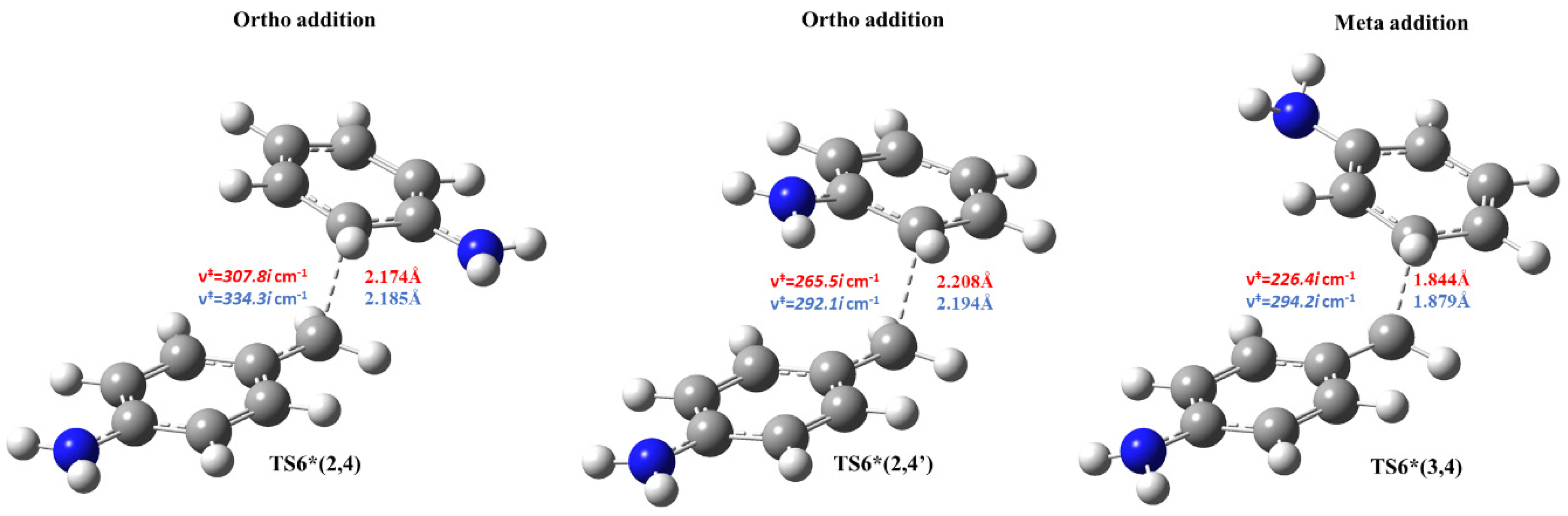
- Aniline addition-type side reactions had been also investigated and it was found that aminal formation is both thermodynamically and kinetically preferable, but it is a kinetic dead-end. Both 2,4- and 4,4′-MDAH+ formation from MCH+ and A has low-lying transition state and TS is submerged in the case of 2,4-MDAH+ making likely the formation of 2,4-MDAH+ beside 4,4-MDAH+.
- Gas phase thermodynamic properties for the reactants, products and intermediates were determined and carefully compared to the literature. Based on our G3MP2B3 and CBS-QB3 calculations, accurate standard enthalpy of formation is recommended for the intermediates.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sonnenschein, M.F. Polyurethanes: Science, Technology, Markets, and Trends; Dow Chemical Company: Midland, MI, USA, 2015; p. 73. [Google Scholar]
- Mustata, F.R.; Tudorachi, N.; Bicu, I. Epoxy Resins Cross-Linked with Bisphenol A/Methylenedianiline Novolac Resin Type: Curing and Thermal Behavior Study. Ind. Eng. Chem. Res. 2012, 51, 8415–8424. [Google Scholar] [CrossRef]
- Perkins, G.T. Process for the Preparation of 4,4′-methylenedianiline. U.S. Patent 3,367,969, 6 February 1968. [Google Scholar]
- Botella, P.; Corma, A.; Carr, R.H.; Mitchell, C.J. Towards an industrial synthesis of diamino diphenyl methane (DADPM) using novel delaminated materials: A breakthrough step in the production of isocyanates for polyurethanes. Appl. Catal. A Gen. 2011, 398, 143–149. [Google Scholar] [CrossRef]
- de Angelis, A.; Ingallina, P.; Perego, C. Solid Acid Catalysts for Industrial Condensations of Ketones and Aldehydes with Aromatics. Ind. Eng. Chem. Res. 2004, 43, 1169–1178. [Google Scholar] [CrossRef]
- Nafziger, J.L.; Rader, L.A.; Seward, I.J. Process for preparing polyamines with ion exchange resin catalysts. U.S. Patent 4,554,378, 19 November 1985. [Google Scholar]
- Corma, A.; Botella, P.; Mitchell, C. Replacing HCl by solid acids in industrial processes: Synthesis of diamino diphenyl methane (DADPM) for producing polyurethanes. Chem. Commun. 2004, 10, 2008–2010. [Google Scholar] [CrossRef] [PubMed]
- Salzinger, M.; Fichtl, M.B.; Lercher, J.A. On the influence of pore geometry and acidity on the activity of parent and modified zeolites in the synthesis of methylenedianiline. Appl. Catal. A Gen. 2011, 393, 189–194. [Google Scholar] [CrossRef]
- Keller, T.C.; Arras, J.; Wershofen, S.; Perez-Ramirez, J. Design of hierarchical zeolite catalysts for the manufacture of polyurethane intermediates. ACS Catal. 2015, 5, 734–743. [Google Scholar] [CrossRef]
- Tian, J.; An, H.; Cheng, X.; Zhao, X.; Wang, Y. Synthesis of 4,4′-methylenedianiline catalyzed by SO3H-functionalized ionic liquids. Ind. Eng. Chem. Res. 2015, 54, 7571–7579. [Google Scholar] [CrossRef]
- Wegener, G.; Brandt, M.; Duda, L.; Hofmann, J.; Klesczewski, B.; Koch, D.; Kumpf, R.J.; Orzesek, H.; Pirkl, H.G.; Six, C.; et al. Trends in industrial catalysis in the polyurethane industry. Appl. Catal. A Gen. 2001, 221, 303–335. [Google Scholar] [CrossRef]
- Moore, W.M. Amines, Aromatic, Methylenedianiline. In Kirk-Othmer Encyclopedia of Chemical Technology; Grayson, M., Ed.; John Wiley and Sons: Hoboken, NJ, USA, 2007; pp. 338–348. [Google Scholar]
- Wang, C.Y.; Li, H.Q.; Wang, L.G.; Cao, Y.; Liu, H.T.; Zhang, Y. Insights on the mechanism for synthesis of methylenedianiline from aniline and formaldehyde through HPLC-MS and isotope tracer studies. Chin. Chem. Lett. 2012, 23, 1254–1258. [Google Scholar] [CrossRef]
- Stow, S.M.; Onifer, T.M.; Forsythe, J.G.; Nefzger, H.; Kwiecien, N.W.; May, J.C.; McLean, J.A.; Hercules, D.M. Structural characterization of methylenedianiline regioisomers by ion mobility-mass spectrometry, tandem mass spectrometry, and computational strategies. 2. Electrospray spectra of 3-ring and 4-ring isomers. Anal. Chem. 2015, 87, 6288–6296. [Google Scholar] [CrossRef] [PubMed]
- Griswold, J. Analysis of aniline-water solutions. Ind. Eng. Chem. Anal. Ed. 1940, 12, 89–90. [Google Scholar] [CrossRef]
- Katayama, M.; Ashiki, S.; Amakasu, T.; Ozutsumi, K. Liquid structure of benzene and its derivatives as studied by means of X-ray scattering. Phys. Chem. Liq. 2010, 48, 797–809. [Google Scholar] [CrossRef]
- Kishore, K.; Santhanalakshmi, K.N. A thermochemical study on the reactions of aniline with formaldehyde in the presence of acid medium. Thermochim. Acta 1983, 68, 59–74. [Google Scholar] [CrossRef]
- Wang, C.; Li, H.; Cao, Y.; Liu, H.; Hou, X.; Zhang, Y. Thermodynamic analysis on synthesis of 4,4-methylenedianiline by reaction of aniline and formaldehyde. CIESC J. 2012, 63, 2348–2355. [Google Scholar]
- Boros, R.Z.; Koós, T.; Wafaa, C.; Nehéz, K.; Farkas, L.; Viskolcz, B.; Szőri, M. A theoretical study on the phosgenation of methylene diphenyl diamine (MDA). Chem. Phys. Lett. 2018, 706, 568–576. [Google Scholar] [CrossRef]
- Marsi, I.; Viskolcz, B.; Seres, L. Application of the group additivity method to alkyl radicals: An ab initio study. J. Phys. Chem. A 2000, 104, 4497–4504. [Google Scholar] [CrossRef]
- Viskolcz, B.; Berces, T. Enthalpy of formation of selected carbonyl radicals from theory and comparison with experiment. Phys. Chem. Chem. Phys. 2002, 2, 5430–5436. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K.; Rassolov, V.; Pople, J.A. Gaussian-3 theory using reduced Møller-Plesset order. J. Chem. Phys. 1999, 110, 4703–4709. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Dennington, R.D.; Keith, T.A.; Millam, J.M. GaussView05; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Montgomery, J.A.; Frisch, M.J.; Ochterski, J.W.; Petersson, G.A. A complete basis set model chemistry. VI. Use of density functional geometries and frequencies. J. Chem. Phys. 1999, 110, 2822–2827. [Google Scholar] [CrossRef]
- Nicolaides, A.; Rauk, A.; Glukhovtsev, M.N.; Radom, L. Heats of formation from G2, G2(MP2), and G2(MP2, SVP) total energies. J. Phys. Chem. 1996, 100, 17460–17464. [Google Scholar] [CrossRef]
- Active Thermochemical Tables (ATcT) Values Based on Ver. 1.122 of the Thermochemical Network. 2016. Available online: https://atct.anl.gov/ (accessed on 17 December 2017).
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on the generalized born approximation with asymmetric descreening. J. Chem. Theory Comput. 2009, 5, 2447–2464. [Google Scholar] [CrossRef] [PubMed]
- Liptak, M.D.; Gross, K.C.; Seybold, P.G.; Feldgus, S.; Shields, G.C. Absolute pKa Determinations for Substituted Phenols. J. Am. Chem. Soc. 2002, 124, 6421–6427. [Google Scholar] [CrossRef] [PubMed]
- McQuarrie, D.A. Statistical Mechanics; Harper & Row: New York, NY, USA, 1970. [Google Scholar]
- Liptak, M.D.; Shields, G.C. Accurate pKa calculations for carboxylic acids using complete basis set and Gaussian-n models combined with CPCM continuum solvation methods. J. Am. Chem. Soc. 2001, 123, 7314–7319. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Extended Third Millennium Ideal Gas and Condensed Phase Thermochemical Database for Combustion with Updates from Active Thermochemical Tables. 2005. Available online: http://garfield.chem.elte.hu/Burcat/hf.doc (accessed on 24 November 2017).
- NIST Computational Chemistry Comparison and Benchmark Database. NIST Standard Reference Database No. 101. 2006. Available online: https://cccbdb.nist.gov/exp1x.asp (accessed on 24 November 2017).
- Benson, S.W. Thermochemical Kinetics: Methods for the Estimation of Thermochemical Data and Rate Parameters, 2nd ed.; John Wiley: New York, NY, USA, 1976. [Google Scholar]
- Heller, S.R. NIST Structures and Properties Database and Estimation Program. J. Chem. Inf. Comput. Sci. 1991, 31, 432–434. [Google Scholar] [CrossRef]
- NIST Chemistry WebBook, SRD 69. Group Additivity Based Estimates. Available online: http://webbook.nist.gov/chemistry/grp-add/ (accessed on 24 November 2017).
- Zhang, J.; Sun, Y.; Mao, C.; Gao, H.; Zhou, W.; Zhou, Z. Theoretical study of pKa for perchloric acid. J. Mol. Struct. THEOCHEM 2009, 906, 46–49. [Google Scholar] [CrossRef]
- Muckerman, J.T.; Skone, J.H.; Ning, M.; Wasada-Tsutsui, Y. Toward the accurate calculation of pKa values in water and acetonitrile. Biochim. Biophys. Acta 2013, 1827, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Ghalami-Choobar, B.; Ghiami-Shomami, A.; Nikparsa, P. Theoretical Calculation of pKb Values for Anilines and Sulfonamide Drugs in Aquous Solution. J. Theor. Comput. Chem. 2012, 11, 283–295. [Google Scholar] [CrossRef]
- Behjatmanesh-Ardakani, R.; Safaeian, N. pKa predictions of some aniline derivatives by ab initio calculations. Iran. Chem. Commun. 2014, 2, 147–156. [Google Scholar]
- Lu, H.; Chen, X.; Zhan, C.G. First-principles calculation of pKa for cocaine, nicotine, neurotransmitters, and anilines in aqueous solution. J. Phys. Chem. B 2007, 111, 10599–10605. [Google Scholar] [CrossRef] [PubMed]
- Perrin, D.D. Dissociation Constants of Organic Bases in Aqueous Solution; Butterworths: London, UK, 1972. [Google Scholar]
- Knjasev, V. Beiträge zur reaktionskinetischen Untersuchung der säureinduzierten Anilin-Formaldehyd-Kondensation. Ph.D. Thesis, Universität Stuttgart, Stuttgart, Germany, 2007. [Google Scholar]
- Wanhua-BorsodChem Zrt (Bolyai tér 1., H-3700 Kazincbarcika, Hungary). Personal Communication, 2019.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | ΔE0 | ΔH(T) | ΔG(T,P) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Gas | Aniline | Water | Gas | Aniline | Water | Gas | Aniline | Water | |
| A2 + F + AH+ | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| IM0 + AH+ | −29.2 | −15.2 | −11.7 | −30.0 | −15.2 | −11.7 | 17.2 | 26.3 | 31.3 |
| TS1 + AH+ | 60.7 | 48.0 | 32.4 | 52.4 | 40.2 | 24.6 | 121.7 | 106.3 | 92.5 |
| IM1 + AH+ | −58.7 | −57.4 | −60.5 | −64.0 | −61.9 | −65.6 | −6.2 | −10.1 | −8.3 |
| IM1H+ + A | −118.1 | −75.9 | −75.9 | −123.3 | −80.3 | −81.0 | −68.6 | −26.4 | −23.8 |
| TS2 + A | −96.4 | −26.8 | −28.3 | −121.6 | −53.0 | −53.8 | 38.2 | 106.2 | 102.3 |
| IM2H+ + A | −144.4 | −65.3 | −57.7 | −162.4 | −83.2 | −75.2 | −18.5 | 58.0 | 64.0 |
| TS3 + A | −103.2 | −26.4 | −15.7 | −124.4 | −46.3 | −35.1 | 30.9 | 104.1 | 114.2 |
| IM3H+ + A | −111.8 | −60.4 | −59.1 | −132.9 | −80.5 | −79.6 | 23.8 | 70.0 | 72.7 |
| TS4 + A | −62.4 | 3.7 | 10.2 | −88.3 | −20.5 | −13.6 | 81.1 | 141.0 | 148.1 |
| IM4H+ + A | −210.0 | −146.4 | −139.1 | −231.8 | −167.5 | −160.5 | −72.9 | −14.6 | −6.9 |
| TS5 + A | −106.0 | −40.5 | −32.3 | −125.0 | −57.7 | −49.9 | 21.0 | 79.9 | 87.8 |
| IM5H+ + A | −111.1 | −43.6 | −34.4 | −127.3 | −58.9 | −49.3 | 15.1 | 72.3 | 83.0 |
| TS6 + A | −104.8 | −14.4 | −2.8 | −123.0 | −29.9 | −20.7 | 28.0 | 108.8 | 128.2 |
| IM6H+ + A | −157.0 | −92.7 | −95.8 | −177.3 | −110.7 | −114.3 | −20.0 | 34.5 | 33.4 |
| TS7 + A | −108.4 | −38.5 | −37.8 | −132.3 | −60.5 | −60.1 | 35.7 | 98.4 | 101.2 |
| IM7H+ + A | −124.6 | −66.7 | −84.5 | −147.3 | −88.1 | −06.3 | 17.0 | 69.0 | 53.1 |
| 4,4′-MDA + H3O+ + A | 79.9 | −22.6 | −60.0 | 60.0 | −41.5 | −79.3 | 176.7 | 67.6 | 32.8 |
| Species | Δf,298.15KH0 (g) | Method | Ref. | S0(g) | Cv(g) | Ref. |
|---|---|---|---|---|---|---|
| kJ/mol | J/molK | J/molK | ||||
| aniline (A) | 86.5 (0.5) | AS(G3MP2B3) | 1 | 319.0 | 96.6 | 1 |
| 96.0 (9.0) | AS(CBS-QB3) | 1 | 317.3 | 97.4 | ||
| 87.0 ± 0.88 | Burcat | [34] | 311.6 | 104.5 | [34] | |
| non-covalent aniline dimer (A2) | 156.2 | AS(G3MP2B3) | 311.7 | 529.6 | 214.3 | 1 |
| formaldehyde (F) | −111.5 (2.3) | AS(G3MP2B3) | 1 | 224.4 | 26.8 | 1 |
| −113.3 (4.1) | AS(CBS-QB3) | 1 | 224.3 | 26.8 | ||
| −109.2 ± 0.11 | Ruscic ATcT | [28] | 218.8 | 35.4 | [35] | |
| 4,4′-methylene diphenyl diamine (4,4′-MDA) | 171.2 | AS(G3MP2B3) | [19] | 500.1 | 221.4 | 1 |
| 191.5 | AS(CBS-QB3) | 503.6 | 223.3 | |||
| 165.6 | additivity rule | [18] | 522.7 | n.a. | [18] | |
| 172 | additivity rule [36,37] | NIST [37,38] | 511.6 | 234.7 | [38] | |
| 2,4-MDA | 159.4 | AS(G3MP2B3) | 1 | 490.4 | 220.6 | 1 |
| 2′,4-MDA | 168 | AS(G3MP2B3) | 1 | 484.0 | 219.8 | 1 |
| 3,4-MDA | 168.3 | AS(G3MP2B3) | 1 | 499.4 | 221.3 | 1 |
| N-(p-aminobenzyl)aniline (PABA) | 202.4 | AS(G3MP2B3) | 1 | 496.3 | 216.5 | 1 |
| 201.3 | additivity rule | [18] | 514.4 | n.a. | [18] | |
| N-hydroxymethylaniline | −71.8 | AS(G3MP2B3) | 1 | 379.3 | 129.3 | 1 |
| protonated aniline (AH+) | 739.4 | AS(G3MP2B3) | 1 | 339.9 | 97.2 | 1 |
| N-methylenebenzeneaminium | 828.6 | AS(G3MP2B3) | 1 | 346.2 | 107.9 | 1 |
| 4-(anilinomethyl)cyclo-hexa-2,5-dien-1-iminium | 858.5 | AS(G3MP2B3) | 1 | 491.2 | 220.0 | 1 |
| p-aminobenzylaniline (PABAH+) | 785.1 | AS(G3MP2B3) | 1 | 501.2 | 219.4 | 1 |
| 4-methylidenecyclohexa-2,5-diene-1-iminium (MCH+) | 802.8 | AS(G3MP2B3) | 1 | 340.9 | 113.7 | 1 |
| 4,4′-MDAH+ | 814.3 | AS(G3MP2B3) | 1 | 494.8 | 225.1 | 1 |
| 2,4-MDAH+ | 812.2 | AS(G3MP2B3) | 1 | 494.7 | 226.4 | 1 |
| 2′,4-MDAH+ | 830.9 | AS(G3MP2B3) | 1 | 487.1 | 224.4 | 1 |
| 3,4-MDAH+ | 858.6 | AS(G3MP2B3) | 1 | 534.2 | 236.5 | 1 |
| Species | pKa,aq | pKa,aq | |
|---|---|---|---|
| AH+ | 4.6 1 | 4.60 [44] | |
| N-methylenebenzeneanilium |  | 4.2 | |
| PABAH+ |  | 6.7 | |
| MCH+ |  | 11.4 | |
| AMH+ |  | 5.1 |
| Reaction | Aminal (AMH+) Formation | IM3H+ Formation | OABA+ Formation | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Gas | Aniline | Water | Gas | Aniline | Water | Gas | Aniline | Water | |
| ΔrE0(kJ/mol) | −90.7 | −75.2 | −77.2 | 32.7 | 4.9 | −1.4 | −61.1 | −24.9 | −26.8 |
| ΔrH0(kJ/mol) | −92.2 | −76.6 | −78.8 | 29.6 | 2.7 | −4.4 | −62.4 | −25.8 | −28.0 |
| ΔrG0(kJ/mol) | -41.1 | −26.5 | −28.6 | 42.2 | 12.0 | 8.7 | −7.7 | 27.1 | 26.0 |
| Reaction | ΔrE0 (kJ/mol) | Δ‡E0 (kJ/mol) | ||
|---|---|---|---|---|
| Water | Aniline | Water | Aniline | |
| IM5H+ + A → TS6 → IM6H+ + A 1 | −49.2 | −61.4 | 29.2 | 31.6 |
| A+MCH+ → TS6 → 4,4′-MDAH+ | −62.2 | −64.4 | 17.8 | 22.5 |
| A+MCH+ → TS6* → 2,4 MDAH+ | −45.1 | −49.0 | 15.5 | 18.9 |
| A+MCH+ → TS6* → 2′,4-MDAH+ | −52.7 | −53.6 | −1.9 | 2.3 |
| A+MCH+ → TS6* → 3,4-MDAH+ | 48.4 | 45.2 | 54.8 | 57.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boros, R.Z.; Farkas, L.; Nehéz, K.; Viskolcz, B.; Szőri, M. An Ab Initio Investigation of the 4,4′-Methlylene Diphenyl Diamine (4,4′-MDA) Formation from the Reaction of Aniline with Formaldehyde. Polymers 2019, 11, 398. https://doi.org/10.3390/polym11030398
Boros RZ, Farkas L, Nehéz K, Viskolcz B, Szőri M. An Ab Initio Investigation of the 4,4′-Methlylene Diphenyl Diamine (4,4′-MDA) Formation from the Reaction of Aniline with Formaldehyde. Polymers. 2019; 11(3):398. https://doi.org/10.3390/polym11030398
Chicago/Turabian StyleBoros, R. Zsanett, László Farkas, Károly Nehéz, Béla Viskolcz, and Milán Szőri. 2019. "An Ab Initio Investigation of the 4,4′-Methlylene Diphenyl Diamine (4,4′-MDA) Formation from the Reaction of Aniline with Formaldehyde" Polymers 11, no. 3: 398. https://doi.org/10.3390/polym11030398
APA StyleBoros, R. Z., Farkas, L., Nehéz, K., Viskolcz, B., & Szőri, M. (2019). An Ab Initio Investigation of the 4,4′-Methlylene Diphenyl Diamine (4,4′-MDA) Formation from the Reaction of Aniline with Formaldehyde. Polymers, 11(3), 398. https://doi.org/10.3390/polym11030398