Fabrication of Phosphate-Imprinted PNIPAM/SiO2 Hybrid Particles and Their Phosphate Binding Property

Abstract
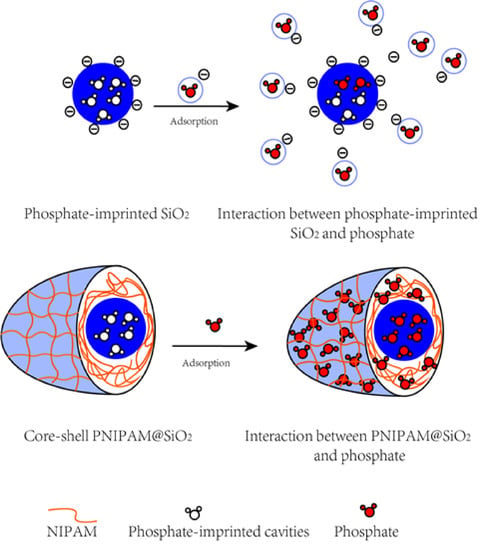
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Phosphate-Imprinted Mesoporous Silica Microsphere
2.3. Preparation of PNIPAM/SiO2 Hybrid Particles
2.4. Phosphate Adsorption Experiment
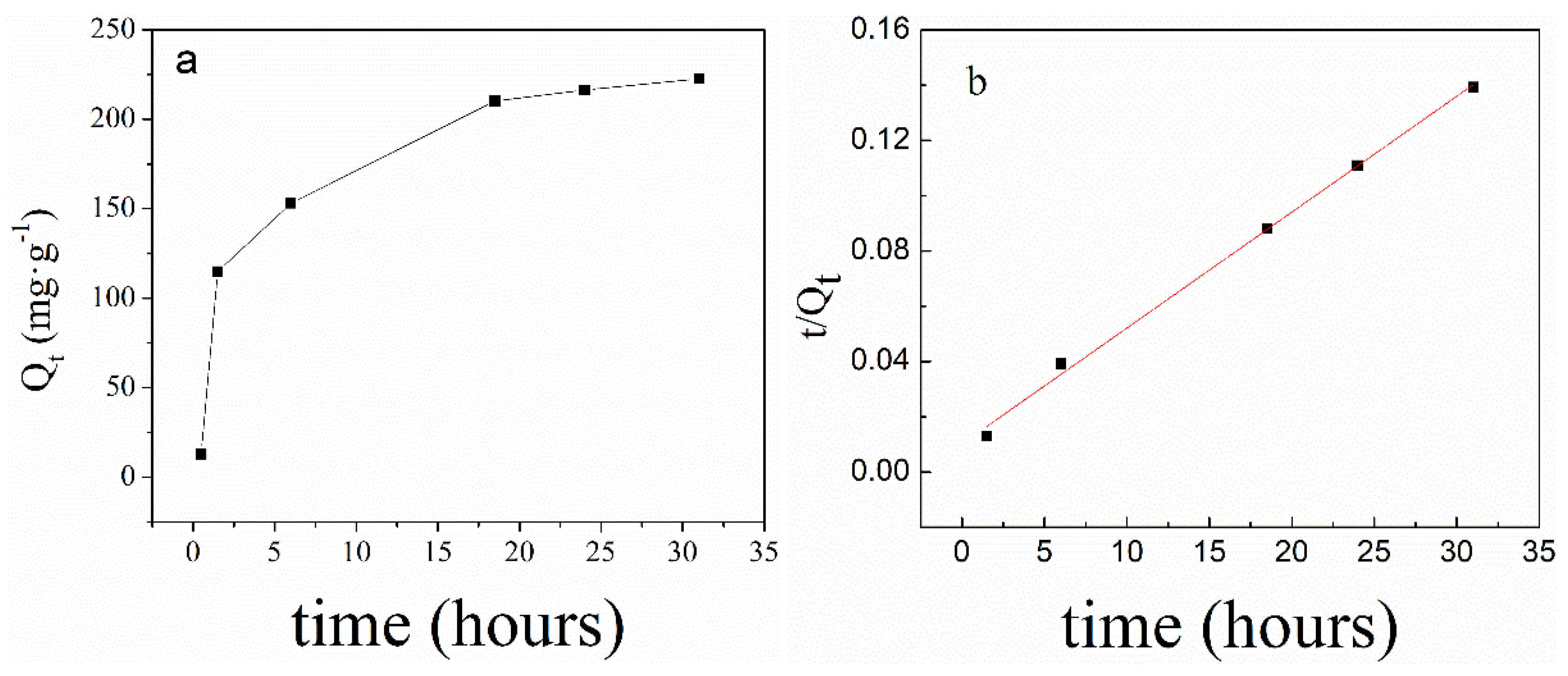
2.4.1. The Adsorption of PNIPAM@SiO2 with Phosphate Anions over Time
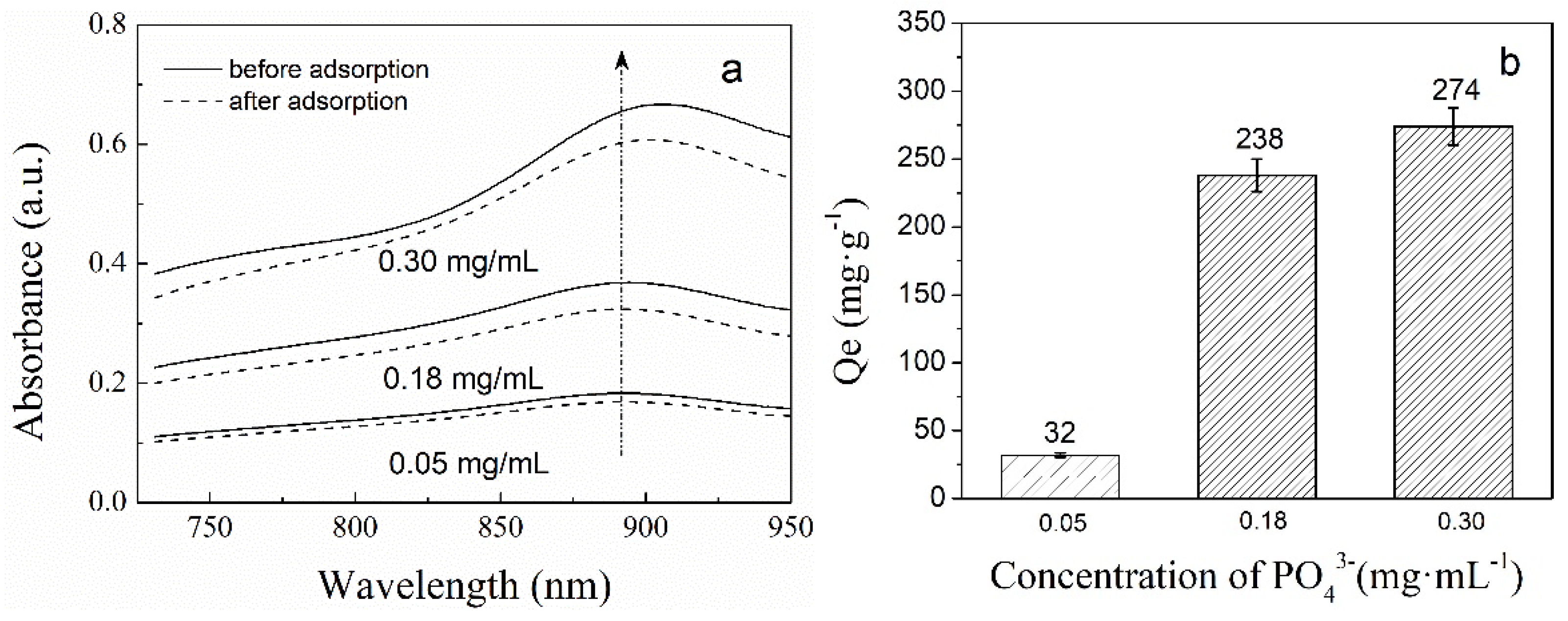
2.4.2. The Phosphate Adsorption of PNIPAM/SiO2 Particles Varying with the Initial Phosphate Concentration
2.4.3. The Effect of Solution pH on the Phosphate Adsorption of PNIPAM/SiO2 Particles
2.5. Characterizations
2.5.1. FT-IR Spectroscopy
2.5.2. Thermogravimetric Analysis
2.5.3. Specific Surface Area and Pore Size Measurements
2.5.4. Particle Size Test
2.5.5. Electron Spectroscopy
2.5.6. The Phosphate Adsorption Performance of PNIPAM/SiO2 Hybrid Particles
3. Results and Discussion
3.1. Preparation and Characterization of Phosphate-Imprinted SiO2 and PNIPAM/SiO2 Particles
3.2. Adsorption of Phosphate Anions in Water by Phosphate Imprinted SiO2 and PNIPAM/SiO2 Hybrid Particles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schroder, J.; Cordell, D.; Smit, A.; Rosemarin, A. Sustainable Use of Phosphorus; Plant Research International, Wageningen University and Research Centre: Wageningen, The Netherlands, 2010. [Google Scholar]
- Hudson, J.J.; Taylor, W.D.; Schindler, D.W. Phosphate Concentrations in Lakes. Nature 2000, 406, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Conley, D.J.; Paerl, H.W.; Howarth, R.W.; Boesch, D.F.; Seitzinger, S.P.; Havens, K.E.; Lancelot, C.; Likens, G.E. Controlling Eutrophication: Nitrogen and Phosphorus. Science 2009, 323, 1014–1015. [Google Scholar] [CrossRef] [PubMed]
- Voulvoulis, N.; Arpon, K.D.; Giakoumis, T. The EU Water Framework Directive: From Great Expectations To Problems with Implementation. Sci. Total Environ. 2017, 575, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Kiriukhin, M.Y.; Collins, K.D. Dynamic Hydration Numbers for Biologically Important Ions. Biophys. Chem. 2002, 99, 155–168. [Google Scholar] [CrossRef]
- Cao, Z.; Gordiichuk, P.I.; Loos, K.; Sudholter, E.J.R.; de Smet, L.C.P.M. The Effect of Guanidinium Functionalization on the Structural Properties and Anion Affinity of Polyelectrolyte Multilayers. Soft Matter 2016, 12, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, H.; Byeon, S.-H. Layered Yttrium Hydroxide l-Y(OH)3 Luminescent Adsorbent for Detection and Recovery of Phosphate from Water over a Wide pH Range. ACS Appl. Mater. Interfaces 2017, 9, 40461–40470. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, J.-K.; Keller, A.A. Removal of Arsenic and Phosphate from Aqueous Solution by Metal (Hydr-)oxide Coated Sand. ACS Sustainable Chem. Eng. 2014, 2, 1128–1138. [Google Scholar] [CrossRef]
- Weng, L.; van Riemsdijk, W.H.; Hiemstra, T. Humic Nanoparticles at the Oxide−Water Interface: Interactions with Phosphate Ion Adsorption. Environ. Sci. Technol. 2008, 42, 8747–8752. [Google Scholar] [CrossRef]
- Karagollu, O.; Gorur, M.; Gode, F.; Sennik, B.; Yilmaz, F. Phosphate ion sensors based on triazole connected ferrocene moieties. Sens. Actuators B 2014, 193, 788–798. [Google Scholar] [CrossRef]
- Paltrinieri, L.; Wang, M.; Sachdeva, S.; Besseling, N.A.M.; Sudholter, E.J.R.; de Smet, L.C.P.M. Fe3O4 Nanoparticles Coated with a Guanidinium-functionalized Polyelectrolyte Extend the pH Range for Phosphate Binding. J. Mater. Chem. A 2017, 5, 18476–18485. [Google Scholar] [CrossRef]
- Alexander, C.; Andersson, H.S.; Andersson, L.I.; Ansell, R.J.; Kirsch, N.; Nicholls, I.A.; O’Mahony, J.; Whitcombe, M.J. Molecular Imprinting Science and Technology: A Survey of the Literature for the Years up to and Including 2003. J. Mol. Recognit. 2006, 19, 106–180. [Google Scholar] [CrossRef] [PubMed]
- Sibrian-Vazquez, M.; Spivak, D.A. Molecular Imprinting Made Easy. J. Am. Chem. Soc. 2004, 126, 7827–7833. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, X.; Lu, W.; Wu, X.; Li, J. Molecular Imprinting: Perspectives and Applications. Chem. Soc. Rev. 2016, 45, 2137–2211. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Fan, F.; Wang, X.; Feng, W.; Hou, Y.; Pei, Z. Fabrication of Hypericin Imprinted Polymer Nanospheres via Thiol-Yne Click Reaction. Polymers 2017, 9, 469. [Google Scholar] [CrossRef]
- Viveiros, R.; Rebocho, S.; Casimiro, T. Green Strategies for Molecularly Imprinted Polymer Development. Polymers 2018, 10, 306. [Google Scholar] [CrossRef]
- Zhang, H.; Ye, L.; Mosbach, K. Non-covalent Molecular Imprinting with Emphasis on its Application in Separation and Drug Development. J. Mol. Recognit. 2006, 19, 248–259. [Google Scholar] [CrossRef]
- Kempe, M.; Mosbach, K. Molecular Imprinting Used for Chiral Separations. J. Chromatogr. A 1995, 694, 3–13. [Google Scholar] [CrossRef]
- Göçenoğlu Sarıkaya, A.; Osman, B.; Çam, T.; Denizli, A. Molecularly Imprinted Surface Plasmon Resonance (SPR) Sensor for Uric Acid Determination. Sens. Actuators B 2017, 251, 763–772. [Google Scholar] [CrossRef]
- Ertürk, G.; Uzun, L.; Tümer, M.A.; Say, R.; Denizli, A. Fab Fragments Imprinted SPR Biosensor for Real-time Human Immunoglobulin G Detection. Biosens. Bioelectron. 2011, 28, 97–104. [Google Scholar] [CrossRef]
- Whitcombe, M.J.; Chianella, I.; Larcombe, L.; Piletsky, S.A.; Noble, J.; Porter, R.; Horgan, A. The Rational Development of Molecularly Imprinted Polymer-based Sensors for Protein Detection. Chem. Soc. Rev. 2011, 40, 1547–1571. [Google Scholar] [CrossRef]
- Rao, T.P.; Kala, R.; Daniel, S. Metal Ion-imprinted Polymers—Novel Materials for Selective Recognition of Inorganics. Anal. Chim. Acta 2006, 578, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Vidyasankar, S.; Arnold, F.H. Molecular Imprinting: Selective Materials for Separations, Sensors and Catalysis. Curr. Opin. Biotechnol. 1995, 6, 218–224. [Google Scholar] [CrossRef]
- Dickert, F.L.; Halikias, K.; Hayden, O.; Piu, L.; Sikorski, R. Sensors Based on Fingerprints of Neutral and Ionic Analytes in Polymeric Materials. Sens. Actuators B 2001, 76, 295–298. [Google Scholar] [CrossRef]
- Hart, B.R.; Shea, K.J. Molecular Imprinting for the Recognition of N-Terminal Histidine Peptides in Aqueous Solution. Macromolecules 2002, 35, 6192–6201. [Google Scholar] [CrossRef]
- Iskierko, Z.; Sharma, P.S.; Prochowicz, D.; Fronc, K.; D’Souza, F.; Toczydłowska, D.; Stefaniak, F.; Noworyta, K. Molecularly Imprinted Polymer (MIP) Film with Improved Surface Area Developed by Using Metal–Organic Framework (MOF) for Sensitive Lipocalin (NGAL) Determination. ACS Appl. Mater. Interfaces 2016, 8, 19860–19865. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.; Brahmbhatt, H.; Watts, J.K.; Turner, N.W. Nucleoside-Tailored Molecularly Imprinted Polymeric Nanoparticles (MIP NPs). Macromolecules 2014, 47, 6322–6330. [Google Scholar] [CrossRef]
- Zhao, Y.; Ma, Y.; Li, H.; Wang, L. Correction to Composite QDs@MIP Nanospheres for Specific Recognition and Direct Fluorescent Quantification of Pesticides in Aqueous Media. Anal. Chem. 2012, 84, 5447. [Google Scholar] [CrossRef]
- Chen, Y.; Li, D.; Bie, Z.; He, X.; Liu, Z. Coupling of Phosphate-Imprinted Mesoporous Silica Nanoparticles-Based Selective Enrichment with Matrix-Assisted Laser Desorption Ionization-Time-of-Flight Mass Spectrometry for Highly Efficient Analysis of Protein Phosphorylation. Anal. Chem. 2016, 88, 1447–1454. [Google Scholar] [CrossRef]
- Rimola, A.; Costa, D.; Sodupe, M.; Lambert, J.-F.; Ugliengo, P. Silica Surface Features and Their Role in the Adsorption of Biomolecules: Computational Modeling and Experiments. Chem. Rev. 2013, 113, 4216–4313. [Google Scholar] [CrossRef]
- Chibowski, E.; Szcześ, A.; Hołysz, L. Changes of Zeta Potential and Particles Size of Silica Caused by DPPC Adsorption and Enzyme Phospholipase A2 Presence. Adsorption 2010, 16, 305–312. [Google Scholar] [CrossRef]
- Ma, M.; Zheng, S.; Chen, H.; Yao, M.; Zhang, K.; Jia, X.; Mou, J.; Xu, H.; Wu, R.; Shi, J. A Combined “RAFT” and “Graft From” Polymerization Strategy for Surface Modification of Mesoporous Silica Nanoparticles: Towards Enhanced Tumor Accumulation and Cancer Therapy Efficacy. J. Mater. Chem. B 2014, 2, 5828–5836. [Google Scholar] [CrossRef]
- Lundqvist, M.; Sethson, I.; Jonsson, B.-H. Protein Adsorption onto Silica Nanoparticles: Conformational Changes Depend on the Particles’ Curvature and the Protein Stability. Langmuir 2004, 20, 10639–10647. [Google Scholar] [CrossRef] [PubMed]
- Rapuano, R.; Carmona-Ribeiro, A.M. Physical Adsorption of Bilayer Membranes on Silica. J. Colloid Interface Sci. 1997, 193, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Wang, Y.; Hao, J.; Caruso, F. Mesoporous Silica-Templated Assembly of Luminescent Polyester Particles. Chem. Mater. 2009, 21, 4310–4315. [Google Scholar] [CrossRef]
- Jun-Hwan, P.; Young-Ho, L.; Seong-Geun, O. Preparation of Thermosensitive PNIPAm-Grafted Mesoporous Silica Particles. Macromol. Chem. Phys. 2007, 208, 2419–2427. [Google Scholar]
- Wu, T.; Zhang, Y.; Wang, X.; Liu, S. Fabrication of Hybrid Silica Nanoparticles Densely Grafted with Thermoresponsive Poly(N-isopropylacrylamide) Brushes of Controlled Thickness via Surface-Initiated Atom Transfer Radical Polymerization. Chem. Mater. 2008, 20, 101–109. [Google Scholar] [CrossRef]
- Liu, C.-H.; Pan, C.-Y. Grafting Polystyrene onto Silica Nanoparticles via RAFT Polymerization. Polymer 2007, 48, 3679–3685. [Google Scholar] [CrossRef]
- Rapuano, R.; Carmona-Ribeiro, A.M. Supported Bilayers on Silica. J. Colloid Interface Sci. 2000, 226, 299–307. [Google Scholar] [CrossRef]
- Moura, S.P.; Carmona-Ribeiro, A.M. Biomimetic Particles: Optimization of Phospholipid Bilayer Coverage on Silica and Colloid Stabilization. Langmuir 2005, 21, 10160–10164. [Google Scholar] [CrossRef] [PubMed]
- Tero, R.; Takizawa, M.; Li, Y.; Yamazaki, M.; Urisu, T. Deposition of Phospholipid Layers on SiO2 Surface Modified by Alkyl-SAM Islands. Appl. Surf. Sci. 2004, 238, 218–222. [Google Scholar] [CrossRef]
- Castellana, E.T.; Cremer, P.S. Solid Supported Lipid Bilayers: From Biophysical Studies to Sensor Design. Surf. Sci. Rep. 2006, 61, 429–444. [Google Scholar] [CrossRef]
- Troutier, A.-L.; Ladaviere, C. An Overview of Lipid Membrane Supported by Colloidal Particles. Adv. Colloid Interface Sci. 2007, 133, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-J.; Liang, Y.-L.; Geng, F.-F.; Lv, F.; Dai, R.-J.; Zhang, Y.-K.; Deng, Y.-L. Preparation of Poly (N-isopropylacrylamide) Brush Grafted Silica Particles via Surface-initiated Atom Transfer Radical Polymerization Used for Aqueous Chromatography. Front. Mater. Sci. 2012, 6, 60–68. [Google Scholar] [CrossRef]
- Schild, H.G. Poly(N-isopropylacrylamide): Experiment, theory and application. Prog. Polym. Sci. 1992, 17, 163–249. [Google Scholar] [CrossRef]
- Cheng, J.; Shan, G.; Pan, P. Temperature and pH-dependent swelling and copper(ii) adsorption of poly(N-isopropylacrylamide) copolymer hydrogel. RSC Adv. 2015, 5, 62091–62100. [Google Scholar] [CrossRef]
- Liu, Y.; Shen, L. From Langmuir Kinetics to First-and Second-order Rate Equations for Adsorption. Langmuir 2008, 24, 11625–11630. [Google Scholar] [CrossRef] [PubMed]
- Federation, W.E.; Association, A.P.H. Standard Methods for the Examination of Water and Wastewater; American Public Health Association (APHA): Washington, DC, USA, 2005. [Google Scholar]
- Chen, Y.; Li, X.; Yin, D.; Li, D.; Bie, Z.; Liu, Z. Dual-template docking oriented molecular imprinting: A facile strategy for highly efficient imprinting within mesoporous materials. Chem. Commun. 2015, 51, 10929–10932. [Google Scholar]
- Zhang, L.; Zhou, N.; Wang, B.; Liu, C.; Zhu, G. Fabrication of Fe3O4/PAH/PSS@ Pd Core–shell Microspheres by Layer-by-layer Assembly and Application in Catalysis. J. Colloid Interface Sci. 2014, 421, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Du, B.; Chen, T.; Nie, J.; Xu, J.; Fan, Z. Preparation and Properties of Thermo-sensitive Organic/Inorganic Hybrid Microgels. Langmuir 2008, 24, 12771–12778. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.A.; Willott, J.D.; Murdoch, T.J.; Webber, G.B.; Wanless, E.J. Specific Ion Modulated Thermoresponse of Poly (N-isopropylacrylamide) Brushes. Phys. Chem. Chem. Phys. 2016, 18, 6037–6046. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorbent | Imprinted SiO2 | PNIPAM Microgel | PNIPAM/SiO2 Particles |
|---|---|---|---|
| Adsorption capacity (mg/g) | 20.0 ± 1.2 | 55.0 ± 2.7 | 271.0 ± 13.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Z.; Chen, Y.; Li, D.; Cheng, J.; Liu, C. Fabrication of Phosphate-Imprinted PNIPAM/SiO2 Hybrid Particles and Their Phosphate Binding Property. Polymers 2019, 11, 253. https://doi.org/10.3390/polym11020253
Cao Z, Chen Y, Li D, Cheng J, Liu C. Fabrication of Phosphate-Imprinted PNIPAM/SiO2 Hybrid Particles and Their Phosphate Binding Property. Polymers. 2019; 11(2):253. https://doi.org/10.3390/polym11020253
Chicago/Turabian StyleCao, Zheng, Yuyuan Chen, Dan Li, Junfeng Cheng, and Chunlin Liu. 2019. "Fabrication of Phosphate-Imprinted PNIPAM/SiO2 Hybrid Particles and Their Phosphate Binding Property" Polymers 11, no. 2: 253. https://doi.org/10.3390/polym11020253
APA StyleCao, Z., Chen, Y., Li, D., Cheng, J., & Liu, C. (2019). Fabrication of Phosphate-Imprinted PNIPAM/SiO2 Hybrid Particles and Their Phosphate Binding Property. Polymers, 11(2), 253. https://doi.org/10.3390/polym11020253