Core-Shell Structure Design of Hollow Mesoporous Silica Nanospheres Based on Thermo-Sensitive PNIPAM and pH-Responsive Catechol-Fe3+ Complex


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
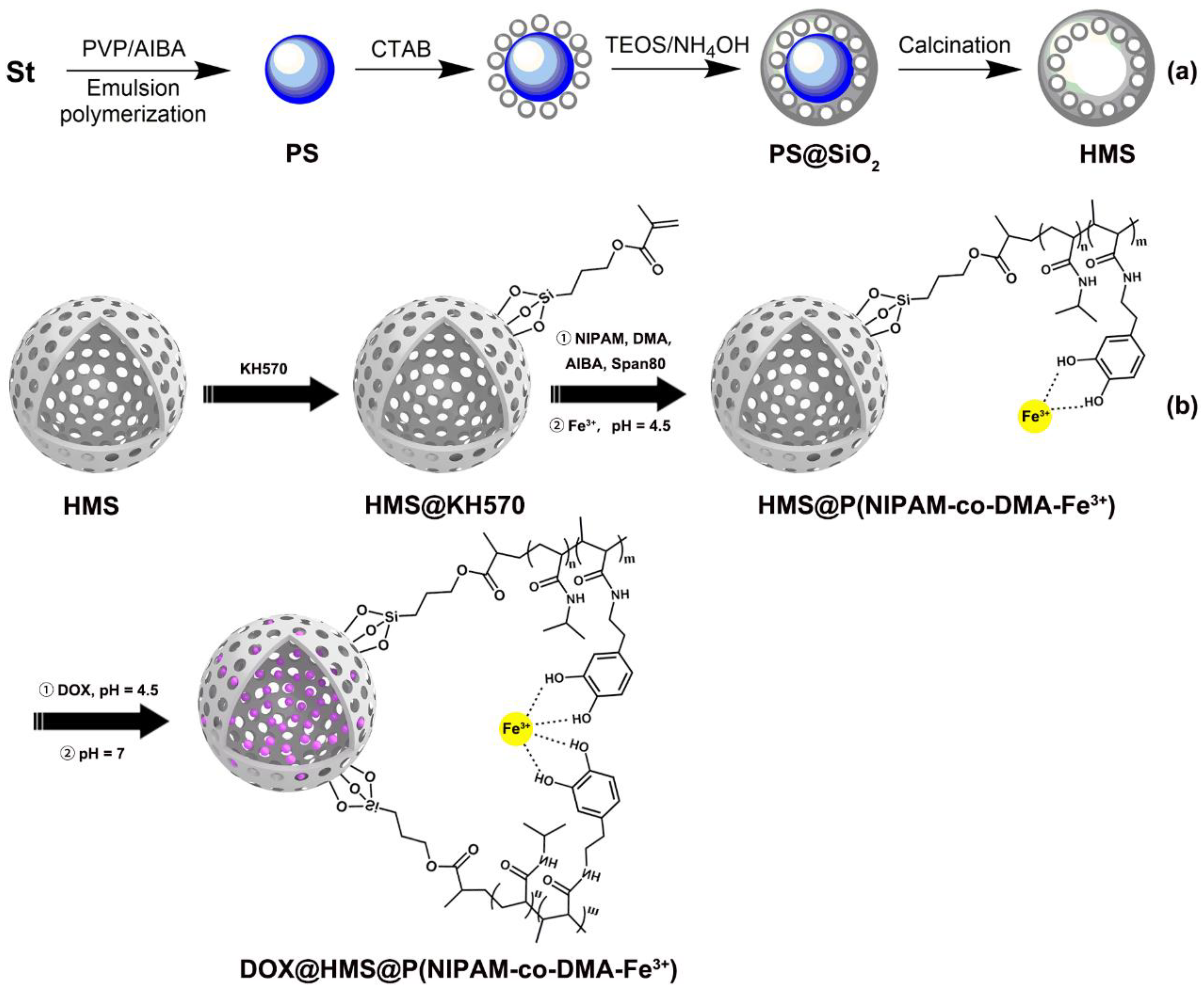
2.2. Synthesis of Polystyrene (PS) Sphere Template
2.3. Synthesis of Hollow Mesoporous Silica Microspheres (HMS)
2.4. Synthesis of N-(3,4-dihydroxyphenethyl) Methacrylamide (DMA)
2.5. Preparation of the KH570-Modified Silica Particle
2.6. Synthesis of HMS Nanoparticles Coated with Sensitive Polymer Shell
2.7. Characterization
2.8. DOX loading capacity and controlled release behaviors in Vito
3. Results
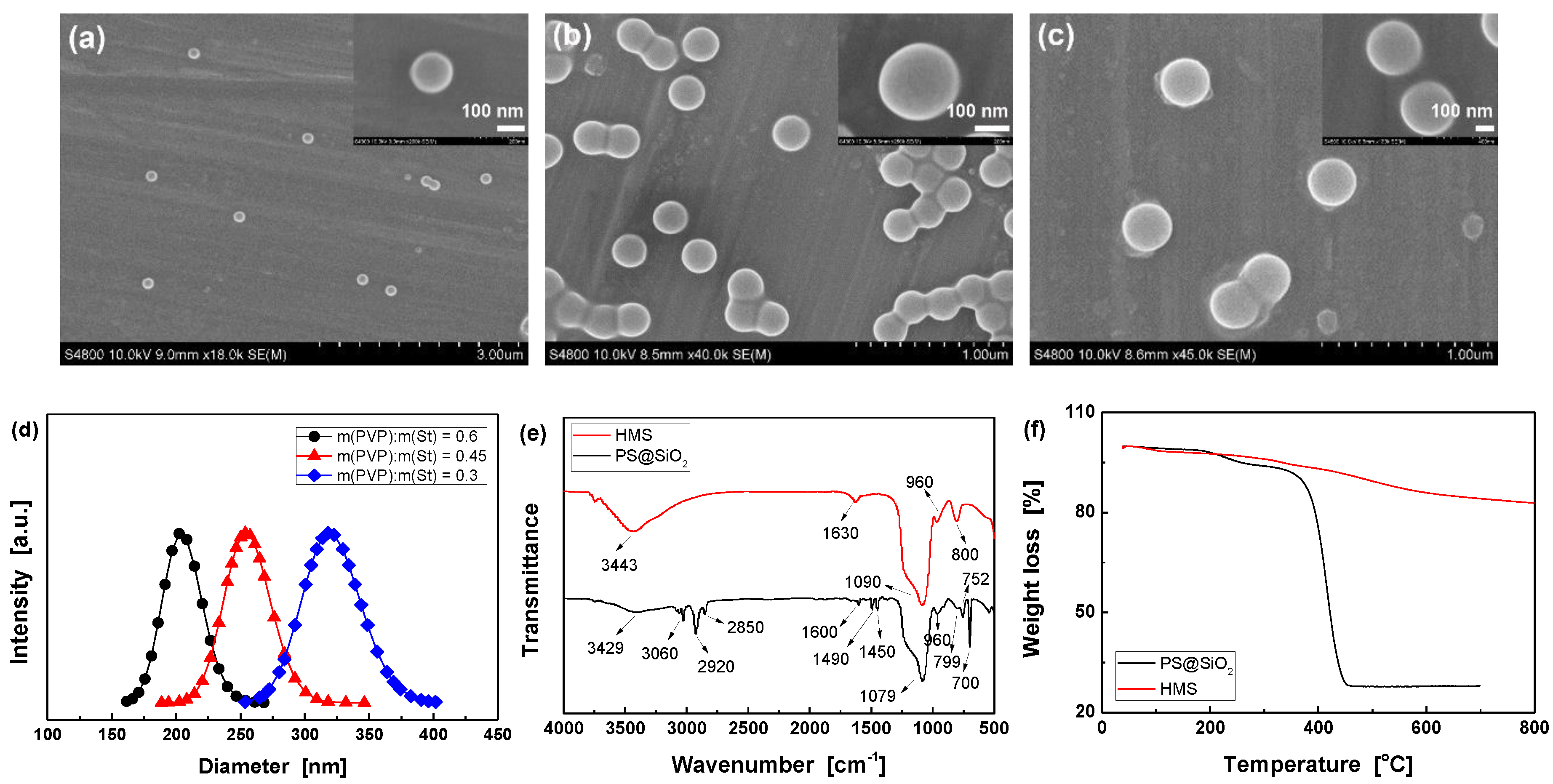
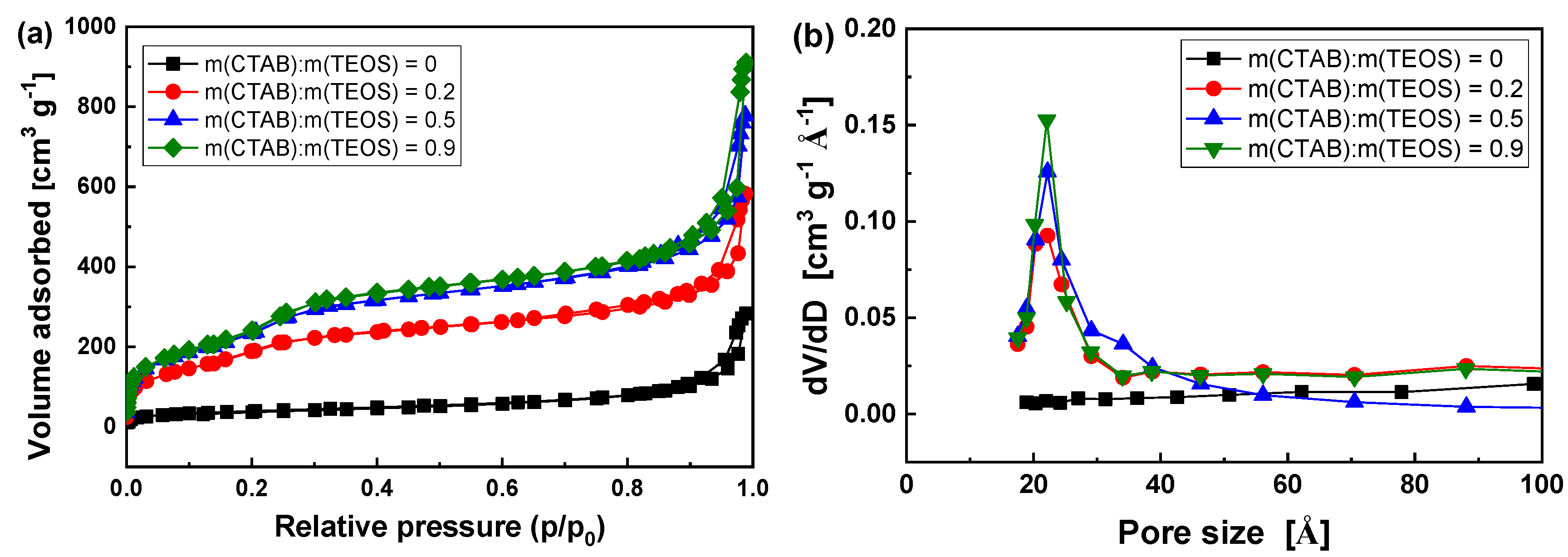
3.1. Optimizing the synthesis of HMS
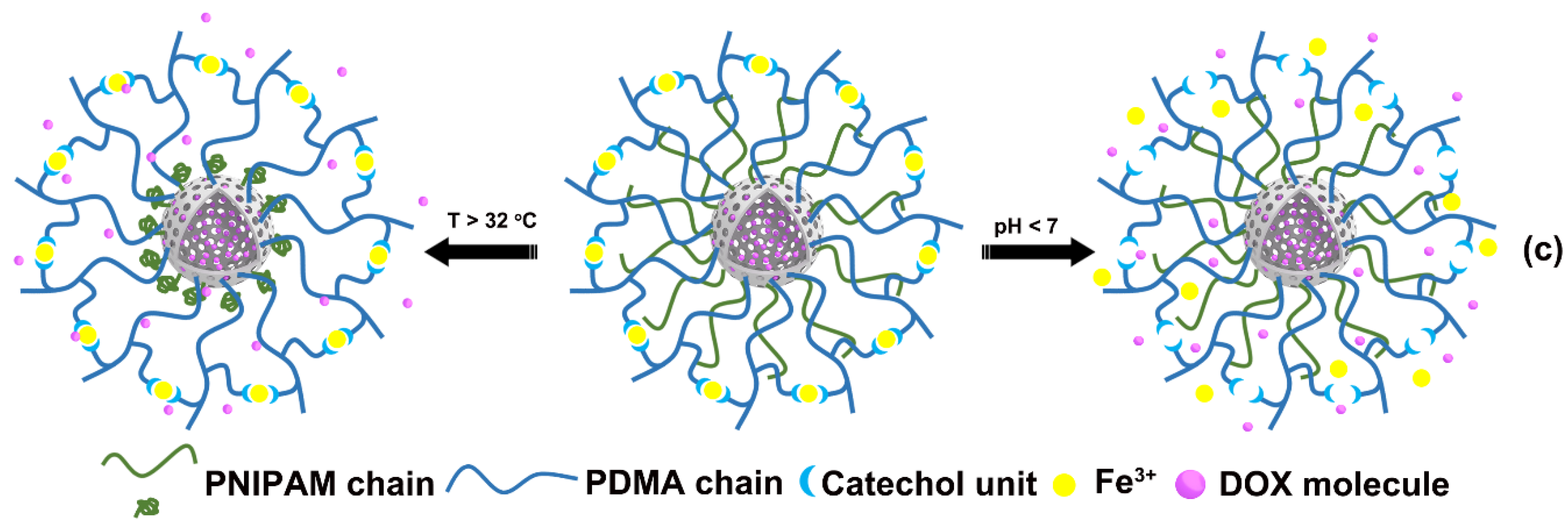
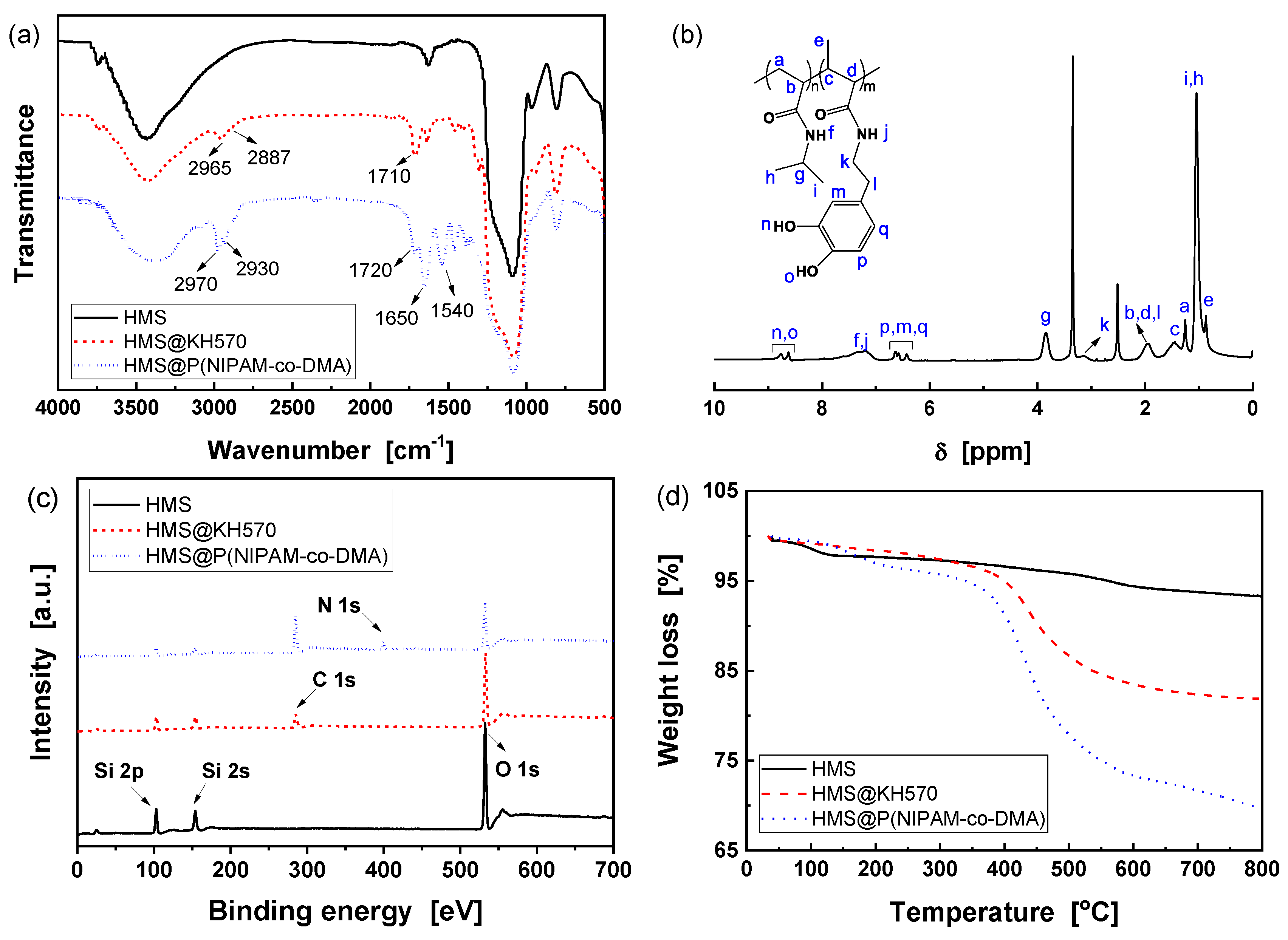
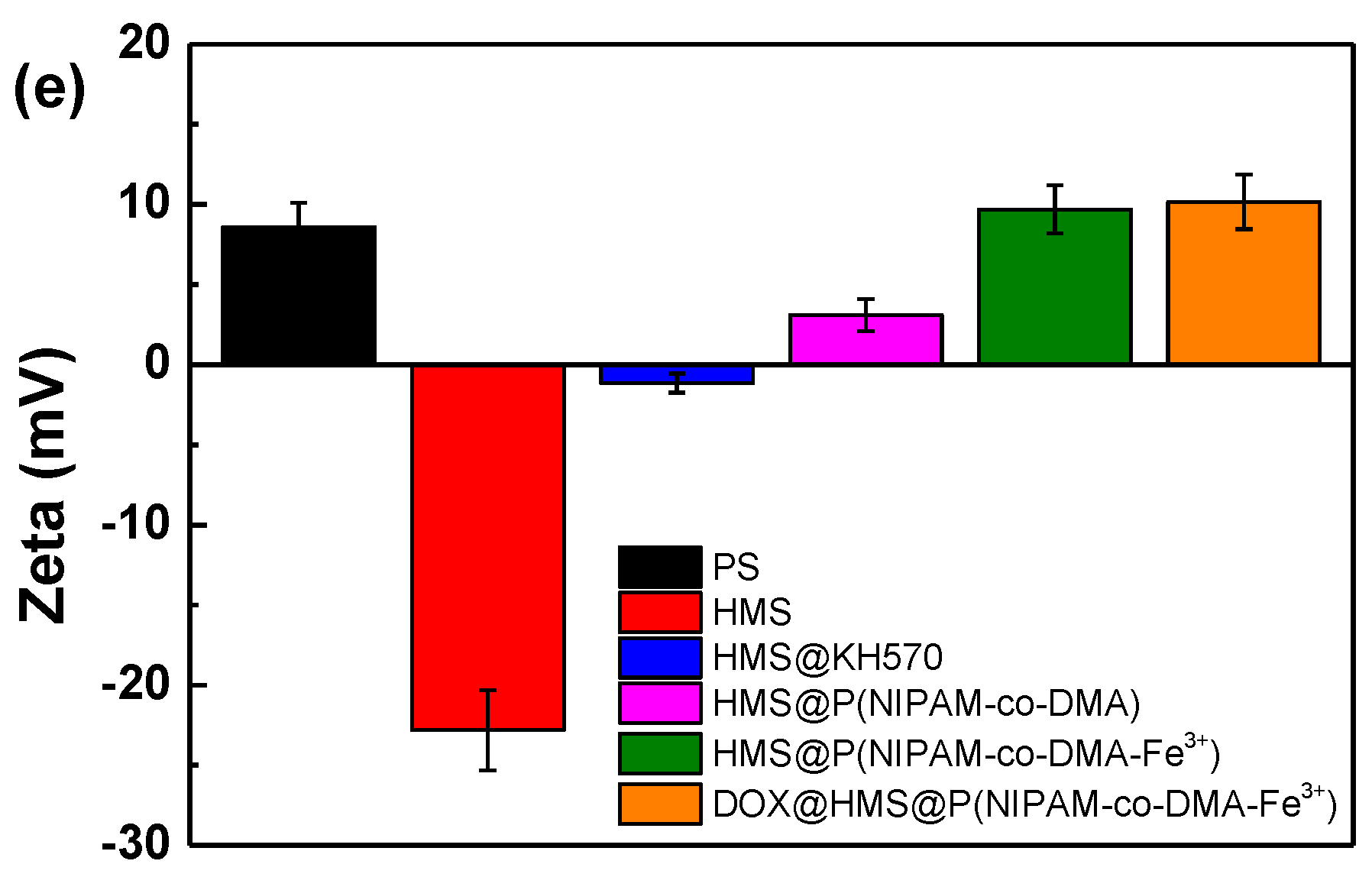
3.2. Preparation of HMS@P(NIPAM-co-DMA)
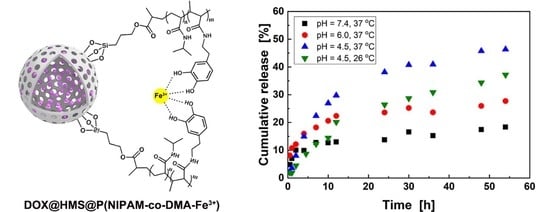
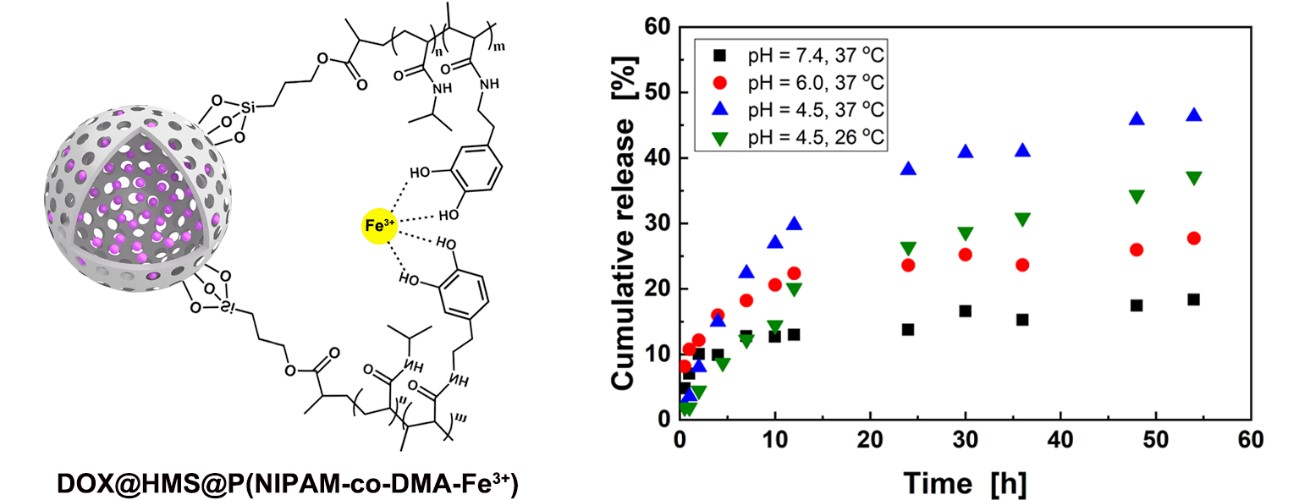
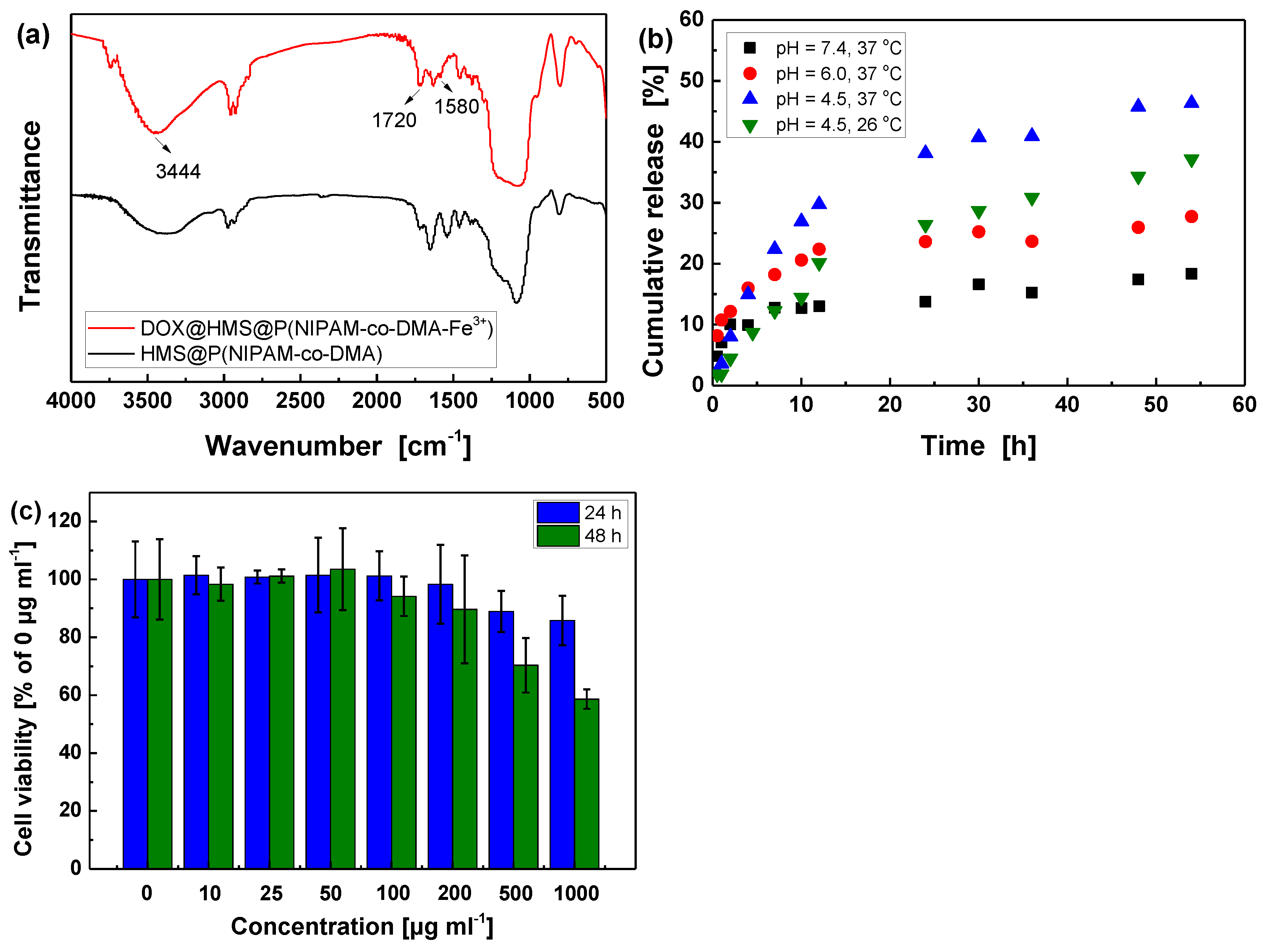
3.3. DOX Loading and Controlled Release in Vitro
3.4. In Vitro Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yavvari, P.S.; Pal, S.; Kumar, S.; Kar, A.; Awasthi, A.K.; Naaz, A.; Srivastava, A.; Bajaj, A. Moldable and removable wound dressing based on dynamic covalent cross-linking of thiol-aldehyde addition. ACS Biomater. Sci. Eng. 2017, 3, 3404–3413. [Google Scholar] [CrossRef]
- Wang, Z.; Deng, X.; Ding, J.; Zhou, W.; Zheng, X.; Tang, G. Mechanisms of drug release in pH-sensitive micelles for tumour targeted drug delivery system: A review. Int. J. Pharm. 2018, 535, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Riaz, M.; Zhang, X.; Lin, C.; Wong, K.H.; Chen, X.; Zhang, G.; Lu, A.; Yang, Z.; Riaz, M.K.; Riaz, M.A. Surface Functionalization and Targeting Strategies of Liposomes in Solid Tumor Therapy: A Review. Int. J. Mol. Sci. 2018, 19, 195. [Google Scholar] [CrossRef] [PubMed]
- Pugazhendhi, A.; Edison, T.N.J.I.; Karuppusamy, I.; Kathirvel, B. Inorganic nanoparticles: A potential cancer therapy for human welfare. Int. J. Pharm. 2018, 539, 104–111. [Google Scholar] [CrossRef]
- Vallet-Regí, M.; Colilla, M.; Gonzalez, B. Medical applications of organic–inorganic hybrid materials within the field of silica-based bioceramics. Chem. Soc. Rev. 2011, 40, 596–607. [Google Scholar] [CrossRef]
- Chen, J.-F.; Ding, H.-M.; Wang, J.-X.; Shao, L. Preparation and characterization of porous hollow silica nanoparticles for drug delivery application. Biomaterials 2004, 25, 723–727. [Google Scholar] [CrossRef]
- Li, Y.; Shi, J. Hollow-Structured Mesoporous Materials: Chemical Synthesis, Functionalization and Applications. Adv. Mater. 2014, 26, 3176–3205. [Google Scholar] [CrossRef]
- Torchilin, V.P. Multifunctional nanocarriers. Adv. Drug Deliv. Rev. 2006, 58, 1532–1555. [Google Scholar] [CrossRef]
- Hartono, S.B.; Phuoc, N.T.; Yu, M.; Jia, Z.; Monteiro, M.J.; Qiao, S.Z.; Yu, C. Functionalized large pore mesoporous silica nanoparticles for gene delivery featuring controlled release and co-delivery. J. Mater. Chem. B 2014, 2, 718–726. [Google Scholar] [CrossRef]
- Spange, S. Silica surface modification by cationic polymerization and carbenium intermediates. Prog. Polym. Sci. 2000, 25, 781–849. [Google Scholar] [CrossRef]
- Bull, S.D.; Davidson, M.G.; Elsen, J.M.H.V.D.; Fossey, J.S.; Jenkins, A.T.A.; Jiang, Y.-B.; Kubo, Y.; Marken, F.; Sakurai, K.; Zhao, J.; et al. Exploiting the Reversible Covalent Bonding of Boronic Acids: Recognition, Sensing, and Assembly. Acc. Chem. Res. 2012, 46, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Cai, K.; Hu, Y.; Zhao, L.; Liu, P.; Duan, L.; Yang, W. Mesoporous silica nanoparticles end-capped with collagen: Redox-responsive nanoreservoirs for targeted drug delivery. Angew. Chem. 2011, 50, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Shen, D.; Zhou, L.; Li, W.; Li, X.; Yao, C.; Wang, R.; El-Toni, A.M.; Zhang, F.; Zhao, D. Spatially Confined Fabrication of Core–Shell Gold Nanocages@Mesoporous Silica for Near-Infrared Controlled Photothermal Drug Release. Chem. Mater. 2013, 25, 3030–3037. [Google Scholar] [CrossRef]
- Huang, S.-H.; Juang, R.-S. Biochemical and biomedical applications of multifunctional magnetic nanoparticles: A review. J. Nanopart. Res. 2011, 13, 4411–4430. [Google Scholar] [CrossRef]
- Tannock, I.F.; Rotin, D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989, 49, 4373–4384. [Google Scholar]
- Van Der Stappen, J.W.J.; Williams, A.C.; Maciewicz, R.A.; Paraskeva, C. Activation of cathepsin B, secreted by a colorectal cancer cell line requires low pH and is mediated by cathepsin D. Int. J. Cancer 1996, 67, 547–554. [Google Scholar] [CrossRef]
- Zhang, L.; Guo, R.; Yang, M.; Jiang, X.; Liu, B. Thermo and pH Dual-Responsive Nanoparticles for Anti-Cancer Drug Delivery. Adv. Mater. 2007, 19, 2988–2992. [Google Scholar] [CrossRef]
- Kaga, S.; Truong, N.P.; Esser, L.; Senyschyn, D.; Sanyal, A.; Sanyal, R.; Quinn, J.F.; Davis, T.P.; Kaminskas, L.M.; Whittaker, M.R. Influence of Size and Shape on the Biodistribution of Nanoparticles Prepared by Polymerization-Induced Self-Assembly. Biomacromolecules 2017, 18, 3963–3970. [Google Scholar] [CrossRef]
- Khor, S.Y.; Vu, M.N.; Pilkington, E.H.; Johnston, A.P.R.; Whittaker, M.R.; Quinn, J.F.; Truong, N.P.; Davis, T.P. Elucidating the Influences of Size, Surface Chemistry, and Dynamic Flow on Cellular Association of Nanoparticles Made by Polymerization-Induced Self-Assembly. Small 2018, 14, 1801702. [Google Scholar] [CrossRef]
- Wang, D.; Huang, H.; Zhou, M.; Lu, H.; Chen, J.; Chang, Y.-T.; Gao, J.; Chai, Z.; Hu, Y. A thermoresponsive nanocarrier for mitochondria-targeted drug delivery. Chem. Commun. 2019, 55, 4051–4054. [Google Scholar] [CrossRef]
- You, Y.-Z.; Kalebaila, K.K.; Brock, S.L.; Oupický, D. Temperature-Controlled Uptake and Release in PNIPAM-Modified Porous Silica Nanoparticles. Chem. Mater. 2008, 20, 3354–3359. [Google Scholar] [CrossRef]
- Vaccaro, E.; Waite, J.H. Yield and post-yield behavior of mussel byssal thread: A self-healing biomolecular material. Biomacromolecules 2001, 2, 906–911. [Google Scholar] [CrossRef]
- Xia, N.N.; Xiong, X.M.; Wang, J.; Rong, M.Z.; Zhang, M.Q. A seawater triggered dynamic coordinate bond and its application for underwater self-healing and reclaiming of lipophilic polymer. Chem. Sci. 2016, 7, 2736–2742. [Google Scholar] [CrossRef]
- Lee, B.P.; Narkar, A.; Wilharm, R. Effect of metal ion type on the movement of hydrogel actuator based on catechol-metal ion coordination chemistry. Sens. Actuators B Chem. 2016, 227, 248–254. [Google Scholar] [CrossRef]
- Gong, C.; Lu, C.; Li, B.; Shan, M.; Wu, G. Injectable dopamine-modified poly(α,β-aspartic acid) nanocomposite hydrogel as bioadhesive drug delivery system. J. Biomed. Mater. Res. Part A 2017, 105, 1000–1008. [Google Scholar] [CrossRef]
- Qi, G.; Wang, Y.; Estevez, L.; Switzer, A.K.; Duan, X.; Yang, X.; Giannelis, E.P. Facile and Scalable Synthesis of Monodispersed Spherical Capsules with a Mesoporous Shell. Chem. Mater. 2010, 22, 2693–2695. [Google Scholar] [CrossRef]
- Zou, H.; Wu, S.; Ran, Q.; Shen, J. A Simple and Low-Cost Method for the Preparation of Monodisperse Hollow Silica Spheres. J. Phys. Chem. C 2008, 112, 11623–11629. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, M.; Wang, D.; Zhang, W.; Liu, S. Synthesis of hollow mesoporous silica microspheres through surface sol–gel process on polystyrene-co-poly(4-vinylpyridine) core–shell microspheres. Microporous Mesoporous Mater. 2011, 139, 1–7. [Google Scholar] [CrossRef]
- White, E.M.; Seppala, J.E.; Rushworth, P.M.; Ritchie, B.W.; Sharma, S.; Locklin, J. Switching the Adhesive State of Catecholic Hydrogels using Phototitration. Macromolecules 2013, 46, 8882–8887. [Google Scholar] [CrossRef]
- Szunerits, S.; Boukherroub, R. Preparation and Characterization of Thin Films of SiOxon Gold Substrates for Surface Plasmon Resonance Studies. Langmuir 2006, 22, 1660–1663. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Wen, Y.; Yuan, Y.; Wu, W.; Liu, C. Tunable wettability of monodisperse core-shell nano-SiO2 modified with poly(methylhydrosiloxane) and allyl-poly(ethylene glycol). Colloids Surf. A Physicochem. Eng. Asp. 2014, 441, 16–24. [Google Scholar] [CrossRef]
- Chen, J.C.; Luo, W.Q.; Wang, H.D.; Xiang, J.M.; Jin, H.F.; Chen, F.; Cai, Z. A versatile method for the preparation of end-functional polymers onto SiO2 nanoparticles by a combination of surface-initiated ATRP and Huisgen [3 + 2] cycloaddition. Appl. Surf. Sci. 2010, 256, 2490–2495. [Google Scholar] [CrossRef]
- Mishra, P.K.; Wimmer, R. Aerosol assisted self-assembly as a route to synthesize solid and hollow spherical lignin colloids and its utilization in layer by layer deposition. Ultrason. Sonochem. 2017, 35, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, X.; Zheng, L.; Yu, L.; Zhang, Q.; Liu, W. Preparation and characterization of monocaprate nanostructured lipid carriers. Colloids Surf. A Physicochem. Eng. Asp. 2007, 311, 106–111. [Google Scholar] [CrossRef]
- Blas, H.; Save, M.; Pasetto, P.; Boissière, C.; Sanchez, C.; Charleux, B. Elaboration of Monodisperse Spherical Hollow Particles with Ordered Mesoporous Silica Shells via Dual Latex/Surfactant Templating: Radial Orientation of Mesopore Channels. Langmuir 2008, 24, 13132–13137. [Google Scholar] [CrossRef]
- Le, Y.; Chen, J.-F.; Wang, W.-C. Study on the silica hollow spheres by experiment and molecular simulation. Appl. Surf. Sci. 2004, 230, 319–326. [Google Scholar] [CrossRef]
- Wu, X.; Tian, Y.; Cui, Y.; Wei, L.; Wang, Q.; Chen, Y. Raspberry-like Silica Hollow Spheres: Hierarchical Structures by Dual Latex−Surfactant Templating Route. J. Phys. Chem. C 2007, 111, 9704–9708. [Google Scholar] [CrossRef]
- Zhu, Y.; Shi, J.; Chen, H.; Shen, W.; Dong, X. A facile method to synthesize novel hollow mesoporous silica spheres and advanced storage property. Microporous Mesoporous Mater. 2005, 84, 218–222. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, X. Carbon dioxide adsorption properties and adsorption/desorption kinetics of amine-functionalized KIT-6. Appl. Energy 2018, 211, 1080–1088. [Google Scholar] [CrossRef]
- Niu, D.; Ma, Z.; Li, Y.; Shi, J. Synthesis of Core−Shell Structured Dual-Mesoporous Silica Spheres with Tunable Pore Size and Controllable Shell Thickness. J. Am. Chem. Soc. 2010, 132, 15144–15147. [Google Scholar] [CrossRef]
- Hong, R.Y.; Chen, L.L.; Li, J.H.; Li, H.Z.; Zheng, Y.; Ding, J. Preparation and application of polystyrene-grafted ZnO nanoparticles. Polym. Adv. Technol. 2007, 18, 901–909. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, Y.; Da Silva, L.P.; Pirraco, R.P.; Ma, M.; Yang, L.; Reis, R.L.; Chen, J. A thermo-/pH-responsive hydrogel (PNIPAM-PDMA-PAA) with diverse nanostructures and gel behaviors as a general drug carrier for drug release. Polym. Chem. 2018, 9, 4063–4072. [Google Scholar] [CrossRef]
- Watts, J.F.; Wolstenholme, J. An Introduction to Surface Analysis by XPS and AES; Wiley: Hoboken, NJ, USA, 2003; pp. 5–6. [Google Scholar]
- Chen, M.; Wu, L.; Zhou, S.; You, B. A Method for the Fabrication of Monodisperse Hollow Silica Spheres. Adv. Mater. 2006, 18, 801–806. [Google Scholar] [CrossRef]
- Whitfield, R.; Parkatzidis, K.; Rolland, M.; Truong, N.P.; Anastasaki, A. Tuning Dispersity by Photo-Induced ATRP: Monomodal Distributions with ppm Copper Concentration. Angew. Chem. Int. Ed. 2019, 58, 13323–13328. [Google Scholar] [CrossRef]
- Whitfield, R.; Truong, N.P.; Messmer, D.; Parkatzidis, K.; Rolland, M.; Anastasaki, A.; Phuoc, N.T.; Parkatzidis, K. Tailoring polymer dispersity and shape of molecular weight distributions: Methods and applications. Chem. Sci. 2019, 10, 8724–8734. [Google Scholar] [CrossRef]
- Zhu, Y.; Shi, J.; Shen, W.; Chen, H.; Dong, X.; Ruan, M. Preparation of novel hollow mesoporous silica spheres and their sustained-release property. Nanotechnology 2005, 16, 2633–2638. [Google Scholar] [CrossRef]
- Wang, Y.; Bi, K.; Shu, J.; Liu, X.; Xu, J.; Deng, G. Ultrasound-controlled DOX-SiO2 nanocomposites enhance the antitumour efficacy and attenuate the toxicity of doxorubicin. Nanoscale 2019, 11, 4210–4218. [Google Scholar] [CrossRef]
- Ardeshirzadeh, B.; Anaraki, N.A.; Irani, M.; Rad, L.R.; Shamshiri, S. Controlled release of doxorubicin from electrospun PEO/chitosan/graphene oxide nanocomposite nanofibrous scaffolds. Mater. Sci. Eng. C 2015, 48, 384–390. [Google Scholar] [CrossRef]
- Chang, B.; Sha, X.; Guo, J.; Jiao, Y.; Wang, C.; Yang, W. Thermo and pH dual responsive, polymer shell coated, magnetic mesoporous silica nanoparticles for controlled drug release. J. Mater. Chem. 2011, 21, 9239–9247. [Google Scholar] [CrossRef]
- Bardajee, G.R.; Hooshyar, Z.; Farsi, M.; Mobini, A.; Sang, G. Synthesis of a novel thermo/pH sensitive nanogel based on salep modified graphene oxide for drug release. Mater. Sci. Eng. C 2017, 72, 558–565. [Google Scholar] [CrossRef]
- He, Q.; Shi, J. Mesoporous silica nanoparticle based nano drug delivery systems: Synthesis, controlled drug release and delivery, pharmacokinetics and biocompatibility. J. Mater. Chem. 2011, 21, 5845–5855. [Google Scholar] [CrossRef]
- Chastek, T.T.; Wadajkar, A.; Nguyen, K.T.; Hudson, S.D.; Chastek, T.Q. Polyglycol-templated synthesis of poly (N-isopropyl acrylamide) microgels with improved biocompatibility. Colloid Polym. Sci. 2010, 288, 105. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Weight Loss < 250 °C [%] | Weight Loss at 250~800 °C [%] | Residue Weight [%] | Grafting Yield [%] |
|---|---|---|---|---|
| HMS | 2.5 | 4.1 | 93.4 | - |
| HMS@KH570 | 2.0 | 16.2 | 81.8 | 14.8 |
| HMS@P(NIPAM-co-DMA) | 3.9 | 26.4 | 69.7 | 14.6 |
| Sample | Drug loading 1 [mg] | Loading capacity 1 (LC) [%] | Mass percentage 1 [%] | Entrapment efficiency 1 (EE) [%] | Mass percentage 2 [%] |
|---|---|---|---|---|---|
| HMS | 4.70 | 9.4 | 8.5 | 94.0 | 8.1 |
| HMS@P(NIPAM-co-DMA-Fe3+) | 4.73 | 9.5 | 8.6 | 94.6 | 8.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, W.; Zhang, Z.; Rong, M.; Zhang, M. Core-Shell Structure Design of Hollow Mesoporous Silica Nanospheres Based on Thermo-Sensitive PNIPAM and pH-Responsive Catechol-Fe3+ Complex. Polymers 2019, 11, 1832. https://doi.org/10.3390/polym11111832
Peng W, Zhang Z, Rong M, Zhang M. Core-Shell Structure Design of Hollow Mesoporous Silica Nanospheres Based on Thermo-Sensitive PNIPAM and pH-Responsive Catechol-Fe3+ Complex. Polymers. 2019; 11(11):1832. https://doi.org/10.3390/polym11111832
Chicago/Turabian StylePeng, Weili, Zeping Zhang, Minzhi Rong, and Mingqiu Zhang. 2019. "Core-Shell Structure Design of Hollow Mesoporous Silica Nanospheres Based on Thermo-Sensitive PNIPAM and pH-Responsive Catechol-Fe3+ Complex" Polymers 11, no. 11: 1832. https://doi.org/10.3390/polym11111832
APA StylePeng, W., Zhang, Z., Rong, M., & Zhang, M. (2019). Core-Shell Structure Design of Hollow Mesoporous Silica Nanospheres Based on Thermo-Sensitive PNIPAM and pH-Responsive Catechol-Fe3+ Complex. Polymers, 11(11), 1832. https://doi.org/10.3390/polym11111832