Surface-Initiated Initiators for Continuous Activator Regeneration (SI ICAR) ATRP of MMA from 2,2,6,6–tetramethylpiperidine–1–oxy (TEMPO) Oxidized Cellulose Nanofibers for the Preparations of PMMA Nanocomposites

 , ,
, , 


 and
and 
Abstract
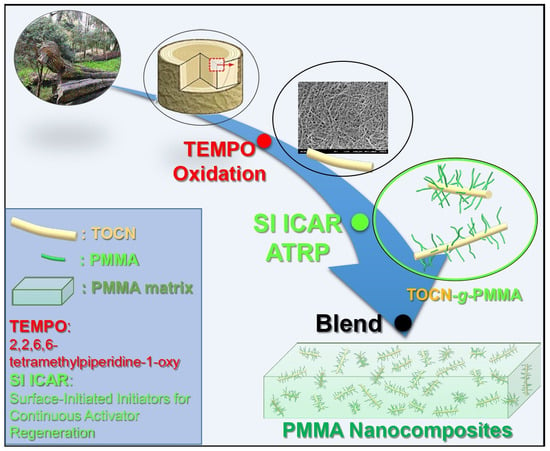
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of 2,2,6,6–tetramethylpiperidine–oxy (TEMPO)-Oxidized Cellulose Nanofibers (TOCNs)
2.3. Surface Modification of 2,2,6,6–tetramethylpiperidine–oxy (TEMPO)-Oxidized Cellulose Nanofibers (TOCN) with Atom Transfer Radical Polymerization (ATRP) Initiating Moiety (TOCN–Br)
2.4. Surface-Initiated Initiators for Continuous Activator Regeneration Atom Transfer Radical Polymerization (SI ICAR) ATRP of Methyl Methyacrylate (MMA) from 2,2,6,6–tetramethylpiperidine–oxy (TEMPO)-Oxidized Cellulose Nanofibers (TOCN) with ATRP Initiating Moiety (TOCN)–Br
2.5. Characterization
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Usuki, A.; Kojima, Y.; Kawasumi, M.; Okada, A.; Fukushima, Y.; Kurauchi, T.; Kamigaito, O. Synthesis of nylon 6-clay hybrid. J. Mater. Res. 1993, 8, 1179–1184. [Google Scholar] [CrossRef]
- Fu, H.-K.; Huang, C.-F.; Huang, J.-M.; Chang, F.-C. Studies on thermal properties of PS nanocomposites for the effect of intercalated agent with side groups. Polymer 2008, 49, 1305–1311. [Google Scholar] [CrossRef]
- O’Connell, D.W.; Birkinshaw, C.; O’Dwyer, T.F. Heavy metal adsorbents prepared from the modification of cellulose: A review. Bioresour. Technol. 2008, 99, 6709–6724. [Google Scholar] [CrossRef]
- Huang, C.-F.; Tu, C.-W.; Lee, R.-H.; Yang, C.-H.; Hung, W.-C.; Lin, K.-Y.A. Study of various diameter and functionality of TEMPO-oxidized cellulose nanofibers on paraquat adsorptions. Polym. Degrad. Stab. 2019, 161, 206–212. [Google Scholar] [CrossRef]
- Lin, K.-Y.A.; Heish, Y.-T.; Tsai, T.-Y.; Huang, C.-F. TEMPO-oxidized pulp as an efficient and recyclable sorbent to remove paraquat from water. Cellulose 2015, 22, 3261–3274. [Google Scholar] [CrossRef]
- Huang, C.-F.; Chen, J.-K.; Tsai, T.-Y.; Hsieh, Y.-A.; Lin, K.-Y.A. Dual-functionalized cellulose nanofibrils prepared through TEMPO-mediated oxidation and surface-initiated ATRP. Polymer 2015, 72, 395–405. [Google Scholar] [CrossRef]
- Nogi, M.; Iwamoto, S.; Nakagaito, A.N.; Yano, H. Optically transparent nanofiber paper. Adv. Mater. 2009, 21, 1595–1598. [Google Scholar] [CrossRef]
- Isogai, A.; Kato, Y. Preparation of polyglucuronic acid from cellulose by TEMPO-mediated oxidation. Cellulose 1998, 5, 153–164. [Google Scholar] [CrossRef]
- Siro, I.; Plackett, D. Microfibrillated cellulose and new nanocomposite materials: A review. Cellulose 2010, 17, 459–494. [Google Scholar] [CrossRef]
- Eichhorn, S.J. Cellulose nanowhiskers: Promising materials for advanced applications. Soft Matter 2011, 7, 303–315. [Google Scholar] [CrossRef]
- Isogai, A.; Saito, T.; Fukuzumi, H. TEMPO-oxidized cellulose nanofibers. Nanoscale 2011, 3, 71–85. [Google Scholar] [CrossRef]
- Huang, T.; Kuboyama, K.; Fukuzumi, H.; Ougizawa, T. PMMA/TEMPO-oxidized cellulose nanofiber nanocomposite with improved mechanical properties, high transparency and tunable birefringence. Cellulose 2018, 25, 2393–2403. [Google Scholar] [CrossRef]
- Endo, R.; Saito, T.; Isogai, A. TEMPO-oxidized cellulose nanofibril/poly(vinyl alcohol) composite drawn fibers. Polymer 2013, 54, 935–941. [Google Scholar] [CrossRef]
- Kurihara, T.; Isogai, A. Properties of poly(acrylamide)/TEMPO-oxidized cellulose nanofibril composite films. Cellulose 2014, 21, 291–299. [Google Scholar] [CrossRef]
- Yokozawa, T.; Ohta, Y. Transformation of step-growth polymerization into living chain-growth polymerization. Chem. Rev. 2016, 116, 1950–1968. [Google Scholar] [CrossRef]
- Sawamoto, M. Modern cationic vinyl polymerization. Prog. Polym. Sci. 1991, 16, 111–172. [Google Scholar] [CrossRef]
- Ito, S.; Goseki, R.; Ishizone, T.; Hirao, A. Synthesis of well-controlled graft polymers by living anionic polymerization towards exact graft polymers. Polym. Chem. 2014, 5, 5523–5534. [Google Scholar] [CrossRef]
- Goseki, R.; Ito, S.; Matsuo, Y.; Higashihara, T.; Hirao, A. Precise synthesis of macromolecular architectures by novel iterative methodology combining living anionic polymerization with specially designed linking chemistry. Polymers 2017, 9, 470. [Google Scholar] [CrossRef]
- Huang, C.F.; Aimi, J.; Lai, K.Y. Synthesis of novel mu-star copolymers with poly(N-octyl benzamide) and poly(epsilon-caprolactone) miktoarms through chain-growth condensation polymerization, styrenics-assisted atom transfer radical coupling, and ring-opening polymerization. Macromol. Rapid Commun. 2017, 38, 1600607. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Pitsikalis, M.; Pispas, S.; Iatrou, H. Polymers with complex architecture by living anionic polymerization. Chem. Rev. 2001, 101, 3747–3792. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Muller, A.H.E. 50 years of living polymerization. Prog. Polym. Sci. 2006, 31, 1039–1040. [Google Scholar] [CrossRef]
- You, J.; Yoon, J.A.; Kim, J.; Huang, C.-F.; Matyjaszewski, K.; Kim, E. Excimer Emission from Self-Assembly of Fluorescent Diblock Copolymer Prepared by Atom Transfer Radical Polymerization. Chem.Mater. 2010, 22, 4426–4434. [Google Scholar] [CrossRef]
- Kamigaito, M.; Ando, T.; Sawamoto, M. Metal-catalyzed living radical polymerization. Chem. Rev. 2001, 101, 3689–3745. [Google Scholar] [CrossRef]
- Huang, C.-F.; Chen, W.-H.; Aimi, J.; Huang, Y.-S.; Venkatesan, S.; Chiang, Y.-W.; Huang, S.-H.; Kuo, S.-W.; Chen, T. Synthesis of well-defined PCL-b-PnBA-b-PMMA ABC-type triblock copolymers: Toward the construction of nanostructures in epoxy thermosets. Polym. Chem. 2018, 9, 5644–5654. [Google Scholar] [CrossRef]
- Aimi, J.; Wang, P.-H.; Shih, C.-C.; Huang, C.-F.; Nakanishi, T.; Takeuchi, M.; Hsuehe, H.-Y.; Chen, W.-C. A star polymer with a metallo-phthalocyanine core as a tunable charge storage material for nonvolatile transistor memory devices. J. Mater. Chem. C 2018, 6, 2724–2732. [Google Scholar] [CrossRef]
- Lu, Y.C.; Chou, L.C.; Huang, C.F. Iron-catalysed atom transfer radical polyaddition for the synthesis and modification of novel aliphatic polyesters displaying lower critical solution temperature and pH-dependent release behaviors. Polym. Chem. 2019, 10, 3912–3921. [Google Scholar] [CrossRef]
- Han, Y.-M.; Chen, H.-H.; Huang, C.-F. Polymerization and degradation of aliphatic polyesters synthesized by atom transfer radical polyaddition. Polym. Chem. 2015, 6, 4565–4574. [Google Scholar] [CrossRef]
- Sathesh, V.; Chen, J.K.; Chang, C.J.; Aimi, J.; Chen, Z.C.; Hsu, Y.C.; Huang, Y.S.; Huang, C.F. Synthesis of poly(epsilon-caprolactone)-based miktoarm star copolymers through ROP, SA ATRC, and ATRP. Polymers 2018, 10, 858. [Google Scholar] [CrossRef]
- Lai, K.-Y.; Huang, Y.-S.; Chu, C.-Y.; Huang, C.-F. Synthesis of poly(N-H benzamide)-b-poly(lauryl methacrylate)-b-poly(N-H benzamide) symmetrical triblock copolymers by combinations of CGCP, SARA ATRP, and SA ATRC. Polymer 2018, 137, 385–394. [Google Scholar] [CrossRef]
- Moad, G.; Chong, Y.K.; Postma, A.; Rizzardo, E.; Thang, S.H. Advances in RAFT polymerization: The synthesis of polymers with defined end-groups. Polymer 2005, 46, 8458–8468. [Google Scholar] [CrossRef]
- Chen, C.; Guo, X.F.; Du, J.H.; Choi, B.; Tang, H.L.; Feng, A.C.; Thang, S.H. Synthesis of multifunctional miktoarm star polymers via an RGD peptide-based RAFT agent. Polym. Chem. 2019, 10, 228–234. [Google Scholar] [CrossRef]
- Huang, Y.-S.; Chen, J.-K.; Chen, T.; Huang, C.-F. Synthesis of PNVP-based copolymers with tunable thermosensitivity by sequential reversible addition–fragmentation chain transfer copolymerization and ring-opening polymerization. Polymers 2017, 9, 231. [Google Scholar] [CrossRef]
- Hawker, C.J.; Bosman, A.W.; Harth, E. New polymer synthesis by nitroxide mediated living radical polymerizations. Chem. Rev. 2001, 101, 3661–3688. [Google Scholar] [CrossRef]
- Bielawski, C.W.; Grubbs, R.H. Living ring-opening metathesis polymerization. Prog. Polym. Sci. 2007, 32, 1–29. [Google Scholar] [CrossRef]
- Kamber, N.E.; Jeong, W.; Waymouth, R.M.; Pratt, R.C.; Lohmeijer, B.G.G.; Hedrick, J.L. Organocatalytic ring-opening polymerization. Chem. Rev. 2007, 107, 5813–5840. [Google Scholar] [CrossRef]
- Huang, C.-F.; Chen, M.-J.; Lin, C.-H.; Chiang, Y.-W. Synthesis of well-defined poly(N-H benzamide-co-N-octyl benzamide)s and the study of their blends with nylon 6. Polymers 2017, 9, 172. [Google Scholar] [CrossRef]
- Van Steenberge, P.H.M.; D’hooge, D.R.; Wang, Y.; Zhong, M.J.; Reyniers, M.F.; Konkolewicz, D.; Matyjaszewski, K.; Marin, G.B. Linear gradient quality of ATRP copolymers. Macromolecules 2012, 45, 8519–8531. [Google Scholar] [CrossRef]
- De Rybel, N.; Van Steenberge, P.H.M.; Reyniers, M.F.; Barner-Kowollik, C.; D’hooge, D.R.; Marin, G.B. An update on the pivotal role of kinetic modeling for the mechanistic understanding and design of bulk and solution RAFT polymerization. Macromol. Theor. Simul. 2017, 26, 1600048. [Google Scholar] [CrossRef]
- Fierens, S.K.; Telitel, S.; Van Steenberge, P.H.M.; Reyniers, M.F.; Marin, G.B.; Lutz, J.F.; D’hooge, D.R. Model-based design to push the boundaries of sequence control. Macromolecules 2016, 49, 9336–9344. [Google Scholar] [CrossRef]
- Fierens, S.K.; D’hooge, D.R.; Van Steenberge, P.H.M.; Reyniers, M.F.; Marin, G.B. MAMA-SG1 initiated nitroxide mediated polymerization of styrene: From arrhenius parameters to model-based design. Chem. Eng. J. 2015, 278, 407–420. [Google Scholar] [CrossRef]
- Van Steenberge, P.H.M.; D’hooge, D.R.; Reyniers, M.F.; Marin, G.B.; Cunningham, M.F. 4-dimensional modeling strategy for an improved understanding of miniemulsion NMP of acrylates initiated by SG1-macroinitiator. Macromolecules 2014, 47, 7732–7741. [Google Scholar] [CrossRef]
- Gigmes, D.; Van Steenberge, P.H.M.; Siri, D.; D’hooge, D.R.; Guillaneuf, Y.; Lefay, C. Simulation of the degradation of cyclic ketene acetal and vinyl-based copolymers synthesized via a radical process: Influence of the reactivity ratios on the degradability properties. Macromol. Rapid Commun. 2018, 39, 1800193. [Google Scholar] [CrossRef]
- Tsai, T.-Y.; Huang, C.-F. Data in support of dualfunctionalized cellulose nanofibrils prepared through TEMPO-mediated oxidation and surface-initiated ATRP. Data in Brief 2015, 3, 195–200. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Jakubowski, W.; Min, K.; Tang, W.; Huang, J.Y.; Braunecker, W.A.; Tsarevsky, N.V. Diminishing catalyst concentration in atom transfer radical polymerization with reducing agents. Prod. Natl. Acad. Sci. USA 2006, 103, 15309–15314. [Google Scholar] [CrossRef]
- D’hooge, D.R.; Van Steenberge, P.H.M.; Reyniers, M.F.; Marin, G.B. Fed-batch control and visualization of monomer sequences of individual ICAR ATRP gradient copolymer chains. Polymers 2014, 6, 1074–1095. [Google Scholar] [CrossRef]
- Fierens, S.K.; Van Steenberge, P.H.M.; Reyniers, M.F.; Marin, G.B.; D’hooge, D.R. How penultimate monomer unit effects and initiator influence ICAR ATRP of n-butyl acrylate and methyl methacrylate. AIChE J. 2017, 63, 4971–4986. [Google Scholar] [CrossRef]
- Porras, C.T.; D’hooge, D.R.; Van Steenberge, P.H.M.; Reyniers, M.F.; Marin, G.B. A theoretical exploration of the potential of ICAR ATRP for one- and two-pot synthesis of well-defined diblock copolymers. Macromol. React. Eng. 2013, 7, 311–326. [Google Scholar] [CrossRef]
- Porras, C.T.; D’hooge, D.R.; Van Steenberge, P.H.M.; Reyniers, M.F.; Marin, G.B. ICAR ATRP for estimation of intrinsic macro-activation/deactivation arrhenius parameters under polymerization conditions. Ind. Eng. Chem. Res. 2014, 53, 9674–9685. [Google Scholar] [CrossRef]
- Cheng, K.-C.; Huang, C.-F.; Wei, Y.; Hsu, S.-H. Novel chitosan–cellulose nanofiber selfhealing hydrogels to correlate self-healing properties of hydrogels with neural regeneration effects. NPG Asia Mater. 2019, 11, 25. [Google Scholar] [CrossRef]
- Chen, R.-D.; Huang, C.-F.; Hsu, S.-H. Composites of waterborne polyurethane and cellulose nanofibers for 3D printing and bioapplications. Carbohydr. Polym. 2019, 212, 75–88. [Google Scholar] [CrossRef]
- Araki, J.; Wada, M.; Kuga, S. Steric stabilization of a cellulose microcrystal suspension by poly(ethylene glycol) grafting. Langmuir 2001, 17, 21–27. [Google Scholar] [CrossRef]
- Iwamoto, S.; Kai, W.H.; Isogai, T.; Saito, T.; Isogai, A.; Iwata, T. Comparison study of TEMPO-analogous compounds on oxidation efficiency of wood cellulose for preparation of cellulose nanofibrils. Polym. Degrad. Stab. 2010, 95, 1394–1398. [Google Scholar] [CrossRef]
- Habibi, Y.; Goffin, A.L.; Schiltz, N.; Duquesne, E.; Dubois, P.; Dufresne, A. Bionanocomposites based on poly(epsilon-caprolactone)-grafted cellulose nanocrystals by ring-opening polymerization. J. Mater. Chem. 2008, 18, 5002–5010. [Google Scholar] [CrossRef]
- Oh, S.Y.; Yoo, D.I.; Shin, Y.; Seo, G. FTIR analysis of cellulose treated with sodium hydroxide and carbon dioxide. Carbohydr. Res. 2005, 340, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Yoo, D.I.; Shin, Y.; Kim, H.C.; Kim, H.Y.; Chung, Y.S.; Park, W.H.; Youk, J.H. Crystalline structure analysis of cellulose treated with sodium hydroxide and carbon dioxide by means of X-ray diffraction and FTIR spectroscopy. Carbohydr. Res. 2005, 340, 2376–2391. [Google Scholar] [CrossRef] [PubMed]
- El-Sakhawy, M.; Kamel, S.; Salama, A.; Tohamy, H.A.S. Preparation and infrared study of cellulose based amphiphilic materials. Cellul. Chem. Technol. 2018, 52, 193–200. [Google Scholar]
- Chen, W.B.; He, H.; Zhu, H.X.; Cheng, M.X.; Li, Y.H.; Wang, S.F. Thermo-responsive cellulose-based material with switchable wettability for controllable oil/water separation. Polymers 2018, 10, 592. [Google Scholar] [CrossRef] [PubMed]
- Tommasini, F.J.; Ferreira, L.D.C.; Tienne, L.G.P.; Aguiar, V.D.; da Silva, M.H.P.; Rocha, L.F.D.; Marques, M.D.V. Poly(methyl methacrylate)-SiC nanocomposites prepared through in situ polymerization. Mater. Res. 2018, 21, 20180086. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Deconvoluted Signal (eV) | Fraction | ||||
|---|---|---|---|---|---|---|
| C–C | C–OH | C–O | O–C=O | fC–OH | fO–C=O | |
| Cellulose | 284.20 | 285.8 | 286.81 | – | 32.7 | – |
| TOCN | 284.16 | 285.77 | 286.79 | 287.96 | 28.6 | 15.0 |
| TOCN–Br | 284.18 | 284.74 | 286.84 | 287.94 | 12.7 | 19.0 |
| Sample | M/I a | Mn | PDI | Grafted PMMA (wt%) b | WCA(°) c |
|---|---|---|---|---|---|
| TOCN–g–PMMA1 | 100 | 12,000 | 1.16 | 25 | 96.3 ± 1.5 |
| TOCN–g–PMMA2 | 200 | 18,800 | 1.17 | 33 | 98.7 ± 2.5 |
| TOCN–g–PMMA3 | 300 | 28,000 | 1.13 | 38 | 92.3 ± 1.5 |
| Sample | Td5 (°C) a | T–(dT/dW),max (°C) b | Tg (°C) c | Strength (MPa) d | Strain (%) d |
|---|---|---|---|---|---|
| PMMA | 417 | 453 | 99 | 17.1 | 0.88 |
| TOCN | 195 | 216 | – | – | – |
| TOCN–g–PMMA2 | 278 | 365 | – | – | – |
| TOCN–g–PMMA2/PMMA Composites | |||||
| 1%TOCN | 327 | 384 | 110 | 37.2 | 1.92 |
| 3%TOCN | 320 | 373 | 105 | 24.9 | 1.36 |
| 5%TOCN | 326 | 381 | 103 | 22.7 | 1.20 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, C.-W.; Tsai, F.-C.; Chang, C.-J.; Yang, C.-H.; Kuo, S.-W.; Zhang, J.; Chen, T.; Huang, C.-F. Surface-Initiated Initiators for Continuous Activator Regeneration (SI ICAR) ATRP of MMA from 2,2,6,6–tetramethylpiperidine–1–oxy (TEMPO) Oxidized Cellulose Nanofibers for the Preparations of PMMA Nanocomposites. Polymers 2019, 11, 1631. https://doi.org/10.3390/polym11101631
Tu C-W, Tsai F-C, Chang C-J, Yang C-H, Kuo S-W, Zhang J, Chen T, Huang C-F. Surface-Initiated Initiators for Continuous Activator Regeneration (SI ICAR) ATRP of MMA from 2,2,6,6–tetramethylpiperidine–1–oxy (TEMPO) Oxidized Cellulose Nanofibers for the Preparations of PMMA Nanocomposites. Polymers. 2019; 11(10):1631. https://doi.org/10.3390/polym11101631
Chicago/Turabian StyleTu, Cheng-Wei, Fang-Chang Tsai, Chi-Jung Chang, Cheng-Han Yang, Shiao-Wei Kuo, Jiawei Zhang, Tao Chen, and Chih-Feng Huang. 2019. "Surface-Initiated Initiators for Continuous Activator Regeneration (SI ICAR) ATRP of MMA from 2,2,6,6–tetramethylpiperidine–1–oxy (TEMPO) Oxidized Cellulose Nanofibers for the Preparations of PMMA Nanocomposites" Polymers 11, no. 10: 1631. https://doi.org/10.3390/polym11101631
APA StyleTu, C.-W., Tsai, F.-C., Chang, C.-J., Yang, C.-H., Kuo, S.-W., Zhang, J., Chen, T., & Huang, C.-F. (2019). Surface-Initiated Initiators for Continuous Activator Regeneration (SI ICAR) ATRP of MMA from 2,2,6,6–tetramethylpiperidine–1–oxy (TEMPO) Oxidized Cellulose Nanofibers for the Preparations of PMMA Nanocomposites. Polymers, 11(10), 1631. https://doi.org/10.3390/polym11101631