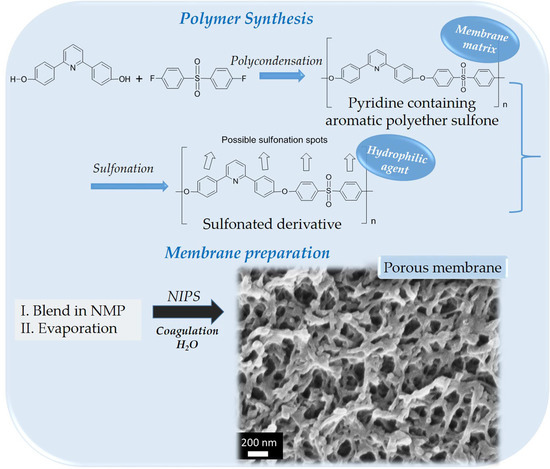
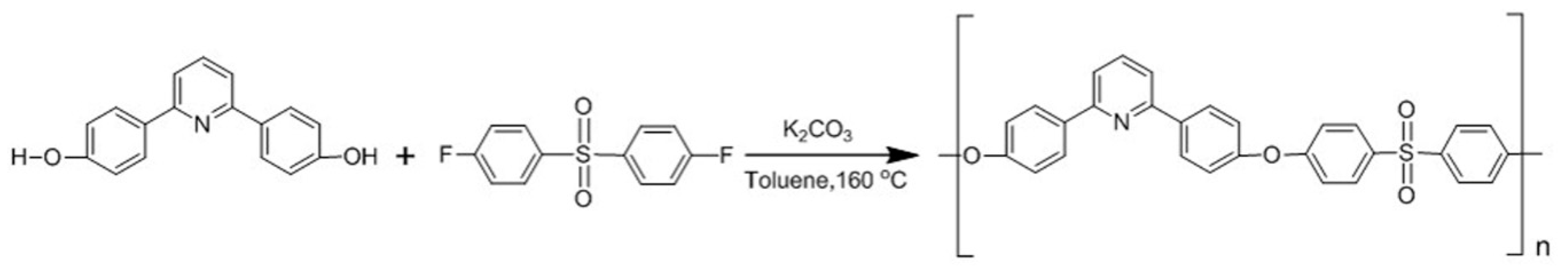
Scheme 1.
Experimental procedure for PDSPP synthesis.
Scheme 1.
Experimental procedure for PDSPP synthesis.
Scheme 2.
Experimental procedure for SPDSPP synthesis.
Scheme 2.
Experimental procedure for SPDSPP synthesis.
Figure 1.
Representative SEM images of the cross-sectional morphologies of the membranes (a) M1’-35NT, and (b) M1’-75NT.
Figure 1.
Representative SEM images of the cross-sectional morphologies of the membranes (a) M1’-35NT, and (b) M1’-75NT.
Figure 2.
Representative SEM images of (a) cross-sectional; (b) top; and (c) bottom surface morphologies of the membrane M2-PVP6.
Figure 2.
Representative SEM images of (a) cross-sectional; (b) top; and (c) bottom surface morphologies of the membrane M2-PVP6.
Figure 3.
ATR-FTIR spectra of the membranes M1-5O and M1-15O on top and bottom surfaces. For comparison, ATR-FTIR spectra of the PDSPP and SPDSPP homopolymers are shown.
Figure 3.
ATR-FTIR spectra of the membranes M1-5O and M1-15O on top and bottom surfaces. For comparison, ATR-FTIR spectra of the PDSPP and SPDSPP homopolymers are shown.
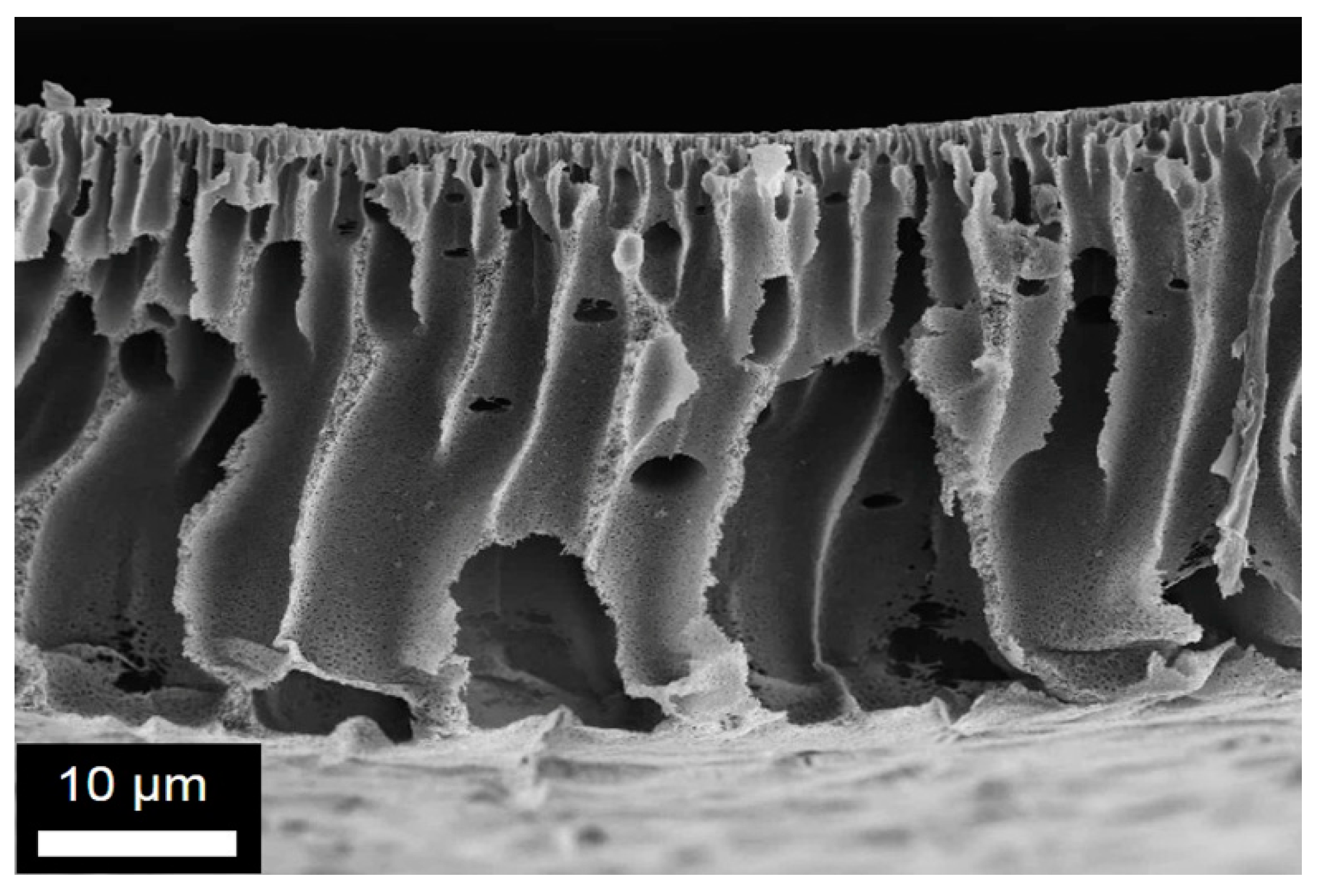
Figure 4.
Representative SEM images obtained for the membrane M1-6: (a) and (b) cross-section at two different magnifications; (c) top and (d) bottom surface structures.
Figure 4.
Representative SEM images obtained for the membrane M1-6: (a) and (b) cross-section at two different magnifications; (c) top and (d) bottom surface structures.
Figure 5.
Representative SEM images obtained for the membrane M2-6: (a) and (b) cross-section at two different magnifications; (c) top and (d) bottom surface structures.
Figure 5.
Representative SEM images obtained for the membrane M2-6: (a) and (b) cross-section at two different magnifications; (c) top and (d) bottom surface structures.
Figure 6.
Representative SEM images obtained for the membrane M2-12: (a) and (b) cross-section at two different magnifications; (c) top and (d) bottom surface structure.
Figure 6.
Representative SEM images obtained for the membrane M2-12: (a) and (b) cross-section at two different magnifications; (c) top and (d) bottom surface structure.
Figure 7.
Representative SEM images of the cross-section, the top and the bottom surfaces of the membranes (a), (c), (e) M0-6 and (b), (d), (f) M0-12.
Figure 7.
Representative SEM images of the cross-section, the top and the bottom surfaces of the membranes (a), (c), (e) M0-6 and (b), (d), (f) M0-12.
Figure 8.
Representative SEM images obtained for the membrane M2-20: (a) and (b) cross-section at two different magnifications; (c) top and (d) bottom surface structure.
Figure 8.
Representative SEM images obtained for the membrane M2-20: (a) and (b) cross-section at two different magnifications; (c) top and (d) bottom surface structure.
Figure 9.
Representative SEM image of the cross-sectional morphology of membrane M2,2-12.
Figure 9.
Representative SEM image of the cross-sectional morphology of membrane M2,2-12.
Figure 10.
TGA curves of the bulk and porous membranes (a) M1-6; and (b) M1,1-12.
Figure 10.
TGA curves of the bulk and porous membranes (a) M1-6; and (b) M1,1-12.
Figure 11.
ATR-FTIR spectra of the bulk and porous membrane M2-6.
Figure 11.
ATR-FTIR spectra of the bulk and porous membrane M2-6.
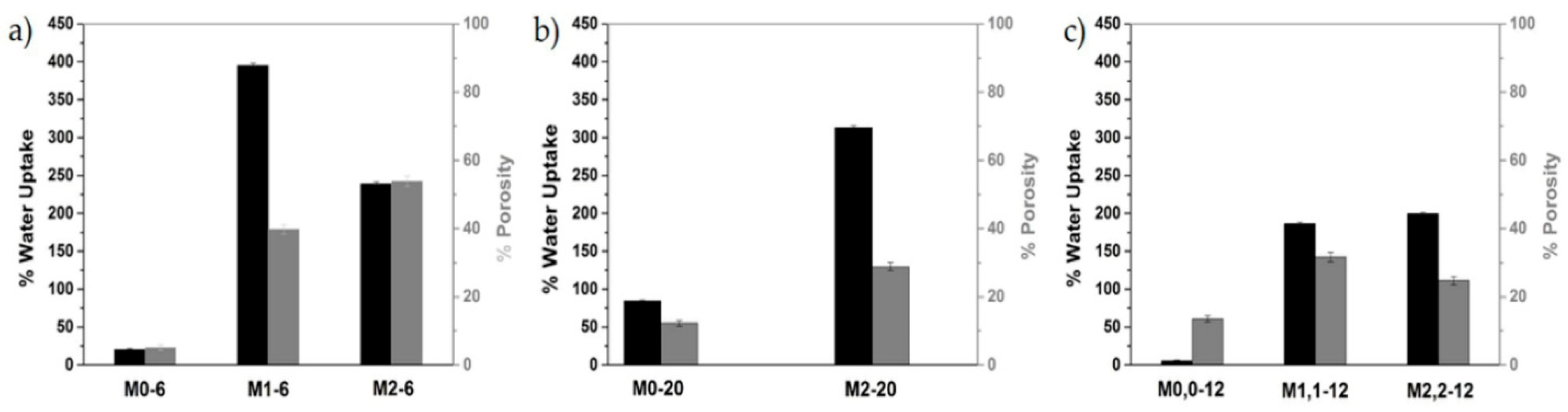
Figure 12.
Schematic representation of WU and porosity values for the membranes (a) M0-6, M1-6, and M2-6; (b) M0-20, and M2-20; and (c) M0,0-12, M1,1-12, and M2,2-12.
Figure 12.
Schematic representation of WU and porosity values for the membranes (a) M0-6, M1-6, and M2-6; (b) M0-20, and M2-20; and (c) M0,0-12, M1,1-12, and M2,2-12.
Table 1.
Preparation of membranes with 90/10 w/w PDSPP/SPDSPP in NMP at 5% or 15% w/w, casting at 80 °C until 35%, 50%, 75%, 90% w/w total final polymers’ concentration and immersion in H2O at 25 °C or 60 °C for 3 h.
Table 1.
Preparation of membranes with 90/10 w/w PDSPP/SPDSPP in NMP at 5% or 15% w/w, casting at 80 °C until 35%, 50%, 75%, 90% w/w total final polymers’ concentration and immersion in H2O at 25 °C or 60 °C for 3 h.
| Membrane | PDSPP Initial/Final Concentration (w/w %) | SPDSPP Initial/Final Concentration (w/w %) | Initial/Final Concentration (w/w %) in NMP | Temperature (°C)/Coagulation Time (h) in H2O | Membrane Thickness (μm) |
|---|
| M1’-35N | 13.5/31.5 | 1.5/3.5 | 85/65 | 25/3 | 200 |
| M1’-50N | 13.5/45 | 1.5/5 | 85/50 | 25/3 | 220 |
| M1’-75N | 13.5/67.5 | 1.5/7.5 | 85/25 | 25/3 | 50 |
| M1’-90N | 13.5/81 | 1.5/9 | 85/10 | 25/3 | 45 |
| M1’-35NT | 13.5/31.5 | 1.5/3.5 | 85/65 | 60/3 | 350 |
| M1’-50NT | 13.5/45 | 1.5/5 | 85/50 | 60/3 | 290 |
| M1’-75NT | 13.5/67.5 | 1.5/7.5 | 85/25 | 60/3 | 90 |
| M1’-90NT | 13.5/81 | 1.5/9 | 85/10 | 60/3 | 70 |
Table 2.
Preparation of membranes with 80/20 w/w PDSPP/SPDSPP in NMP at 5% or 15% w/w, casting at 80 °C until 75% w/w total final polymers’ concentration and immersion in H2O at 60 °C for 3 h or 24 h.
Table 2.
Preparation of membranes with 80/20 w/w PDSPP/SPDSPP in NMP at 5% or 15% w/w, casting at 80 °C until 75% w/w total final polymers’ concentration and immersion in H2O at 60 °C for 3 h or 24 h.
| Membrane | PDSPP Initial/Final Concentration (w/w %) | SPDSPP Initial/Final Concentration (w/w %) | Initial/Final Concentration (w/w %) in NMP | Temperature (°C)/Coagulation Time (h) in H2O | Membrane Thickness (μm) |
|---|
| M1-5O | 4/60 | 1/15 | 95/25 | 60/24 | 20 |
| M1-5 | 4/60 | 1/15 | 95/25 | 60/3 | 20 |
| M1-15O | 12/60 | 3/15 | 85/25 | 60/24 | 85 |
| M1-15 | 12/60 | 3/15 | 85/25 | 60/3 | 90 |
Table 3.
Preparation of membranes with 70/30 w/w PDSPP/SPDSPP in NMP at 5% or 15% w/w, addition of PVP or PEG at 12.5% w/w final concentration, casting at 80 °C until 75% w/w total final polymers’ concentration and immersion in H2O at 60 °C for 3 h.
Table 3.
Preparation of membranes with 70/30 w/w PDSPP/SPDSPP in NMP at 5% or 15% w/w, addition of PVP or PEG at 12.5% w/w final concentration, casting at 80 °C until 75% w/w total final polymers’ concentration and immersion in H2O at 60 °C for 3 h.
| Membrane | PDSPP Initial/Final Concentration (w/w %) | SPDSPP Initial/Final Concentration (w/w %) | Initial/Final Concentration (w/w %) in NMP | Porogen Initial/Final Concentration (w/w %) | Temperature (°C)/Coagulation Time (h) in H2O | Membrane Thickness (μm) |
|---|
| M0-5 | 3.5/52.5 | 1.5/22.5 | 95/25 | - | 60/3 | 20 |
| M0-15 | 10.5/52.5 | 4.5/22.5 | 85/25 | - | 60/3 | 60 |
| M2-PVP6 | 3.5/43.7 | 1.5/18.8 | 94/25 | 1/12.5 | 60/3 | 15 |
| M2-PVP18 | 10.5/43.7 | 4.5/78.8 | 82/25 | 3/12.5 | 60/3 | 80 |
| M2-PEG6 | 3.5/43.7 | 1.5/18.8 | 94/25 | 1/12.5 | 60/3 | 25 |
| M2-PEG18 | 10.5/43.7 | 4.5/18.8 | 82/25 | 3/12.5 | 60/3 | 55 |
Table 4.
WU and porosity values for the membranes M1’-35NT, M1’-75NT, M2-PVP6, and M2-PEG6.
Table 4.
WU and porosity values for the membranes M1’-35NT, M1’-75NT, M2-PVP6, and M2-PEG6.
| Membrane | WU (%) | Porosity (%) |
|---|
| M1’-35NT | 12.7 ± 0.8 | 4.3 ± 0.5 |
| M1’-75NT | 13.4 ± 1.0 | 15.2 ± 0.9 |
| M2-PVP6 | 21.2 ± 1.3 | 21.0 ± 1.2 |
| M2-PEG6 | 28.7 ± 1.1 | 29.1 ± 1.3 |
Table 5.
PWF and PWP values for the membranes M2-PVP6, M2-PEG6, and M2-PVP18.
Table 5.
PWF and PWP values for the membranes M2-PVP6, M2-PEG6, and M2-PVP18.
| Membrane | PWF [L/(m2∗h)] | PWP [L/(m2∗h∗MPa)] |
|---|
| M2-PVP6 | 12.1 ± 2.2 | 50 ± 3.2 |
| M2-PEG6 | 82.4 ± 3.6 | 3300 ± 9.1 |
| M2-PVP18 | 0.2 ± 0.1 | 8.7 ± 1.6 |
Table 6.
Preparation of membranes with 80/20 w/w or 70/30 w/w PDSPP/SPDSPP in NMP at 5% and 15% w/w, addition of PVP at 3.3% w/w final concentration, casting at 80 °C until 20% w/w total final polymers’ concentration and immersion in H2O at 60 °C for 3 h.
Table 6.
Preparation of membranes with 80/20 w/w or 70/30 w/w PDSPP/SPDSPP in NMP at 5% and 15% w/w, addition of PVP at 3.3% w/w final concentration, casting at 80 °C until 20% w/w total final polymers’ concentration and immersion in H2O at 60 °C for 3 h.
| Membrane | PDSPP Initial/Final Concentration (w/w %) | SPDSPP Initial/Final Concentration (w/w %) | Initial/Final Concentration (w/w %) in NMP | PVP Initial/Final Concentration (w/w %) | Temperature (°C)/Coagulation Time (h) in H2O | Membrane Thickness (μm) |
|---|
| M0-6 | 5/16.7 | - | 94/80 | 1/3.3 | 60/3 | 50 |
| M1-6 | 4/13.3 | 1/3.3 | 94/80 | 1/3.3 | 60/3 | 70 |
| M2-6 | 3.5/11.7 | 1.5/5 | 94/80 | 1/3.3 | 60/3 | 70 |
| M0-12 | 10/16.7 | - | 88/80 | 2/3.3 | 60/3 | 170 |
| M2-12 | 7/11.7 | 3/5 | 88/80 | 2/3.3 | 60/3 | 280 |
| Μ2-20 | 7/7 | 3/3 | 80/80 | 10/10 | 60/3 | 265 |
Table 7.
Preparation of membranes with 80/20 w/w or 70/30 w/w PDSPP/SPDSPP in NMP at 10%, addition of PVP at 3.3% w/w final concentration, casting at 80 °C until 20% w/w total final polymers’ concentration and immersion in H2O/NMP 50/50 v/v at 25 °C for 1 h.
Table 7.
Preparation of membranes with 80/20 w/w or 70/30 w/w PDSPP/SPDSPP in NMP at 10%, addition of PVP at 3.3% w/w final concentration, casting at 80 °C until 20% w/w total final polymers’ concentration and immersion in H2O/NMP 50/50 v/v at 25 °C for 1 h.
| Membrane | PDSPP Initial/Final Concentration (w/w %) | SPDSPP Initial/Final Concentration (w/w %) | Initial/Final Concentration (w/w %) in NMP | PVP Initial/Final Concentration (w/w %) | Temperature (°C)/Coagulation Time (h) in H2O/NMP 50/50 v/v | Membrane Thickness (μm) |
|---|
| M0,0-12 | 10 /16.7 | - | 88/80 | 2/3.3 | 25/1 | 80 |
| M1,1-12 | 8/13.3 | 2/3.3 | 88/80 | 2/3.3 | 25/1 | 180 |
| M2,2-12 | 7/11.7 | 3/5 | 88/80 | 2/3.3 | 25/1 | 260 |
Table 8.
WU and porosity values for membranes M0-6, M1-6, M2-6, M0,0-12, M1,1-12, and M2,2-12.
Table 8.
WU and porosity values for membranes M0-6, M1-6, M2-6, M0,0-12, M1,1-12, and M2,2-12.
| Membrane | WU (%) | Porosity (%) |
|---|
| M0-6 | 20.5 ± 1.2 | 5.1 ± 0.8 |
| M1-6 | 395.2 ± 2.9 | 39.8 ± 1.4 |
| M2-6 | 239.2 ± 2.4 | 53.9 ± 1.5 |
| M0-20 | 84.6 ± 1.1 | 12.2 ± 0.8 |
| M2-20 | 313.3 ± 2.3 | 28.8 ± 1.2 |
| M0,0-12 | 5.2 ± 0.8 | 13.5 ± 0.9 |
| M1,1-12 | 186.4 ± 2.3 | 31.6 ± 1.4 |
| M2,2-12 | 199.6 ± 2.1 | 24.7 ± 1.2 |
Table 9.
PWF and PWP values for membranes M2-6 and M2-12.
Table 9.
PWF and PWP values for membranes M2-6 and M2-12.
| Membrane | PWF [(L/(m2∗h)] | PWP [L/(m2∗h∗MPa)] |
|---|
| M2-6 | 827.1 ± 10.6 | 33084.7 ± 36.2 |
| M2-12 | 1820.2 ± 11.5 | 72794.2 ± 37.8 |

 and
and 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}