Cavity Closure of 2-Hydroxypropyl-β-Cyclodextrin: Replica Exchange Molecular Dynamics Simulations

 , , , and
, , , and
Abstract

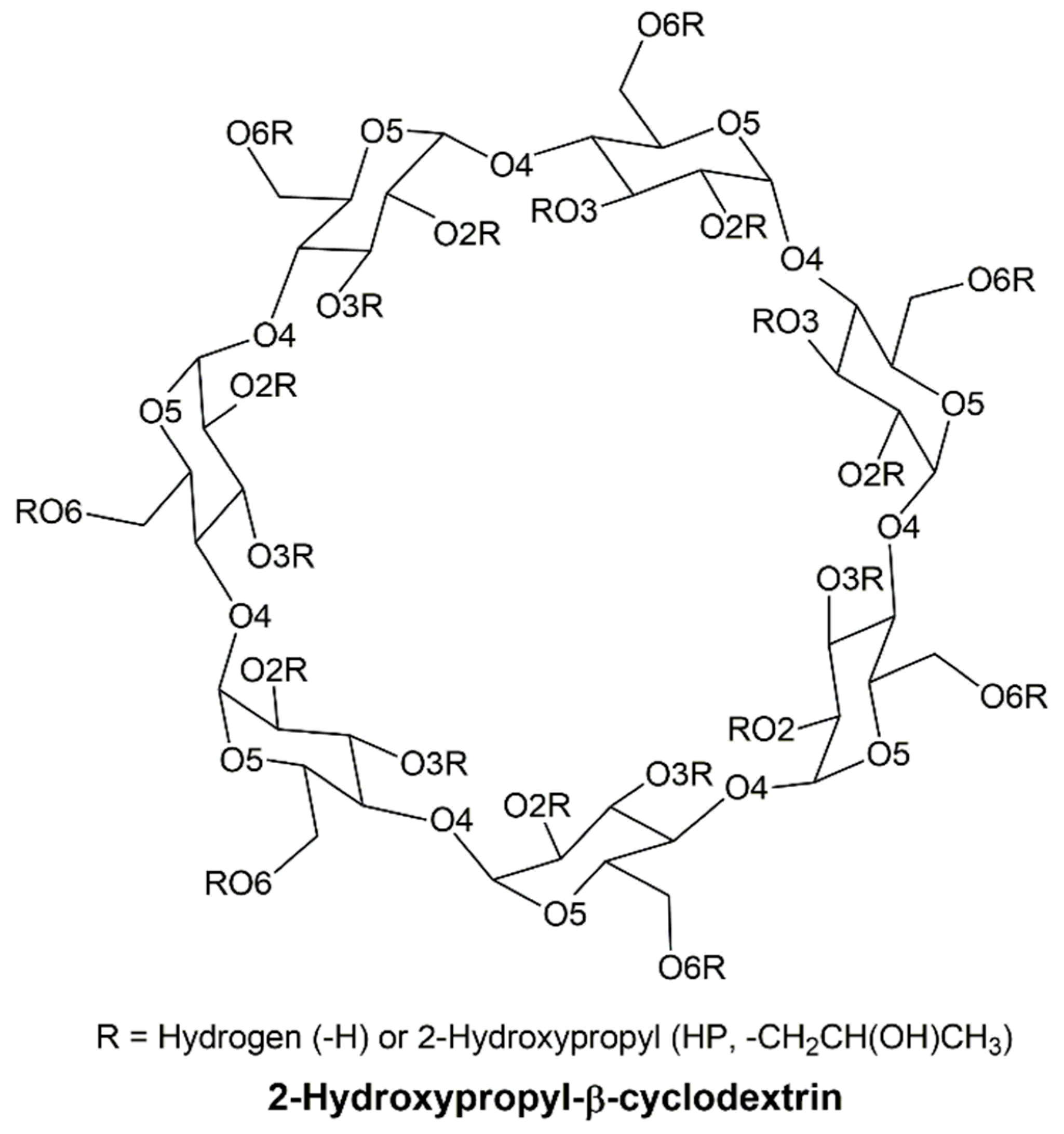
1. Introduction
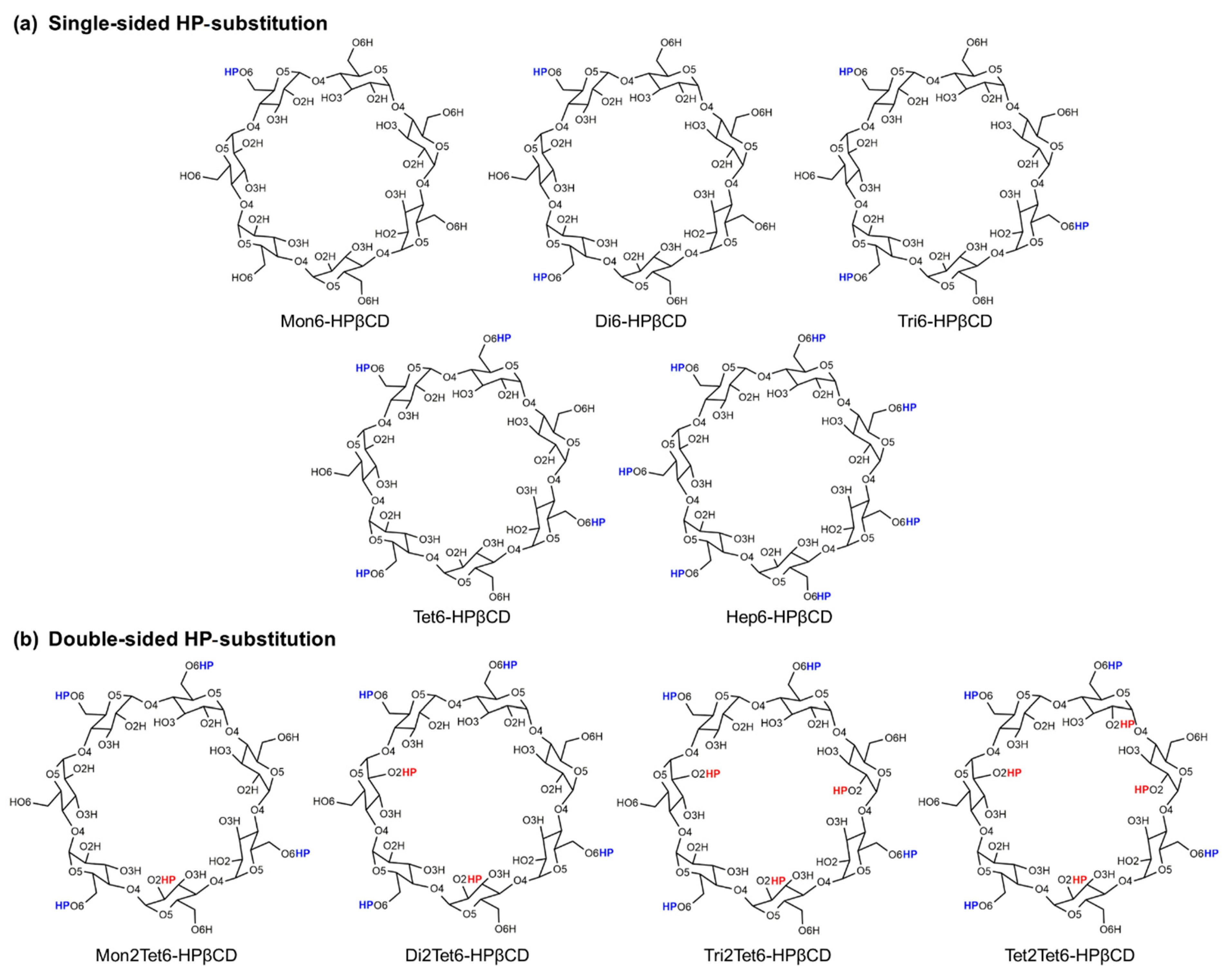
2. Computational Methods
3. Results and Discussion
3.1. Structural Analysis
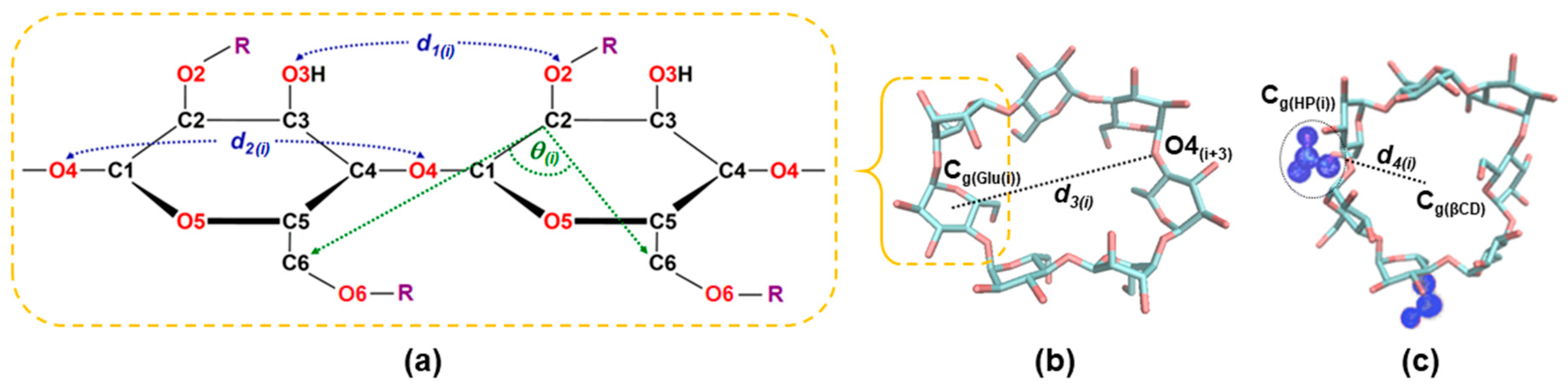
3.1.1. Structural Distortion of Glucose Units in the HPβCDs
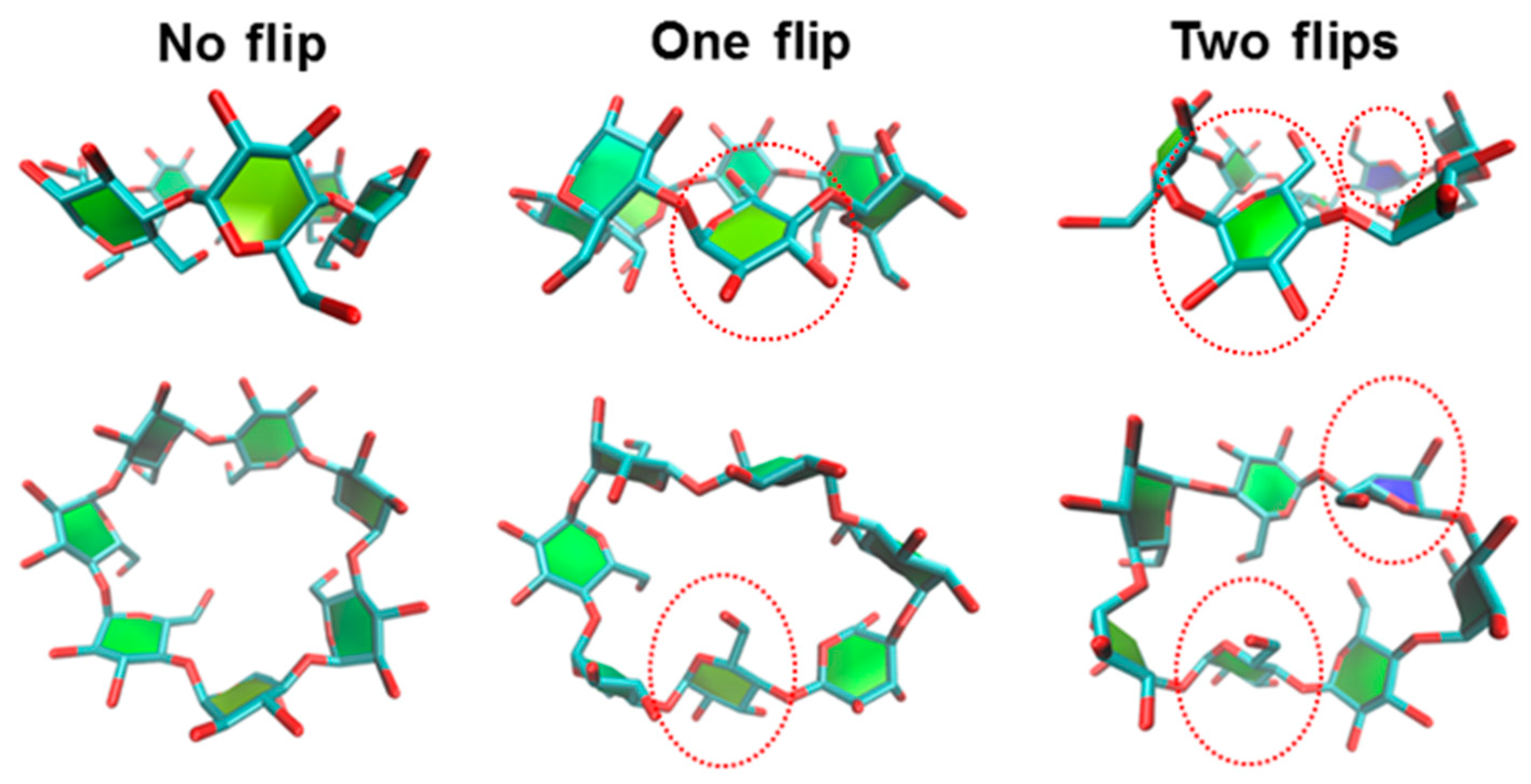
3.1.2. Flipping of the HPβCDs
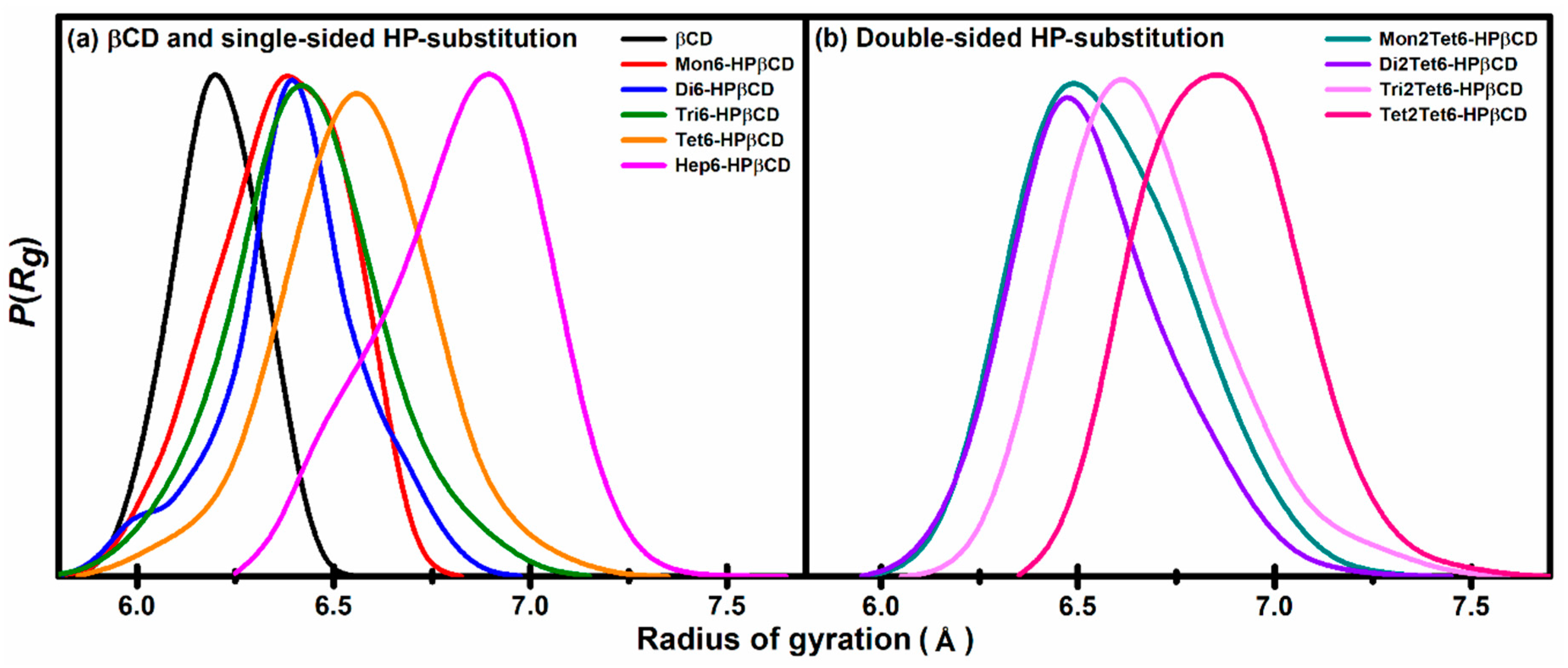
3.1.3. Radius of Gyration
3.1.4. Circularity
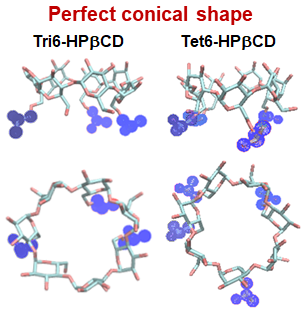
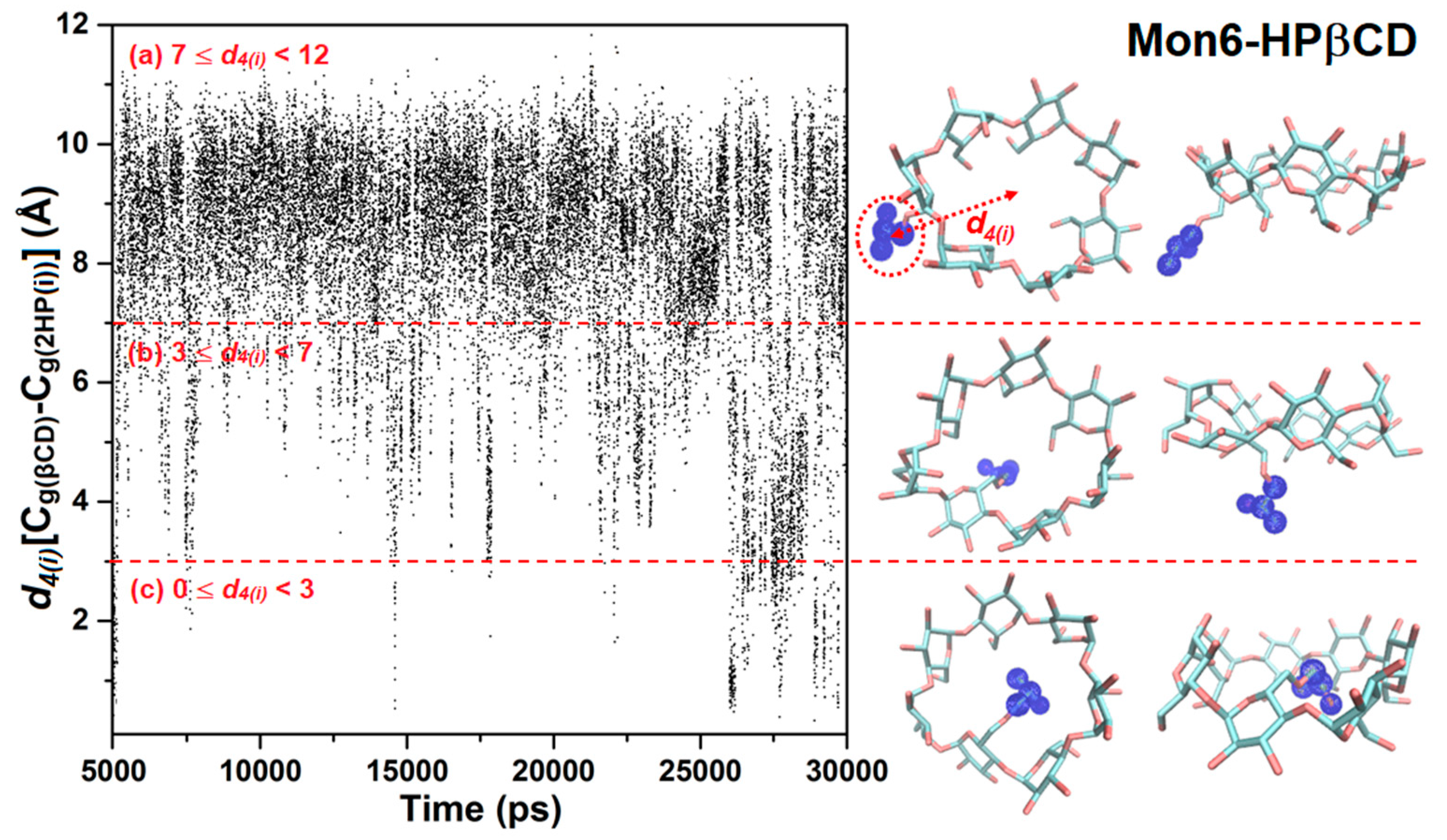
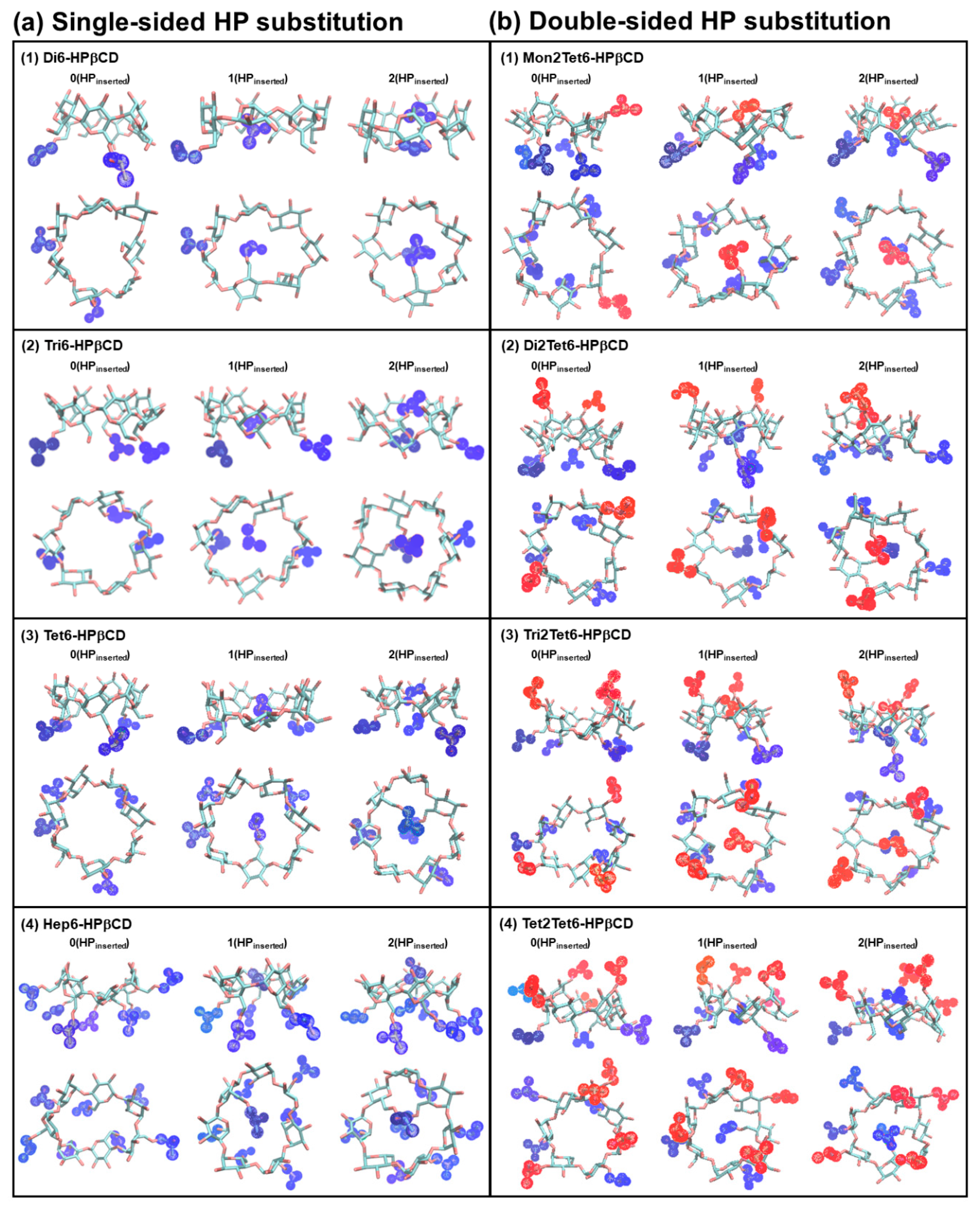
3.2. Cavity Self-Closure
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Uekama, K.; Hirayama, F.; Irie, T. Cyclodextrin Drug Carrier Systems. Chem. Rev. 1998, 98, 2045–2076. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, F.; Uekama, K. Cyclodextrin-based controlled drug release system. Adv. Drug Deliv. Rev. 1999, 36, 125–141. [Google Scholar] [CrossRef]
- Aachmann, F.L.; Otzen, D.E.; Larsen, K.L.; Wimmer, R. Structural background of cyclodextrin-protein interactions. Protein Eng. 2003, 16, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Brewster, M.E. Cyclodextrin-based pharmaceutics: Past, present and future. Nat. Rev. Drug Discov. 2004, 3, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Challa, R.; Ahuja, A.; Ali, J.; Khar, R.K. Cyclodextrins in drug delivery: An updated review. AAPS PharmSciTech 2005, 6, E329–E357. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Jarho, P.; Másson, M.; Järvinen, T. Cyclodextrins in drug delivery. Expert Opin. Drug Deliv. 2005, 2, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Brewster, M.E.; Loftsson, T. Cyclodextrins as pharmaceutical solubilizers. Adv. Drug Deliv. Rev. 2007, 59, 645–666. [Google Scholar] [CrossRef]
- Marques, H.M.C. A review on cyclodextrin encapsulation of essential oils and volatiles. Flavour Frag. J. 2010, 25, 313–326. [Google Scholar] [CrossRef]
- Crini, G. Review: A History of Cyclodextrins. Chem. Rev. 2014, 114, 10940–10975. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.; Scott, R.C. 2-Hydroxypropyl-β-cyclodextrin (HP-β-CD): A toxicology review. Food Chem. Toxicol. 2005, 43, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Schönbeck, C.; Westh, P.; Madsen, J.C.; Larsen, K.L.; Städe, L.W.; Holm, R. Hydroxypropyl-Substituted β-Cyclodextrins: Influence of Degree of Substitution on the Thermodynamics of Complexation with Tauroconjugated and Glycoconjugated Bile Salts. Langmuir 2010, 26, 17949–17957. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Reyes, M.L.; Roa-Morales, G.; Melgar-Fernández, R.; Reyes-Pérez, H.; Gómez-Oliván, L.; Gonzalez-Rivas, N.; Bautista-Renedo, J.; Balderas-Hernández, P. Chiral recognition of abacavir enantiomers by (2-hydroxy)propyl-β-cyclodextrin: UHPLC, NMR and DFT studies. J. Incl. Phenom. Macrocycl. Chem. 2015, 82, 373–382. [Google Scholar] [CrossRef]
- Yuan, C.; Liu, B.; Liu, H. Characterization of hydroxypropyl-β-cyclodextrins with different substitution patterns via FTIR, GC–MS, and TG–DTA. Carbohydr. Polym. 2015, 118, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Buvári-Barcza, Á.; Barcza, L. Influence of the guests, the type and degree of substitution on inclusion complex formation of substituted β-cyclodextrins. Talanta 1999, 49, 577–585. [Google Scholar] [CrossRef]
- Yuan, C.; Jin, Z.; Li, X. Evaluation of complex forming ability of hydroxypropyl-β-cyclodextrins. Food Chem. 2008, 106, 50–55. [Google Scholar] [CrossRef]
- Concha-Santos, S.; Pérez-Casas, S.; Brocos, P.; Piñeiro, Á. Testing the effect of the cavity size and the number of molecular substitutions in host–guest complexes formed by 2-hydroxypropyl-cyclodextrins and n-octyl-β-D-glucopyranoside. J. Chem. Thermodyn. 2013, 67, 112–119. [Google Scholar] [CrossRef]
- Yong, C.; Washington, C.; Smith, W. Structural Behaviour of 2-Hydroxypropyl-β-Cyclodextrin in Water: Molecular Dynamics Simulation Studies. Pharm. Res. 2008, 25, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Pitha, J.; Trinadha Rao, C.; Lindberg, B.; Seffers, P. Distribution of substituents in 2-hydroxypropyl ethers of cyclomaltoheptaose. Carbohydr. Res. 1990, 200, 429–435. [Google Scholar] [CrossRef]
- Trinadha Rao, C.; Pitha, J.; Lindberg, B.; Lindberg, J. Distribution of substituents in O-(2-hydroxypropyl) derivatives of cyclomalto-oligosaccharides (cyclodextrins): Influence of increasing substitution, of the base used in the preparation, and of macrocyclic size. Carbohydr. Res. 1992, 223, 99–107. [Google Scholar] [CrossRef]
- Buvári-Barcza, Á.; Bodnár-Gyarmathy, D.; Barcza, L. Hydroxypropyl-β-cyclodextrins: Correlation between the stability of their inclusion complexes with phenolphthalein and the degree of substitution. J. Incl. Phenom. Mol. Recognit. Chem. 1994, 18, 301–306. [Google Scholar] [CrossRef]
- Malanga, M.; Szemán, J.; Fenyvesi, É.; Puskás, I.; Csabai, K.; Gyémánt, G.; Fenyvesi, F.; Szente, L. “Back to the Future”: A New Look at Hydroxypropyl Beta-Cyclodextrins. J. Pharm. Sci. 2016, 105, 2921–2931. [Google Scholar] [CrossRef] [PubMed]
- Nutho, B.; Nunthaboot, N.; Wolschann, P.; Kungwan, N.; Rungrotmongkol, T. Metadynamics supports molecular dynamics simulation-based binding affinities of eucalyptol and beta-cyclodextrin inclusion complexes. RSC Adv. 2017, 7, 50899–50911. [Google Scholar] [CrossRef]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Hukushima, K.; Nemoto, K. Exchange Monte Carlo Method and Application to Spin Glass Simulations. J. Phys. Soc. Jpn. 1996, 65, 1604–1608. [Google Scholar] [CrossRef]
- Hansmann, U.H.E. Parallel tempering algorithm for conformational studies of biological molecules. Chem. Phys. Lett. 1997, 281, 140–150. [Google Scholar] [CrossRef]
- Nymeyer, H.; Gnanakaran, S.; García, A.E. Atomic Simulations of Protein Folding, Using the Replica Exchange Algorithm. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2004; Volume 383, pp. 119–149. [Google Scholar]
- Cheng, X.; Cui, G.; Hornak, V.; Simmerling, C. Modified Replica Exchange Simulation Methods for Local Structure Refinement. J. Phys. Chem. B 2005, 109, 8220–8230. [Google Scholar] [CrossRef] [PubMed]
- Snor, W.; Liedl, E.; Weiss-Greiler, P.; Karpfen, A.; Viernstein, H.; Wolschann, P. On the structure of anhydrous β-cyclodextrin. Chem. Phys. Lett. 2007, 441, 159–162. [Google Scholar] [CrossRef]
- Kicuntod, J.; Khuntawee, W.; Wolschann, P.; Pongsawasdi, P.; Chavasiri, W.; Kungwan, N.; Rungrotmongkol, T. Inclusion complexation of pinostrobin with various cyclodextrin derivatives. J. Mol. Graph. Model. 2016, 63, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14; University of California, San Francisco: San Francisco, CA, USA, 2014. [Google Scholar]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeiriño, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef]
- Tessier, M.B.; DeMarco, M.L.; Yongye, A.B.; Woods, R.J. Extension of the GLYCAM06 biomolecular force field to lipids, lipid bilayers and glycolipids. Mol. Simul. 2008, 34, 349–364. [Google Scholar] [CrossRef]
- Khuntawee, W.; Rungrotmongkol, T.; Wolschann, P.; Pongsawasdi, P.; Kungwan, N.; Okumura, H.; Hannongbua, S. Conformation study of ε-cyclodextrin: Replica exchange molecular dynamics simulations. Carbohydr. Polym. 2016, 141, 99–105. [Google Scholar] [CrossRef]
- Khuntawee, W.; Kunaseth, M.; Rungnim, C.; Intagorn, S.; Wolschann, P.; Kungwan, N.; Rungrotmongkol, T.; Hannongbua, S. Comparison of Implicit and Explicit Solvation Models for Iota-Cyclodextrin Conformation Analysis from Replica Exchange Molecular Dynamics. J. Chem. Inf. Model. 2017, 57, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Wongpituk, P.; Nutho, B.; Panman, W.; Kungwan, N.; Wolschann, P.; Rungrotmongkol, T.; Nunthaboot, N. Structural dynamics and binding free energy of neral-cyclodextrins inclusion complexes: Molecular dynamics simulation. Mol. Simul. 2017, 43, 1356–1363. [Google Scholar] [CrossRef]
- Mahalapbutr, P.; Nutho, B.; Wolschann, P.; Chavasiri, W.; Kungwan, N.; Rungrotmongkol, T. Molecular insights into inclusion complexes of mansonone E and H enantiomers with various β-cyclodextrins. J. Mol. Graph. Model. 2018, 79, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Mahalapbutr, P.; Thitinanthavet, K.; Kedkham, T.; Nguyen, H.; thi ha Theu, L.; Dokmaisrijan, S.; Huynh, L.; Kungwan, N.; Rungrotmongkol, T. A theoretical study on the molecular encapsulation of luteolin and pinocembrin with various derivatized beta-cyclodextrins. J. Mol. Struct. 2018, in press. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | Degree of Substitution | O2 Substitution | O6 Substitution |
|---|---|---|---|
| βCD | 0.00 | None | None |
| Single-sided HP-substitution | |||
| Mon6-HPβCD | 0.14 | None | 1 (At glucose unit 1) |
| Di6-HPβCD | 0.28 | None | 2 (At glucose units 1 and 3) |
| Tri6-HPβCD | 0.43 | None | 3 (At glucose units 1, 3, and 5) |
| Tet6-HPβCD | 0.57 | None | 4 (At glucose units 1, 3, 5, and 7) |
| Hep6-HPβCD | 1.00 | None | 7 (At all glucose units) |
| Double-sided HP-substitution | |||
| Mon2Tet6-HPβCD | 0.71 | 1 (At glucose unit 4) | 4 (At glucose units 1, 3, 5, and 7) |
| Di2Tet6-HPβCD | 0.85 | 2 (At glucose units 2 and 6) | 4 (At glucose units 1, 3, 5, and 7) |
| Tri2Tet6-HPβCD | 1.00 | 3 (At glucose units 2, 4, and 6) | 4 (At glucose units 1, 3, 5, and 7) |
| Tet2Tet6-HPβCD | 1.14 | 4 (At glucose units 2, 4, 6, and 7) | 4 (At glucose units 1, 3, 5, and 7) |
| Models | The Percentage of the Flip Angle (%) | ||
|---|---|---|---|
| No Flip | One Flip | Two Flips | |
| βCD | 58 | 35 | 7 |
| Single-sided HP-substitution | |||
| Mon6-HPβCD | 69 | 28 | 3 |
| Di6-HPβCD | 74 | 23 | 3 |
| Tri6-HPβCD | 75 | 24 | 1 |
| Tet6-HPβCD | 78 | 21 | 1 |
| Hep6-HPβCD | 73 | 25 | 2 |
| Double-sided HP-substitution | |||
| Mon2Tet6-HPβCD | 77 | 22 | 1 |
| Di2Tet6-HPβCD | 75 | 24 | 1 |
| Tri2Tet6-HPβCD | 70 | 28 | 2 |
| Tet2Tet6-HPβCD | 74 | 24 | 2 |
| Models | |
|---|---|
| βCD | 0.727 ± 0.087 |
| Single-sided HP-substitution | |
| Mon6-HPβCD | 0.746 ± 0.085 |
| Di6-HPβCD | 0.751 ± 0.086 |
| Tri6-HPβCD | 0.814 ± 0.075 |
| Tet6-HPβCD | 0.815 ± 0.076 |
| Hep6-HPβCD | 0.773 ± 0.088 |
| Double-sided HP-substitution | |
| Mon2Tet6-HPβCD | 0.803 ± 0.075 |
| Di2Tet6-HPβCD | 0.767 ± 0.076 |
| Tri2Tet6-HPβCD | 0.785 ± 0.074 |
| Tet2Tet6-HPβCD | 0.742 ± 0.080 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerdpol, K.; Kicuntod, J.; Wolschann, P.; Mori, S.; Rungnim, C.; Kunaseth, M.; Okumura, H.; Kungwan, N.; Rungrotmongkol, T. Cavity Closure of 2-Hydroxypropyl-β-Cyclodextrin: Replica Exchange Molecular Dynamics Simulations. Polymers 2019, 11, 145. https://doi.org/10.3390/polym11010145
Kerdpol K, Kicuntod J, Wolschann P, Mori S, Rungnim C, Kunaseth M, Okumura H, Kungwan N, Rungrotmongkol T. Cavity Closure of 2-Hydroxypropyl-β-Cyclodextrin: Replica Exchange Molecular Dynamics Simulations. Polymers. 2019; 11(1):145. https://doi.org/10.3390/polym11010145
Chicago/Turabian StyleKerdpol, Khanittha, Jintawee Kicuntod, Peter Wolschann, Seiji Mori, Chompoonut Rungnim, Manaschai Kunaseth, Hisashi Okumura, Nawee Kungwan, and Thanyada Rungrotmongkol. 2019. "Cavity Closure of 2-Hydroxypropyl-β-Cyclodextrin: Replica Exchange Molecular Dynamics Simulations" Polymers 11, no. 1: 145. https://doi.org/10.3390/polym11010145
APA StyleKerdpol, K., Kicuntod, J., Wolschann, P., Mori, S., Rungnim, C., Kunaseth, M., Okumura, H., Kungwan, N., & Rungrotmongkol, T. (2019). Cavity Closure of 2-Hydroxypropyl-β-Cyclodextrin: Replica Exchange Molecular Dynamics Simulations. Polymers, 11(1), 145. https://doi.org/10.3390/polym11010145