Viscoelastic Properties of Unentangled Multicyclic Polystyrenes



Abstract
1. Introduction
2. Experimental Section
2.1. Synthesis and Characterization of Cyclic Structures
2.2. Sample Treatment and Rheological Measurements
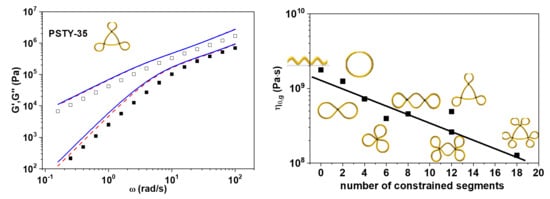
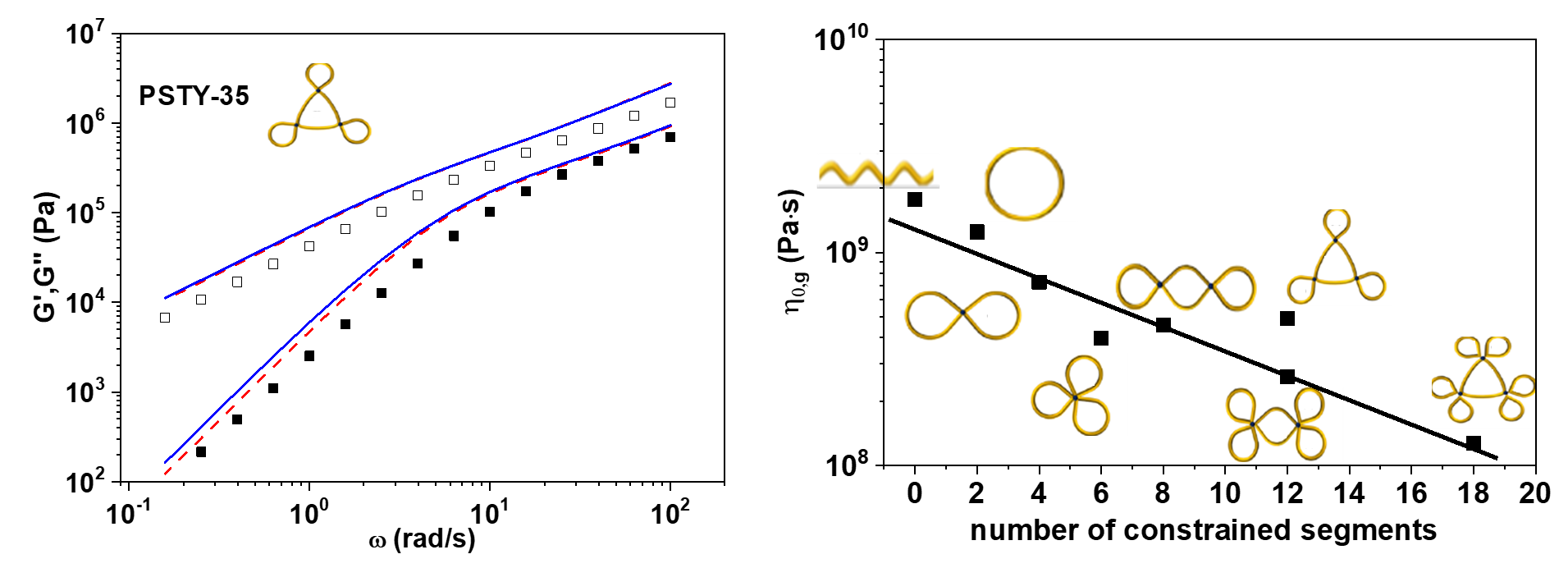
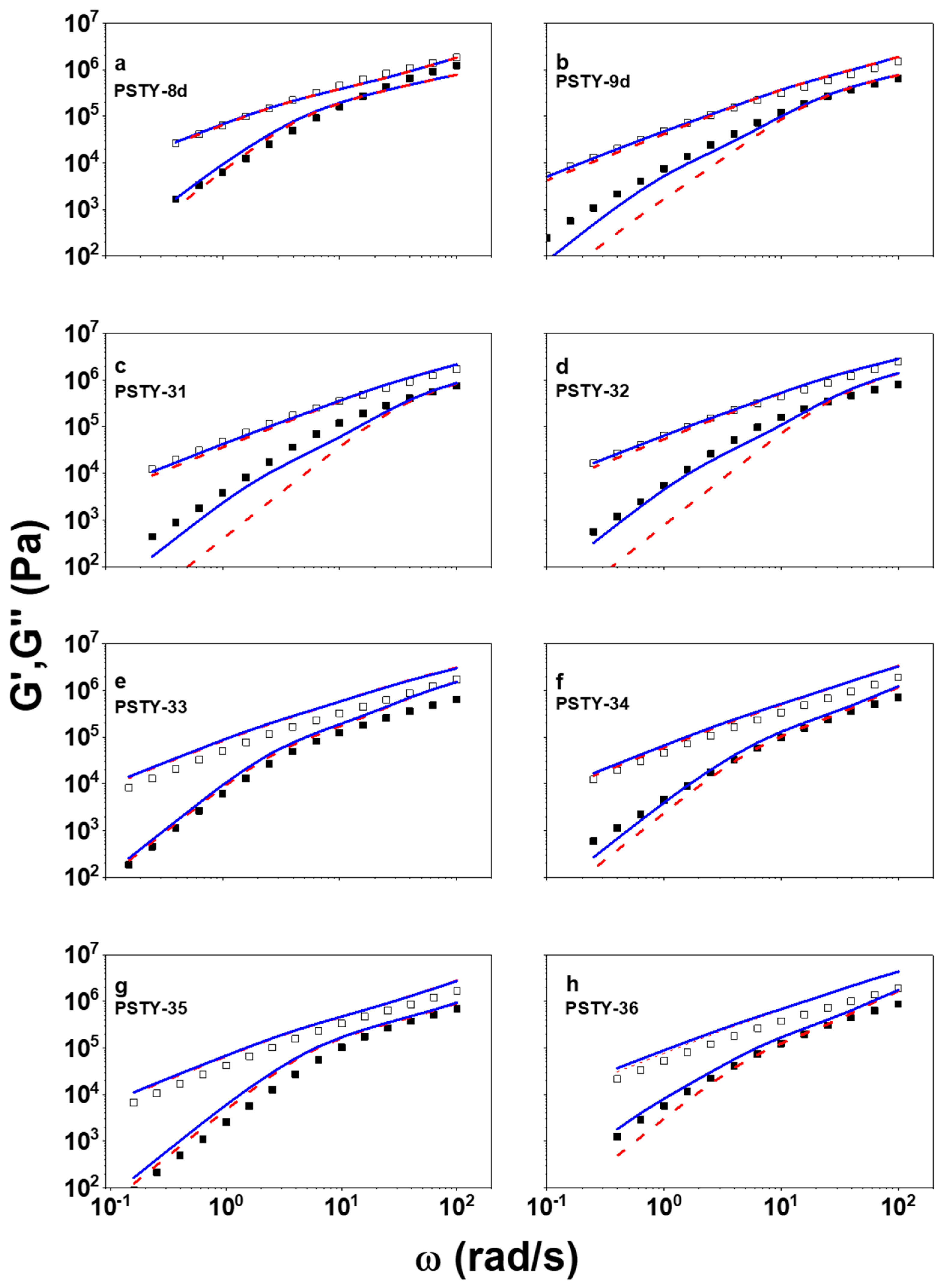
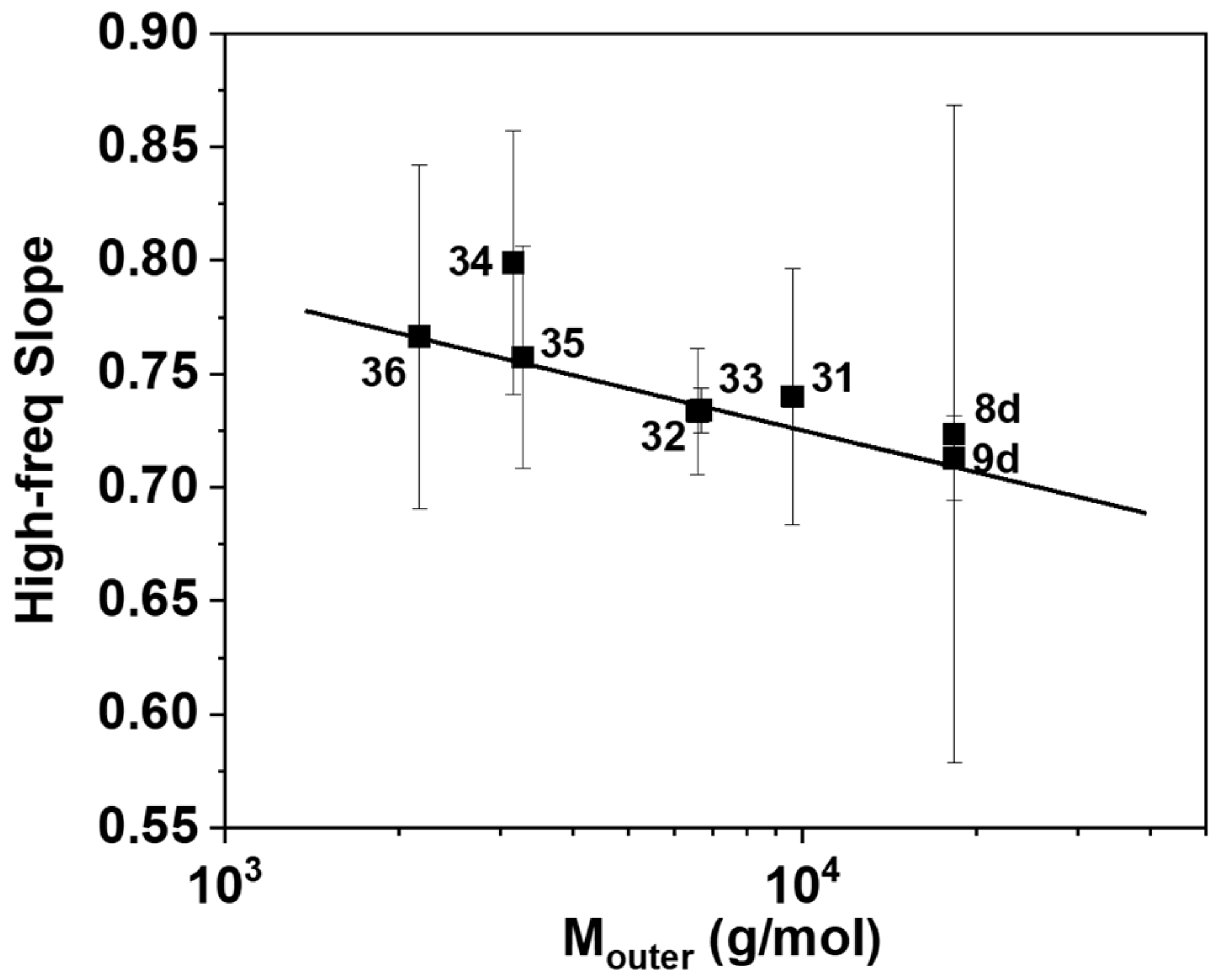
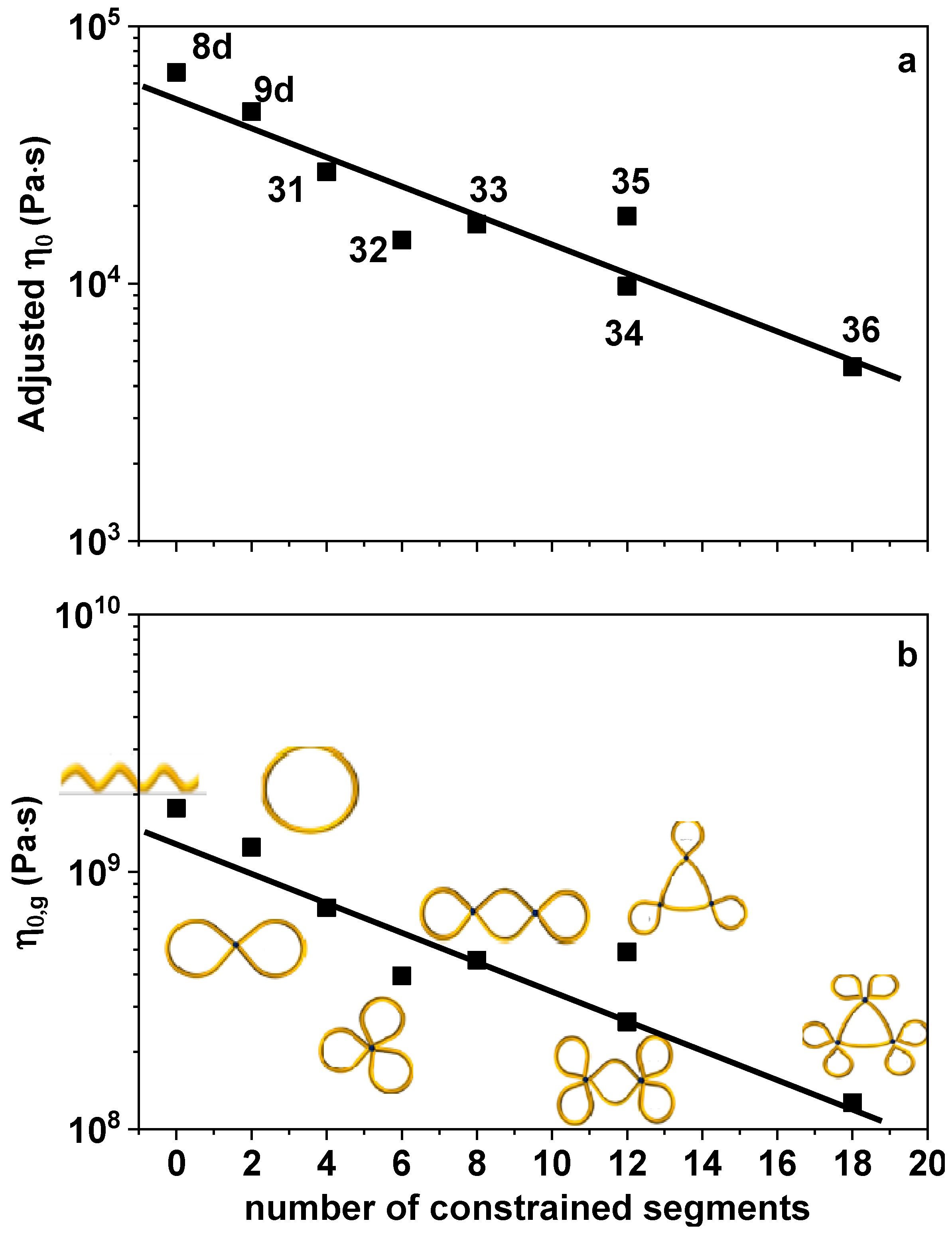
3. Results and Discussion
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Witz, G.; Rechendorff, K.; Adamcik, J.; Dietler, G. Conformation of ring polymers in 2D constrained environments. Phys. Rev. Lett. 2011, 106, 248301. [Google Scholar] [CrossRef] [PubMed]
- Halverson, J.D.; Smrek, J.; Kremer, K.; Grosberg, A.Y. From a melt of rings to chromosome territories: The role of topological constraints in genome folding. Rep. Prog. Phys. 2014, 77, 022601. [Google Scholar] [CrossRef] [PubMed]
- Dean, F.B.; Stasiak, A.; Koller, T.; Cozzarelli, N.R. Duplex DNA knots produced by Escherichia coli topoisomerase I. Structure and requirements for formation. J. Biol. Chem. 1985, 260, 4975–4983. [Google Scholar] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Daly, N.L.; Bond, T.; Waine, C. Plant cyclotides: A unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J. Mol. Biol. 1999, 294, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J. Seamless proteins tie up their loose ends. Science 2006, 311, 1563–1564. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Liu, Y. Cyclic polysiloxanes with linked cyclotetrasiloxane subunits. Angew. Chem. Int. Ed. 2017, 56, 8706–8710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Lackey, M.A.; Cui, J.; Tew, G.N. Gels based on cyclic polymers. J. Am. Chem. Soc. 2011, 133, 4140–4148. [Google Scholar] [CrossRef] [PubMed]
- Morgese, G.; Trachsel, L.; Romio, M.; Divandari, M.; Ramakrishna, S.N.; Benetti, E.M. Topological polymer chemistry enters surface science: Linear versus cyclic polymer brushes. Angew. Chem. Int. Ed. 2016, 55, 15583–15588. [Google Scholar] [CrossRef] [PubMed]
- Verbraeken, B.; Hoogenboom, R. Cyclic polymers: From scientific curiosity to advanced materials for gene delivery and surface modification. Angew. Chem. Int. Ed. 2017, 56, 7034–7036. [Google Scholar] [CrossRef] [PubMed]
- Gooßen, S.; Brás, A.R.; Krutyeva, M.; Sharp, M.; Falus, P.; Feoktystov, A.; Gasser, U.; Pyckhout-Hintzen, W.; Wischnewski, A.; Richter, D. Molecular scale dynamics of large ring polymers. Phys. Rev. Lett. 2014, 113, 168302. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Tang, Q.; Yang, J.; Zhang, K.; Zhao, J. Interfacial diffusion of a single cyclic polymer chain. Soft Matter 2016, 12, 9520–9526. [Google Scholar] [CrossRef] [PubMed]
- Halverson, J.D.; Lee, W.B.; Grest, G.S.; Grosberg, A.Y.; Kremer, K. Molecular dynamics simulation study of nonconcatenated ring polymers in a melt. I. Statics. J. Chem. Phys. 2011, 134, 204904. [Google Scholar] [CrossRef] [PubMed]
- Halverson, J.D.; Lee, W.B.; Grest, G.S.; Grosberg, A.Y.; Kremer, K. Molecular dynamics simulation study of nonconcatenated ring polymers in a melt. II. Dynamics. J. Chem. Phys. 2011, 134, 204905. [Google Scholar] [CrossRef] [PubMed]
- Zardalidis, G.; Mars, J.; Allgaier, J.; Mezger, M.; Richter, D.; Floudas, G. Influence of chain topology on polymer crystallization: Poly(ethylene oxide) (PEO) rings vs. linear chains. Soft Matter 2016, 12, 8124–8134. [Google Scholar] [CrossRef] [PubMed]
- Kapnistos, M.; Lang, M.; Vlassopoulos, D.; Pyckhout-Hintzen, W.; Richter, D.; Cho, D.; Chang, T.; Rubinstein, M. Unexpected power-law stress relaxation of entangled ring polymers. Nat. Mater. 2008, 7, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Halverson, J.D.; Grest, G.S.; Grosberg, A.Y.; Kremer, K. Rheology of ring polymer melts: From linear contaminants to ring-linear blends. Phys. Rev. Lett. 2012, 108, 038301. [Google Scholar] [CrossRef] [PubMed]
- Gooßen, S.; Krutyeva, M.; Sharp, M.; Feoktystov, A.; Allgaier, J.; Pyckhout-Hintzen, W.; Wischnewski, A.; Richter, D. Sensing polymer chain dynamics through ring topology: A neutron spin echo study. Phys. Rev. Lett. 2015, 115, 148302. [Google Scholar] [CrossRef] [PubMed]
- Kruteva, M.; Allgaier, J.; Richter, D. Direct observation of two distinct diffusive modes for polymer rings in linear polymer matrices by pulsed field gradient (PFG) NMR. Macromolecules 2017, 50, 9482–9493. [Google Scholar] [CrossRef]
- Papadopoulos, G.D.; Tsalikis, D.G.; Mavrantzas, V.G. Microscopic dynamics and topology of polymer rings immersed in a host matrix of longer linear polymers: Results from a detailed molecular dynamics simulation study and comparison with experimental data. Polymers 2016, 8, 283. [Google Scholar] [CrossRef]
- Vlassopoulos, D.; Pasquino, R.; Snijkers, F. Progress in the rheology of cyclic polymers. In Topological Polymer Chemistry; Tezuka, Y., Ed.; World Scientific Publishing: Singapore, 2014; pp. 291–316. [Google Scholar]
- Vlassopoulos, D. Macromolecular topology and rheology: Beyond the tube model. Rheol. Acta 2016, 55, 613–632. [Google Scholar] [CrossRef]
- McKenna, G.B.; Plazek, D.J. The viscosity of blends of linear and cyclic molecules of similar molecular mass. Polym. Commun. 1986, 27, 304–306. [Google Scholar]
- Roovers, J. Viscoelastic properties of polybutadiene rings. Macromolecules 1988, 21, 1517–1521. [Google Scholar] [CrossRef]
- Endo, K. Synthesis and properties of cyclic polymers. In New Frontiers in Polymer Synthesis; Kobayashi, S., Ed.; Springer: Berlin, Germany, 2008; pp. 121–183. [Google Scholar]
- Roovers, J. The melt properties of ring polystyrenes. Macromolecules 1985, 18, 1359–1361. [Google Scholar] [CrossRef]
- Cates, M.; Deutsch, J. Conjectures on the statistics of ring polymers. J. Phys. 1986, 47, 2121–2128. [Google Scholar] [CrossRef]
- Gooßen, S.; Brás, A.R.; Pyckhout-Hintzen, W.; Wischnewski, A.; Ricther, D.; Rubinstein, M.; Roovers, R.; Lutz, P.; Jeong, Y.; Chang, T.; et al. Influence of Solvent Quality on Ring Polymer Dimensions. Macromolecules 2015, 48, 1598–1605. [Google Scholar]
- Rubinstein, M. Dynamics of ring polymers in the presence of fixed obstacles. Phys. Rev. Lett. 1986, 57, 3023–3026. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Inoue, T.; Matsumiya, Y. Transient conformational change of bead-spring ring chain during creep process. Macromolecules 2006, 39, 5419–5426. [Google Scholar] [CrossRef]
- Pasquino, R.; Vasilakopoulos, T.C.; Jeong, Y.C.; Lee, H.; Rogers, S.; Sakellariou, G.; Allgaier, J.; Takano, A.; Brás, A.R.; Chang, T.; et al. Viscosity of ring polymer melts. ACS Macro Lett. 2013, 2, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Matsubara, K.; Ohta, Y.; Nakano, T.; Kawaguchi, D.; Takahashi, Y.; Takano, A.; Matsushita, Y. Melt rheology of ring polystyrenes with ultrahigh purity. Macromolecules 2015, 48, 3140–3147. [Google Scholar] [CrossRef]
- Bras, A.R.; Goossen, S.; Krutyeva, M.; Radulescu, A.; Farago, B.; Allgaier, J.; Pyckhout-Hintzen, W.; Wischnewski, A.; Richter, D. Compact structure and non-gaussian dynamics of ring polymer melts. Soft Matter 2014, 10, 3649–3655. [Google Scholar] [CrossRef] [PubMed]
- Tsolou, G.; Stratikis, N.; Baig, C.; Stephanou, P.S.; Mavrantzas, V.G. Melt structure and dynamics of unentangled polyethylene rings rouse theory, atomistic molecular dynamics simulation, and comparison with the linear analogues. Macromolecules 2010, 43, 10692–10713. [Google Scholar] [CrossRef]
- Yan, Z.C.; Costanzo, S.; Jeong, Y.; Chang, T.; Vlassopoulos, D. Linear and nonlinear shear rheology of a marginally entangled ring polymer. Macromolecules 2016, 49, 1444–1453. [Google Scholar] [CrossRef]
- Grosberg, A.Y.; Nechaev, S.K.; Shakhnovich, E.I. The role of topological constraints in the kinetics of collapsed of macromolecules. J. Phys. 1988, 49, 2095–2100. [Google Scholar] [CrossRef]
- Tsalikis, D.G.; Mavrantzas, V.G. Threading of ring poly(ethylene oxide) molecules by linear chains in the melt. ACS Macro Lett. 2014, 3, 763–766. [Google Scholar] [CrossRef]
- Lee, H.C.; Lee, H.; Lee, W.; Chang, T.; Roovers, J. Fractionation of cyclic polystyrene from linear precursor by HPLC at the chromatographic critical condition. Macromolecules 2000, 33, 8119–8121. [Google Scholar] [CrossRef]
- Ge, T.; Panyukoy, S.; Rubinstein, M. Self-similar conformations and dynamics in entangled melts and solutions of nonconcatenated ring polymers. Macromolecules 2016, 49, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.D.; Lu, D.R.; Jia, Z.F.; Monteiro, M.J. Glass transition temperature of cyclic stars. ACS Macro Lett. 2014, 3, 1254–1257. [Google Scholar] [CrossRef]
- Hossain, M.D.; Reid, J.C.; Lu, D.; Jia, Z.; Searles, D.J.; Monteiro, M.J. Influence of constraints within a cyclic polymer on solution properties. Biomacromolecules 2018, 19, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Pipertzis, A.; Hossain, M.D.; Monteiro, M.J.; Floudas, G. Segmental dynamics in multicyclic polystyrenes. Macromolecules 2018, 51, 1488–1497. [Google Scholar] [CrossRef]
- Kapnistos, M.; Koutalas, G.; Hadjichristidis, N.; Roovers, J.; Lohse, D.J.; Vlassopoulos, D. Linear rheology of comb polymers with star-like backbones: Melts and solutions. Rheol. Acta 2006, 46, 273–286. [Google Scholar] [CrossRef]
- Read, D.J.; Auhl, D.; Das, C.; Den Doelder, J.; Kapnistos, M.; Vittorias, I.; McLeish, T.C. Linking models of polymerization and dynamics to predict branched polymer structure and flow. Science 2011, 333, 1871–1874. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Ohta, Y.; Nakamura, M.; Takano, A.; Takahashi, Y.; Matsushita, Y. Precise synthesis and characterization of tadpole-shaped polystyrenes with high purity. Macromolecules 2013, 46, 1075–1081. [Google Scholar] [CrossRef]
- Doi, Y.; Takano, A.; Takahashi, Y.; Matsushita, Y. Melt rheology of tadpole-shaped polystyrenes. Macromolecules 2015, 48, 8667–8674. [Google Scholar] [CrossRef]
- Doi, Y.; Takano, A.; Matsushita, Y. Synthesis and characterization of dumbbell-shaped polystyrene. Polymer 2016, 106, 8–13. [Google Scholar] [CrossRef]
- Doi, Y.; Iwasa, Y.; Watanabe, K.; Nakamura, M.; Takano, A.; Takahashi, Y.; Matsushita, Y. Synthesis and characterization of comb-shaped ring polystyrenes. Macromolecules 2016, 49, 3109–3115. [Google Scholar] [CrossRef]
- Hossain, M.D.; Jia, Z.F.; Monteiro, M.J. Complex polymer topologies built from tailored multifunctional cyclic polymers. Macromolecules 2014, 47, 4955–4970. [Google Scholar] [CrossRef]
- Singla, S.; Zhao, T.; Beckham, H.W. Purification of cyclic polymers prepared from linear precursors by inclusion complexation of linear byproducts with cyclodextrins. Macromolecules 2003, 36, 6945–6948. [Google Scholar] [CrossRef]
- Pasch, H.; Deffieux, A.; Henze, I.; Schappacher, M.; Rique-Lurbet, L. Analysis of macrocyclic polystyrenes. 1. Liquid chromatographic investigations. Macromolecules 1996, 29, 8776–8782. [Google Scholar] [CrossRef]
- Monteiro, M.J. Fitting molecular weight distributions using a log-normal distribution model. Eur. Polym. J. 2015, 65, 197–201. [Google Scholar] [CrossRef]
- Jia, Z.; Monteiro, M.J. Cyclic polymers: Methods and strategies. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 2085–2097. [Google Scholar] [CrossRef]
- Ruymbeke, E.V.; Muliawan, E.B.; Vlassopoulos, D.; Gao, H.; Matyjaszewski, K. Melt rheology of star polymers with large number of small arms, prepared by crosslinking poly(n-butyl acrylate) macromonomers via ATRP. Eur. Polym. J. 2011, 47, 746–751. [Google Scholar] [CrossRef]
- Dorgan, J.R.; Knauss, D.M.; Al-Muallum, H.A.; Huang, T.; Vlassopoulos, D. Melt Rheology of Dendritically Branched Polystyrenes. Macromolecules 2003, 36, 380–388. [Google Scholar] [CrossRef]
- Inoue, T.; Matsui, H.; Osaki, K. Molecular origin of viscoelasticity and chain orientation of glassy polymers. Rheol. Acta 1997, 36, 239–244. [Google Scholar] [CrossRef]
- Inoue, T.; Okamoto, H.; Osaki, K. Birefringence of amorphous polymers. 1. Dynamic measurement on polystyrene. Macromolecules 1991, 24, 5670–5675. [Google Scholar] [CrossRef]
- Ferry, J.D. Viscoelastic Properties of Polymers, 3rd ed.; Wiley: New York, NY, USA, 1980. [Google Scholar]
- Colby, R.H.; Fetters, L.J.; Graessley, W.W. Melt viscosity molecular-weight relationship for linear-polymers. Macromolecules 1987, 20, 2226–2237. [Google Scholar] [CrossRef]
- Santangelo, P.G.; Roland, C.M. Interrupted shear flow of unentangled polymer melts. J. Rheol. 2001, 45, 583–594. [Google Scholar] [CrossRef]
- Bird, R.B.; Armstrong, R.C.; Hassager, O. Dynamics of Polymeric Liquids: Fluid Mechanics, 2nd ed.; Wiley: New York, NY, USA, 1987; Volume I. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PSTY | Structure | τseg × 104 [s] | Mn [g/mol] | Mouter [g/mol] | τouter [s] | Minner [g/mol] | τinner [s] | ϕlinearb | Ml,outer | τl,outer [s] | Ml,inner [g/mol] | τl,inner [s] | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8d |  | 3.74 | 18,300 | 18,300 a | 1 a | 0.25 a | - | - | - | 0.09 | 36,600 a | 0.98 | - | - |
| 9d |  | 4.32 | 18,300 | 18,300 | 1 | 0.068 | - | - | - | ~0.09 | 36,600 | 1.1 | - | - |
| 31 |  | 6.80 | 19,200 | 9600 | 1 | 0.030 | - | - | - | ~0.09 | 19,200 | 0.48 | - | - |
| 32 |  | 16.7 | 19,700 | 6570 | 1 | 0.035 | - | - | - | ~0.09 | 13,200 | 0.56 | - | - |
| 33 |  | 11.5 | 19,100 | 6670 | 0.72 | 0.025 | 5750 | 0.28 | 0.39 | ~0.09 | 13,300 | 0.39 | 11,500 | 0.29 |
| 34 |  | 19.0 | 18,900 | 3150 | 0.66 | 0.0091 | 6300 | 0.34 | 0.17 | ~0.09 | 6300 | 0.14 | 12,600 | 0.58 |
| 35 |  | 8.68 | 19,680 | 3280 | 0.49 | 0.0044 | 9840 | 0.51 | 0.21 | ~0.09 | 6600 | 0.072 | 19,700 | 0.65 |
| 36 |  | 42.6 | 19,440 | 2170 | 0.65 | 0.0097 | 6400 | 0.35 | 0.19 | ~0.09 | 4300 | 0.15 | 12,800 | 1.35 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Z.-C.; Hossain, M.D.; Monteiro, M.J.; Vlassopoulos, D. Viscoelastic Properties of Unentangled Multicyclic Polystyrenes. Polymers 2018, 10, 973. https://doi.org/10.3390/polym10090973
Yan Z-C, Hossain MD, Monteiro MJ, Vlassopoulos D. Viscoelastic Properties of Unentangled Multicyclic Polystyrenes. Polymers. 2018; 10(9):973. https://doi.org/10.3390/polym10090973
Chicago/Turabian StyleYan, Zhi-Chao, Md. D. Hossain, Michael J. Monteiro, and Dimitris Vlassopoulos. 2018. "Viscoelastic Properties of Unentangled Multicyclic Polystyrenes" Polymers 10, no. 9: 973. https://doi.org/10.3390/polym10090973
APA StyleYan, Z.-C., Hossain, M. D., Monteiro, M. J., & Vlassopoulos, D. (2018). Viscoelastic Properties of Unentangled Multicyclic Polystyrenes. Polymers, 10(9), 973. https://doi.org/10.3390/polym10090973