Rubber-Filler Interactions in Polyisoprene Filled with In Situ Generated Silica: A Solid State NMR Study

 , , ,
, , ,  and
and 
Abstract
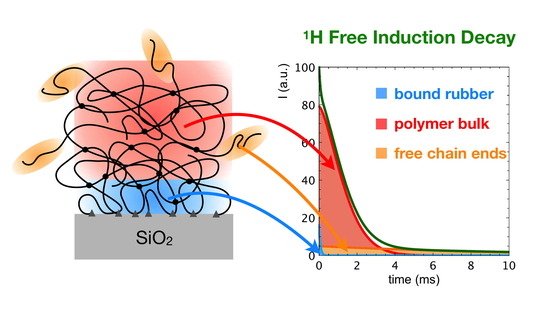
:
1. Introduction
2. Materials and Methods
2.1. Samples Preparation
2.2. Solid State NMR
3. Results
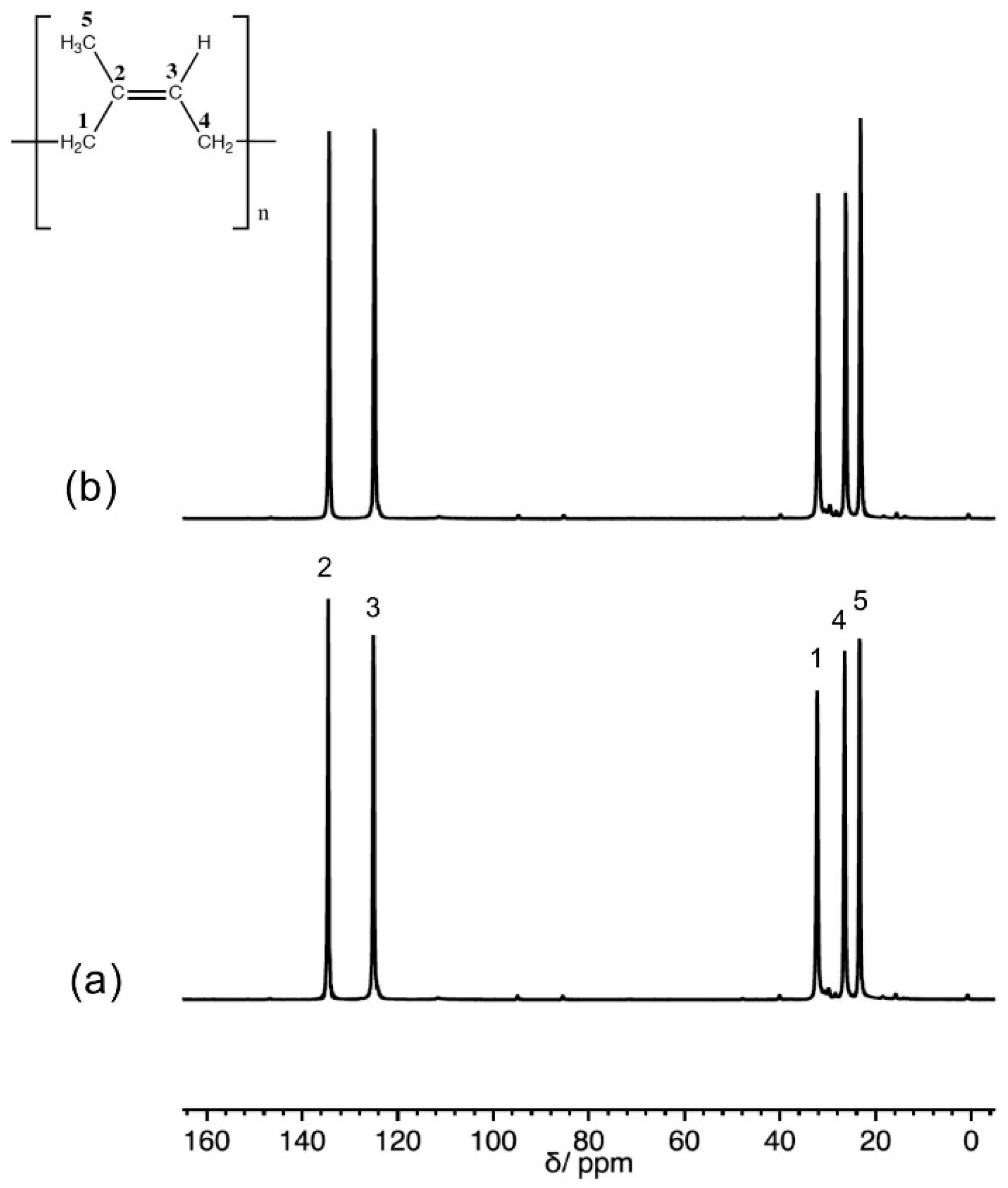
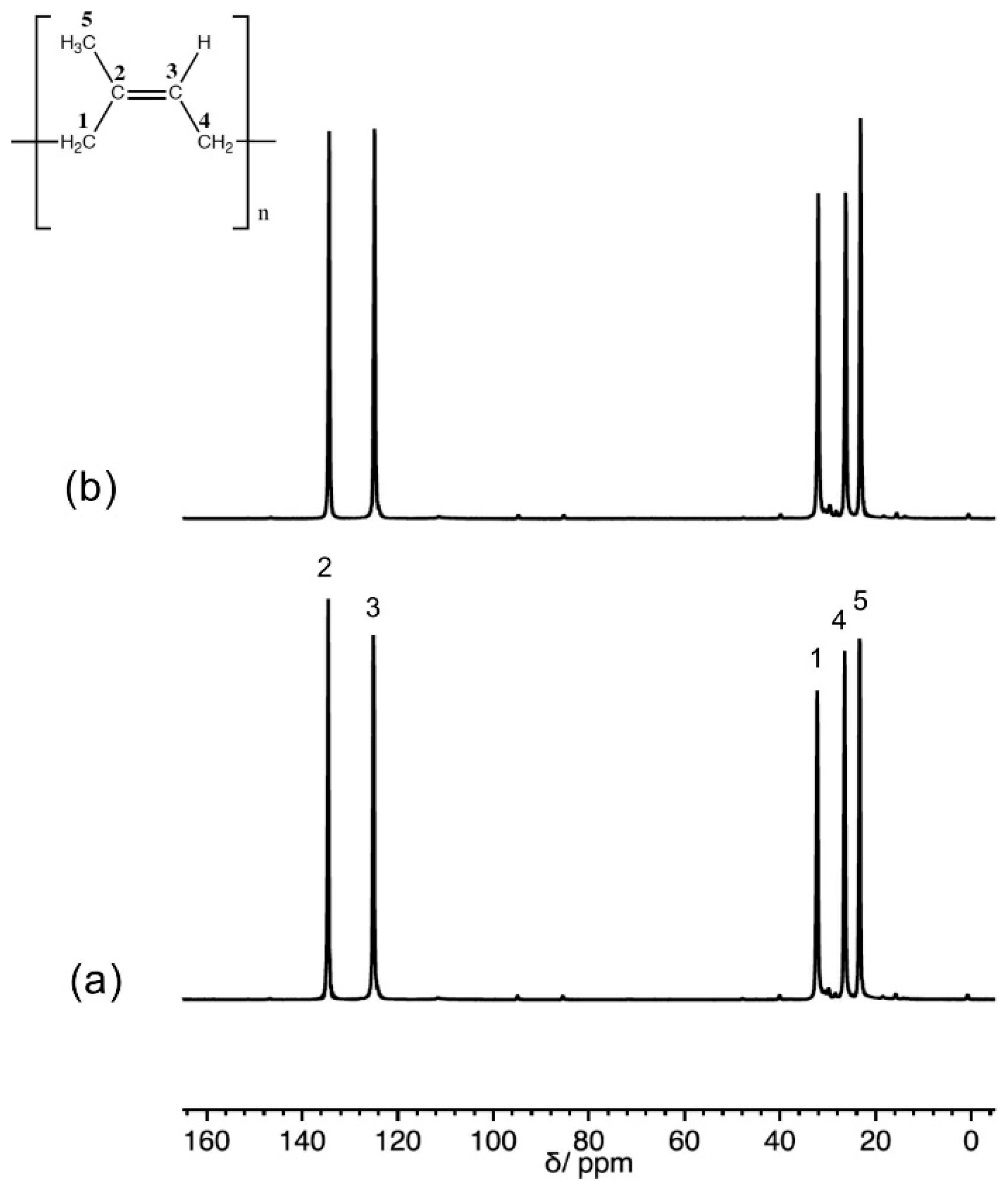
3.1. 13C Solid State NMR Spectra
3.2. 1H Spin-Lattice Relaxation Times T1 and T1ρ
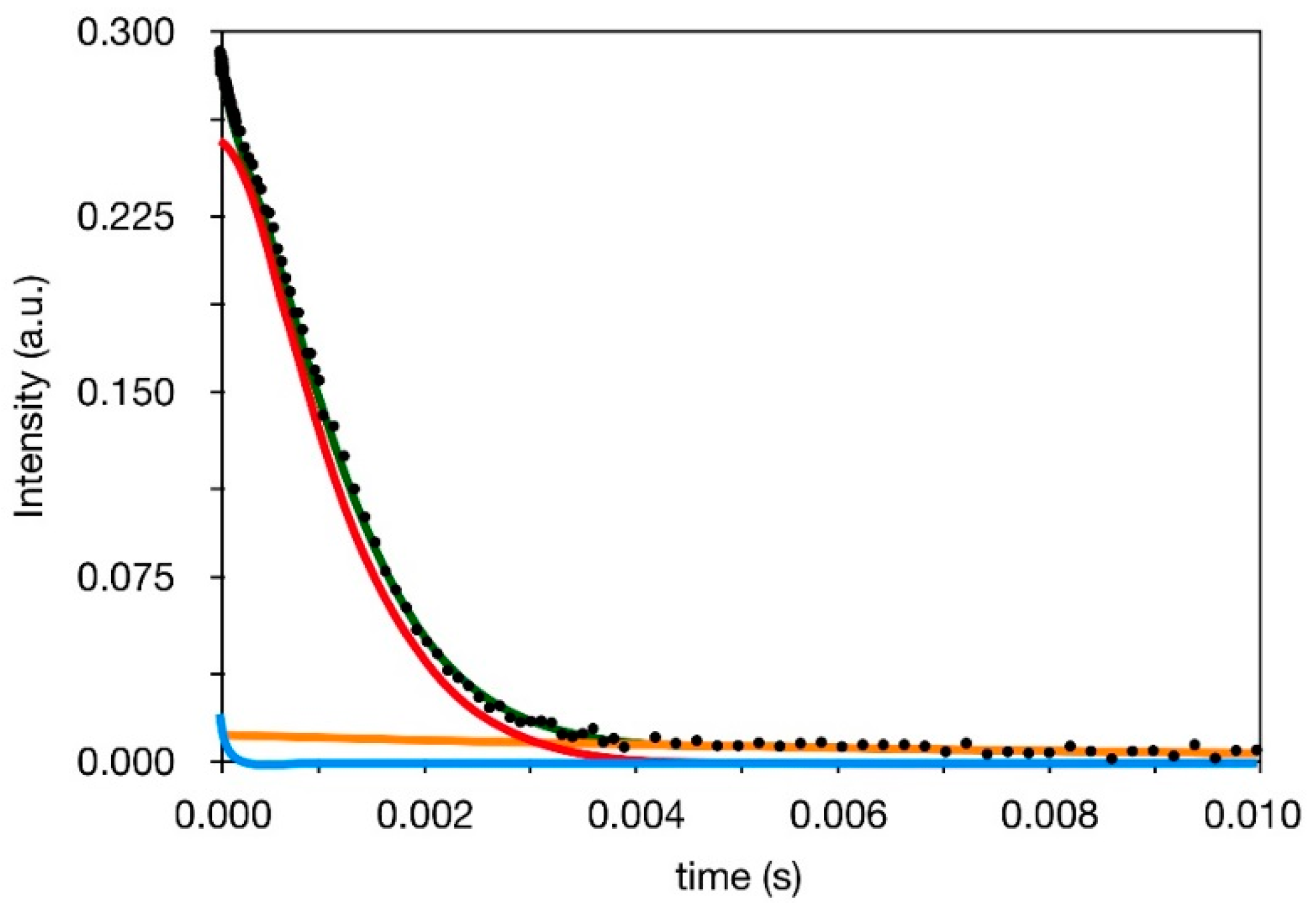
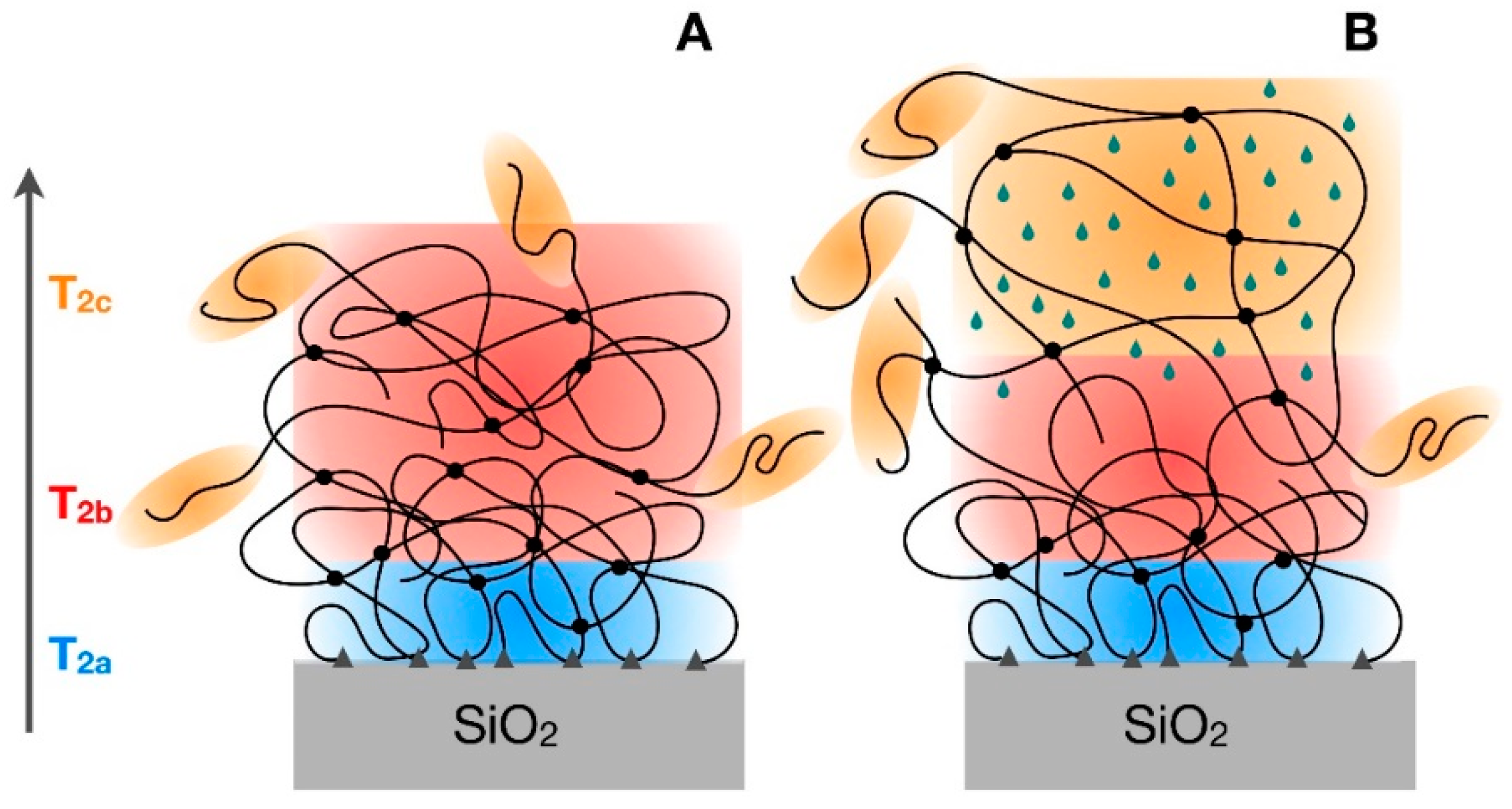
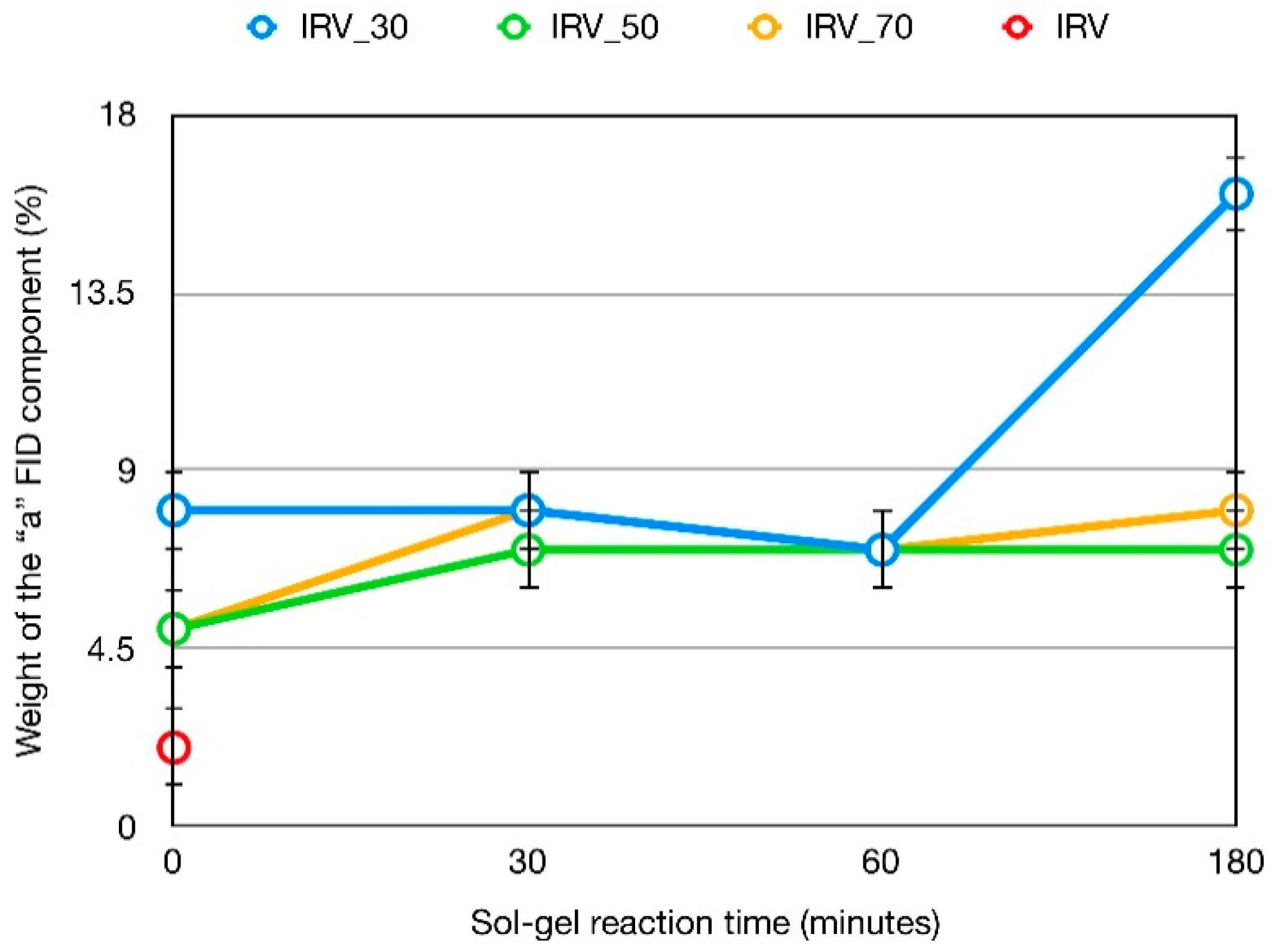
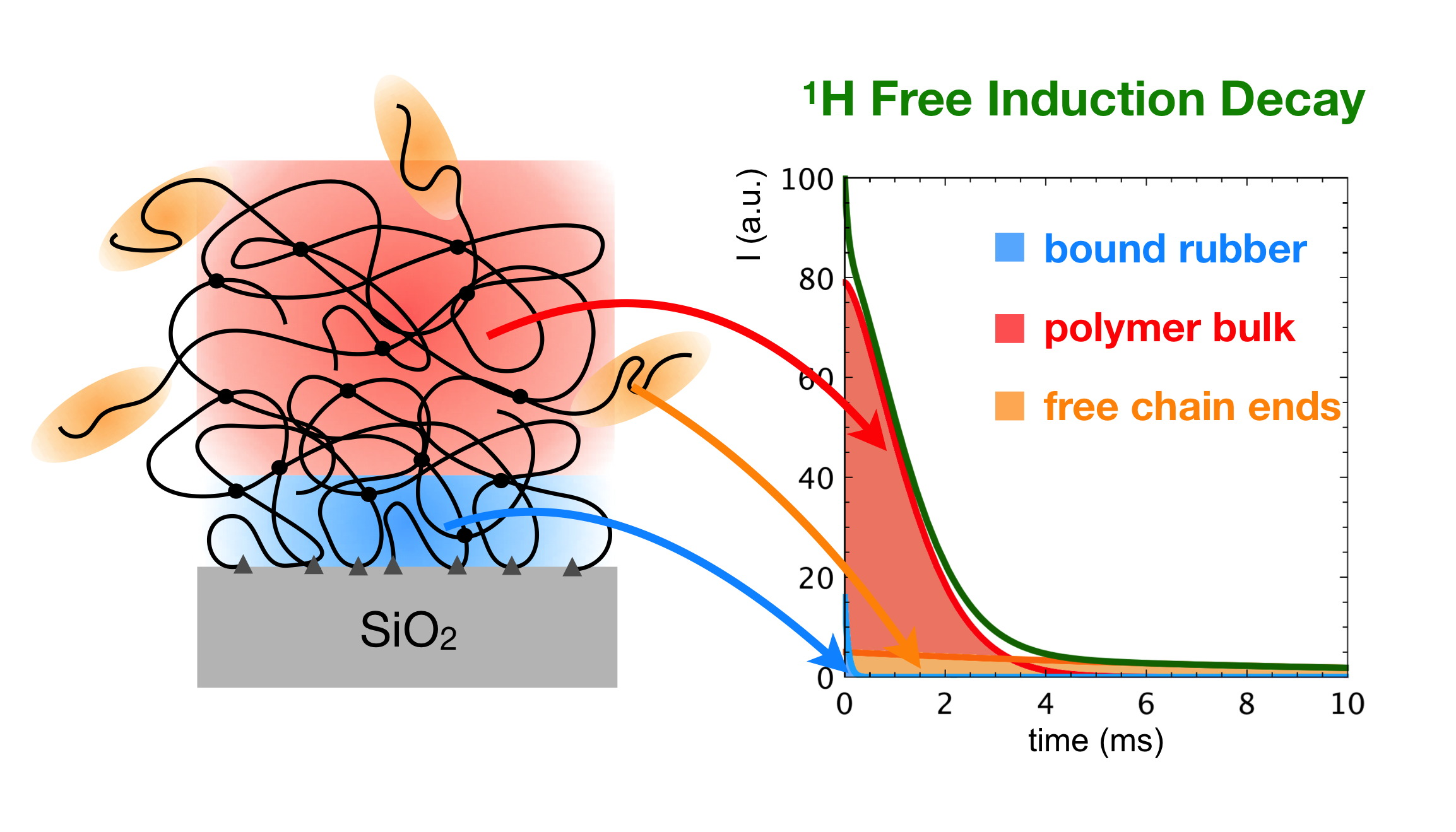
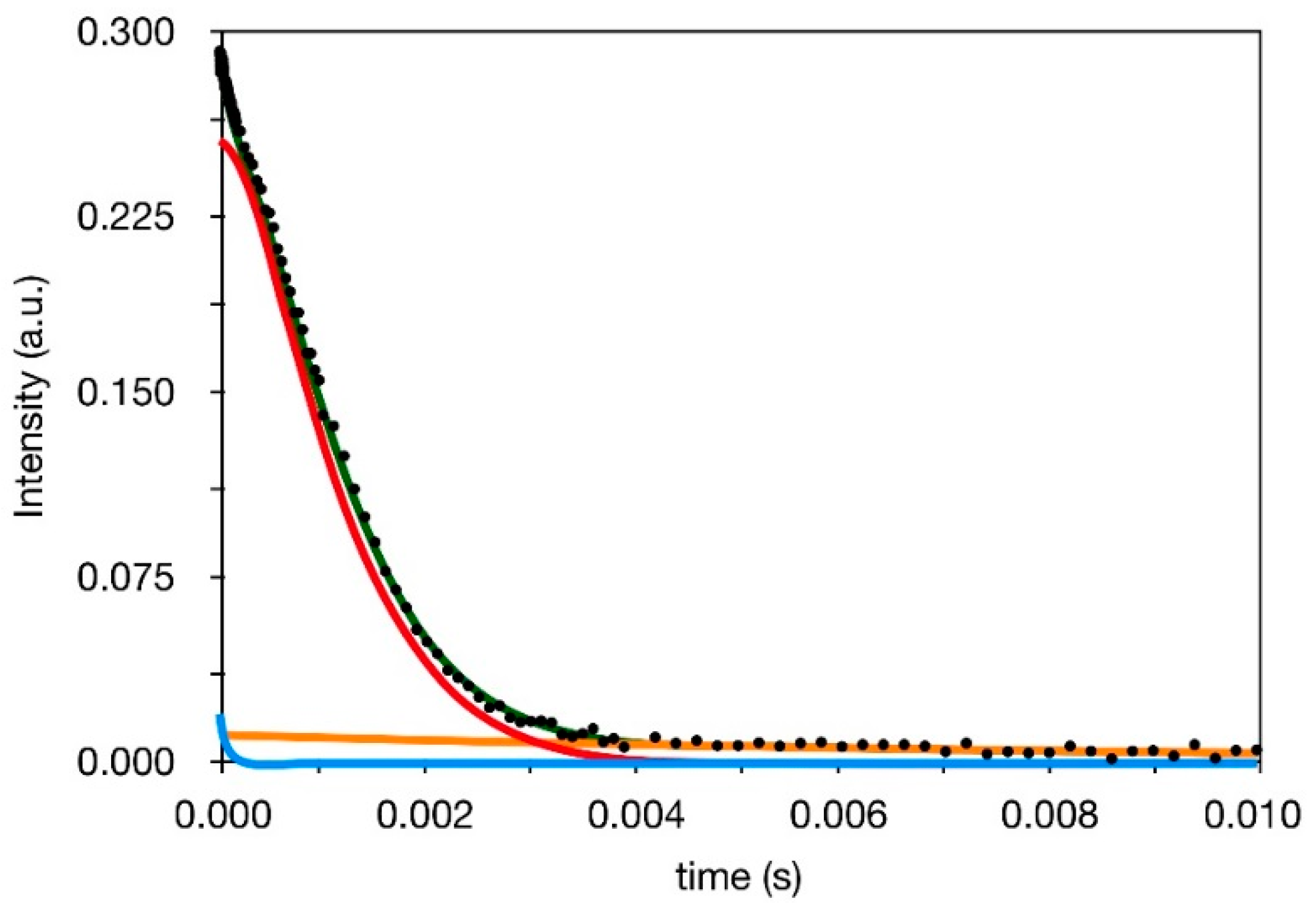
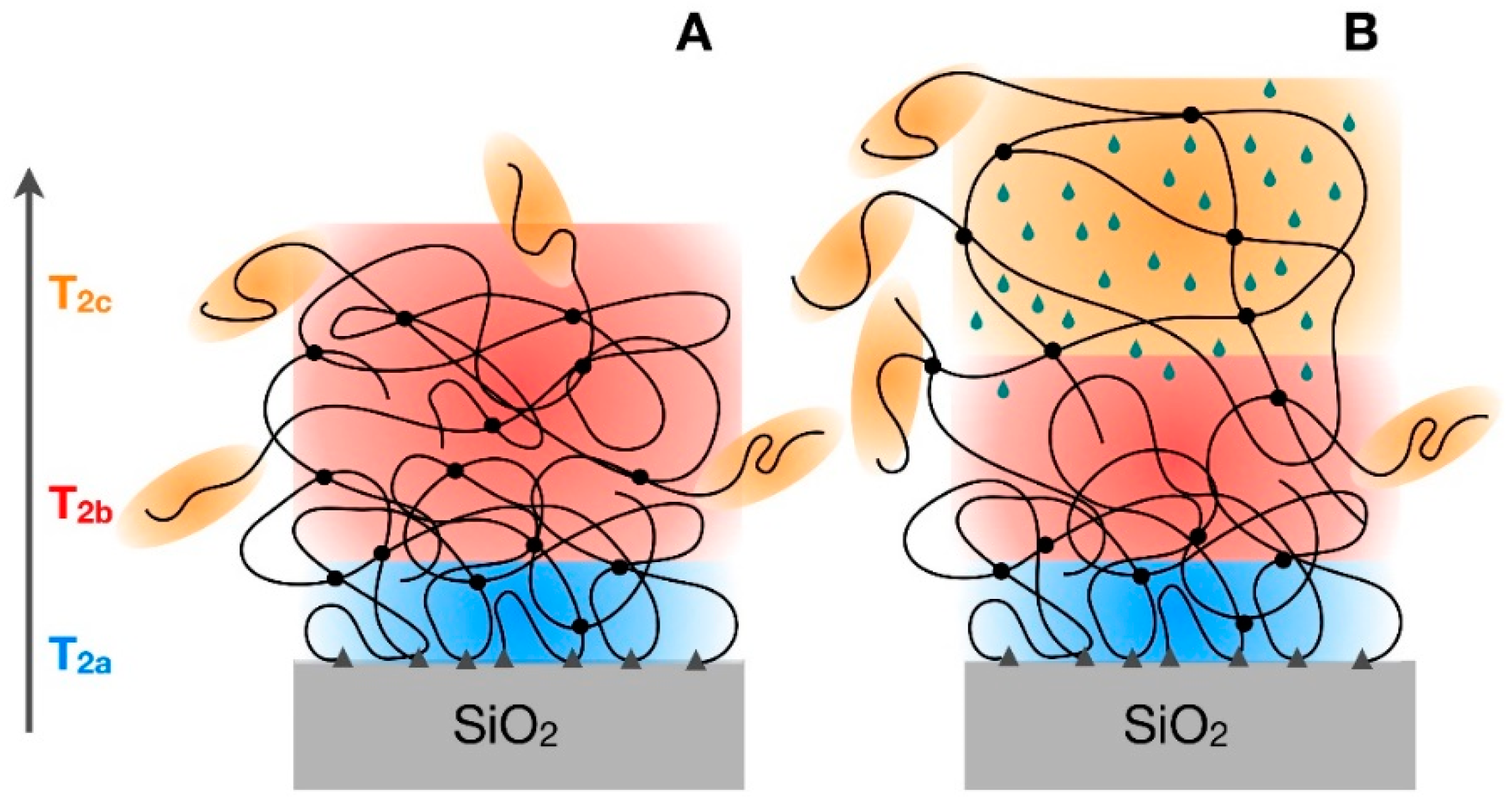
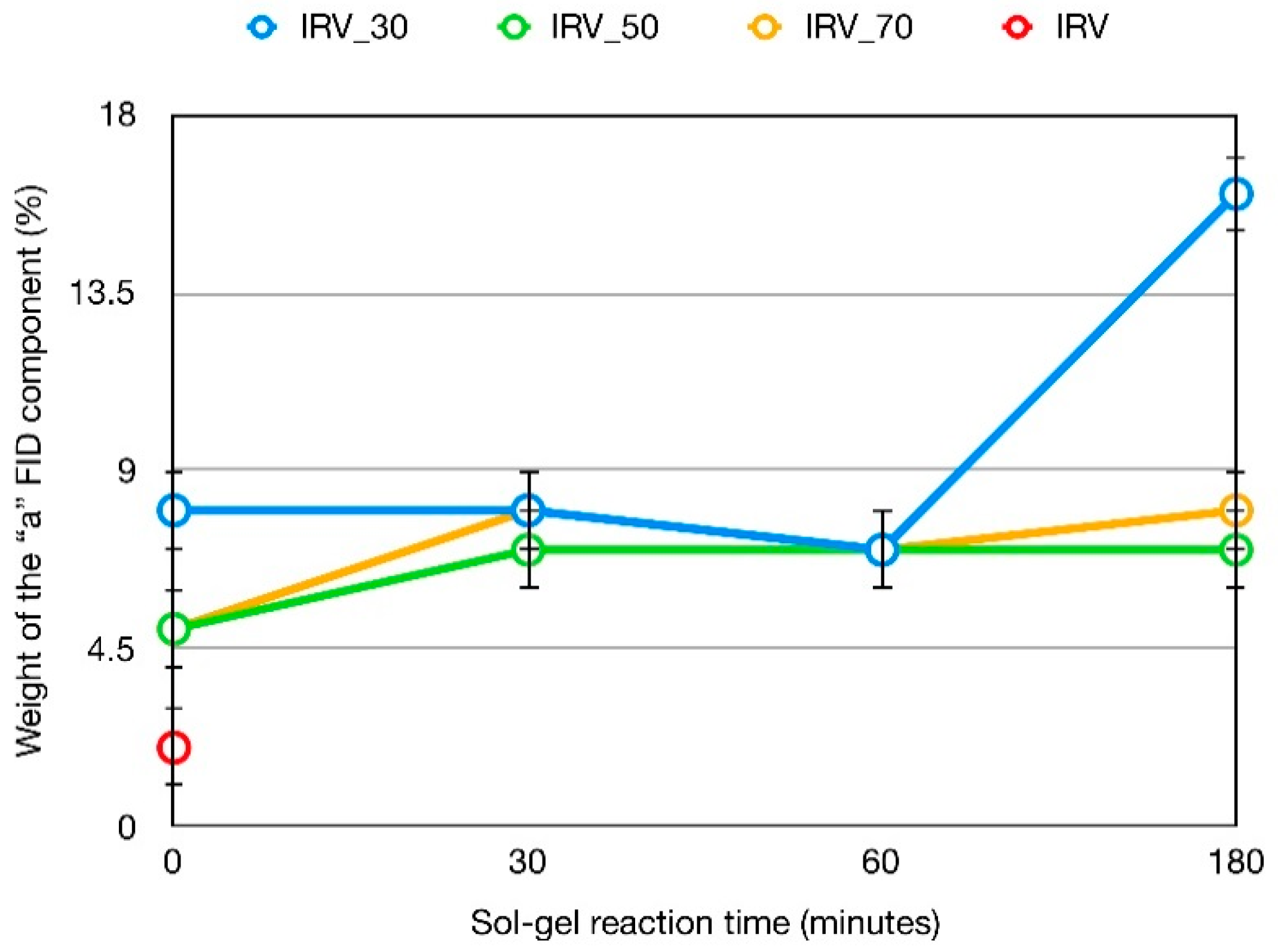
3.3. 1H Spin-Spin Relaxation Times T2
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References and Note
- Kumar, S.K.; Benicewicz, B.C.; Vaia, R.A.; Winey, K.I. 50th Anniversary perspective: Are polymer nanocomposites practical for applications? Macromolecules 2017, 50, 714–731. [Google Scholar] [CrossRef]
- Mouri, H.; Akutagawa, K. Improved tire wet traction through the use of mineral fillers. Rubber Chem. Technol. 1999, 72, 960–968. [Google Scholar] [CrossRef]
- Mallakpour, S.; Naghdi, M. Polymer/SiO2 nanocomposites: Production and applications. Prog. Mater. Sci. 2018, 97, 409–447. [Google Scholar] [CrossRef]
- He, F.; Yuan, T.; Li, C.; Sun, L.; Liao, S. Interfacial interactions and properties of natural rubber–silica composites with liquid natural rubber as a compatibilizer and prepared by a wet-compounding method. J. Appl. Polym. Sci. 2018, 135, 46457. [Google Scholar] [CrossRef]
- Yatsuyanagi, F.; Suzuki, N.; Ito, M.; Kaidou, H. Effects of secondary structure of fillers on the mechanical properties of silica filled rubber systems. Polymer 2001, 42, 9523–9529. [Google Scholar] [CrossRef]
- Vaikuntam, S.R.; Stöckelhuber, K.W.; Bhagavatheswaran, E.S.; Wießner, S.; Scheler, U.; Saalwächter, K.; Formanek, P.; Heinrich, G.; Das, A. Entrapped styrene butadiene polymer chains by sol–gel-derived silica nanoparticles with hierarchical raspberry structures. J. Phys. Chem. B 2018, 122, 2010–2022. [Google Scholar] [CrossRef] [PubMed]
- Messori, M. In situ synthesis of rubber nanocomposites. Adv. Struct. Mater. 2011, 9, 57–85. [Google Scholar]
- Morselli, D.; Bondioli, F.; Luyt, A.S.; Mokhothu, T.H.; Messori, M. Preparation and characterization of EPDM Rubber Modified with in situ generated silica. J. Appl. Polym. Sci. 2013, 128, 2525–2532. [Google Scholar] [CrossRef]
- Bignotti, F.; Borsacchi, S.; de Santis, R.; Geppi, M.; Messori, M.; Sudhakaran, U.P. Interrelation between preparation conditions, structure, and mechanical reinforcement in isoprene rubber filled with in situ generated silica. J. Appl. Polym. Sci. 2012, 125, E398–E412. [Google Scholar] [CrossRef]
- Golitsyn, Y.; Schneider, G.J.; Saalwächter, K. Reduced-mobility layers with high internal mobility in poly(ethylene oxide)-silica nanocomposites. J. Chem. Phys. 2017, 146, 203303. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Cashell, E.; Wardell, G.E.; McBrierty, V.J. An NMR investigation of the interaction between carbon black and cis-polybutadiene. Macromolecules 1976, 9, 653–660. [Google Scholar] [CrossRef]
- Mujtaba, A.; Keller, M.; Ilisch, S.; Radusch, H.-J.; Beiner, M.; Thurn-Albrecht, T.; Saalwächter, K. Detection of surface-immobilized components and their role in viscoelastic reinforcement of rubber–silica nanocomposites. ACS Macro Lett. 2014, 3, 481–485. [Google Scholar] [CrossRef]
- Pérez-Aparicio, R.; Schiewek, M.; Valentín, J.L.; Schneider, H.; Long, D.R.; Saphiannikova, M.; Sotta, P.; Saalwächter, K.; Ott, M. Local chain deformation and overstrain in reinforced elastomers: An NMR study. Macromolecules 2013, 46, 5549–5560. [Google Scholar] [CrossRef]
- Tadiello, L.; D’Arienzo, M.; Di Credico, B.; Hanel, T.; Matejka, L.; Mauri, M.; Morazzoni, F.; Simonutti, R.; Spirkova, M.; Scotti, R. The filler–rubber interface in styrene butadiene nanocomposites with anisotropic silica particles: Morphology and dynamic properties. Soft Matter 2015, 11, 4022–4033. [Google Scholar] [CrossRef] [PubMed]
- Simonutti, R.; Comotti, A.; Negroni, F.; Sozzani, P. 13C and 29Si solid-state NMR of rubber-silica composite materials. Chem. Mater. 1999, 11, 822–828. [Google Scholar] [CrossRef]
- Redaelli, M.; D’Arienzo, M.; Brus, J.; Di Credico, B.; Geppi, M.; Giannini, L.; Matejka, L.; Martini, F.; Panattoni, F.; Spirkova, M.; et al. On the key role of SiO2@POSS hybrid filler in tailoring networking and interfaces in rubber nanocomposites. Polym. Test. 2018, 65, 429–439. [Google Scholar] [CrossRef]
- Borsacchi, S.; Geppi, M.; Veracini, C.A.; Fallani, F.; Ricci, L.; Ruggeri, G. Improving compatibility in LDPE–silica dispersions by photo-grafting reaction. Preparation and solid state NMR investigation. J. Mater. Chem. 2006, 16, 4581–4591. [Google Scholar] [CrossRef]
- Litvinov, V.M.; Orza, R.A.; Klüppel, M.; van Duin, M.; Magusin, P.C.M.M. Rubber-filler interactions and network structure in relation to stress-strain behavior of vulcanized, carbon black filled EPDM. Macromolecules 2011, 44, 4887–4900. [Google Scholar] [CrossRef]
- Wang, M.; Bertmer, M.; Demco, D.E.; Blümich, B.; Litvinov, V.M.; Barthel, H. Indication of heterogeneity in chain-segment order of a PDMS layer grafted onto a silica surface by 1H multiple-quantum NMR. Macromolecules 2003, 36, 4411–4413. [Google Scholar] [CrossRef]
- Litvinov, V.M.; Barthel, H.; Weis, J. Structure of a PDMS layer grafted onto a silica surface studied by means of DSC and solid-state NMR. Macromolecules 2002, 35, 4356–4364. [Google Scholar] [CrossRef]
- ten Brinke, J.W.; Litvinov, V.M.; Wijnhoven, J.E.G.J.; Noordermeer, J.W.M. Interactions of Stöber silica with natural rubber under the influence of coupling agents, studied by 1H NMR T2 relaxation analysis. Macromolecules 2002, 35, 10026–10037. [Google Scholar] [CrossRef]
- Geppi, M.; Borsacchi, S.; Mollica, G.; Veracini, C.A. Applications of solid-state NMR to the study of organic/inorganic multicomponent materials. Appl. Spectrosc. Rev. 2009, 44, 1–89. [Google Scholar] [CrossRef]
- Mathematica, Version 11.1.0.0, 2017; Trademark of Wolfram Research Inc.: Champaign, IL, USA, 2018.
- McBrierty, V.J.; Packer, K.J. Nuclear Magnetic Resonance in Solid Polymers; Cambridge University Press: Cambridge, UK, 1993. [Google Scholar]
- Geppi, M.; Harris, R.K.; Kenwright, A.M.; Say, B.J. A method for analysing proton NMR relaxation data from motionally heterogeneous polymer systems. Solid State Nucl. Magn. Reson. 1998, 12, 15–20. [Google Scholar] [CrossRef]
- Kenwright, A.M.; Say, B.J. Analysis of spin-diffusion measurements by iterative optimisation of numerical models. Solid State Nucl. Magn. Reson. 1996, 7, 85–93. [Google Scholar] [CrossRef]
- Actually, a small amount of protons—From 2 to 5% (see Section 3.2) is also present in silica in silanol groups, the T2 value of which cannot be selectively determined in composites. On the basis of previous 1H FID analysis of silica, however, we expect that most of silanols contribute to the Weibullian function. In any case, due to their small amount, as in the case of T1 and T1ρ, their contribution to reconstructed FIDs was neglected.
- Kristiansen, P.E.; Hansen, E.W.; Pedersen, B. Phase distribution during isothermal crystallization of polyethylene probed by solid-state proton NMR free induction decay. J. Phys. Chem. B 1999, 103, 3552–3558. [Google Scholar] [CrossRef]
- Borsacchi, S.; Paderni, K.; Messori, M.; Toselli, M.; Pilati, F.; Geppi, M. Insights into shape-memory poly(ε-caprolactone) materials by solid-state NMR. Macromolecules 2014, 47, 3544–3552. [Google Scholar] [CrossRef]
- Litvinov, V.M.; Steeman, P.A. EPDM-carbon black Interactions and the reinforcement mechanisms, as studied by low-resolution 1H NMR. Macromolecules 1999, 32, 8476–8490. [Google Scholar] [CrossRef]
- Ou, Y.-C.; Yu, Z.-Z.; Vidal, A.; Donnet, J.B. Effects of alkylation of silicas on interfacial interaction and molecular motions between silicas and rubbers. J. Appl. Polym. Sci. 2012, 125, E398–E412. [Google Scholar] [CrossRef]
- Berriot, J.; Lequeux, F.; Monnerie, L.; Montes, H.; Long, D.; Sotta, P. Filler-elastomer interaction in model filled rubbers, a 1H NMR study. J. Non-Cryst. Sol. 2002, 307–310, 719–724. [Google Scholar] [CrossRef]
- Berriot, J.; Montes, H.; Lequeux, F.; Long, D.; Sotta, P. Evidence for the shift of the glass transition near the particles in silica-filled elastomers. Macromolecules 2002, 35, 9756–9762. [Google Scholar] [CrossRef]
- Papon, A.; Saalwächter, K.; Schäler, K.; Guy, L.; Lequeux, F.; Montes, H. Low-field NMR investigations of nanocomposites: Polymer dynamics and network effects. Macromolecules 2011, 44, 913–922. [Google Scholar] [CrossRef]
- Kim, S.Y.; Meyer, H.W.; Saalwächter, K.; Zukoski, C. Polymer dynamics in PEG-silica nanocomposites: Effects of polymer molecular weight, temperature and solvent dilution. Macromolecules 2012, 45, 4225–4237. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | T1 (s) | T1ρ,1 (ms) | w1 | T1ρ,2 (ms) | w2 | 1/PWRA (ms) |
|---|---|---|---|---|---|---|
| IRV | 0.67 | 4.5 | 15 | 13.2 | 85 | 10 |
| IRV_30_180 | 0.65 | 3.4 | 14 | 13.2 | 86 | 9 |
| IRV_50_180 | 0.66 | 3.3 | 15 | 13.0 | 85 | 9 |
| IRV_70_180 | 0.65 | 4.5 | 19 | 14.2 | 81 | 10 |
| Sample | Silica Content (phr) b | wa (%) | T2b (ms) | wb (%) | wc (%) |
|---|---|---|---|---|---|
| IRV | 0 | 2 | 1.33 | 91 | 7 |
| IRV_30_0 | 10.9 | 8 | 1.72 | 84 | 8 |
| IRV_30_30 | 16.1 | 8 | 1.72 | 86 | 6 |
| IRV_30_60 | 17.9 | 7 | 1.52 | 87 | 6 |
| IRV_30_180 | 23.9 | 16 | 1.56 | 79 | 5 |
| IRV_50_0 | 9.6 | 5 | 1.41 | 88 | 7 |
| IRV_50_30 | 34.5 | 7 | 1.35 | 90 | 3 |
| IRV_50_60 | 39.0 | 7 | 1.34 | 89 | 4 |
| IRV_50_180 | 43.0 | 7 | 1.36 | 87 | 6 |
| IRV_70_0 | 14.3 | 5 | 1.35 | 86 | 9 |
| IRV_70_30 | 48.2 | 8 | 1.44 | 90 | 2 |
| IRV_70_60 | 49.7 | 7 | 1.35 | 87 | 6 |
| IRV_70_180 | 49.5 | 8 | 1.38 | 88 | 4 |
| IRV_30_180sw a | 23.9 | 21 | 2.2 | 46 | 33 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borsacchi, S.; Sudhakaran, U.P.; Calucci, L.; Martini, F.; Carignani, E.; Messori, M.; Geppi, M. Rubber-Filler Interactions in Polyisoprene Filled with In Situ Generated Silica: A Solid State NMR Study. Polymers 2018, 10, 822. https://doi.org/10.3390/polym10080822
Borsacchi S, Sudhakaran UP, Calucci L, Martini F, Carignani E, Messori M, Geppi M. Rubber-Filler Interactions in Polyisoprene Filled with In Situ Generated Silica: A Solid State NMR Study. Polymers. 2018; 10(8):822. https://doi.org/10.3390/polym10080822
Chicago/Turabian StyleBorsacchi, Silvia, Umayal Priyadharsini Sudhakaran, Lucia Calucci, Francesca Martini, Elisa Carignani, Massimo Messori, and Marco Geppi. 2018. "Rubber-Filler Interactions in Polyisoprene Filled with In Situ Generated Silica: A Solid State NMR Study" Polymers 10, no. 8: 822. https://doi.org/10.3390/polym10080822
APA StyleBorsacchi, S., Sudhakaran, U. P., Calucci, L., Martini, F., Carignani, E., Messori, M., & Geppi, M. (2018). Rubber-Filler Interactions in Polyisoprene Filled with In Situ Generated Silica: A Solid State NMR Study. Polymers, 10(8), 822. https://doi.org/10.3390/polym10080822