One-Pot Synthesis of P(O)-N Containing Compounds Using N-Chlorosuccinimide and Their Influence in Thermal Decomposition of PU Foams



Abstract
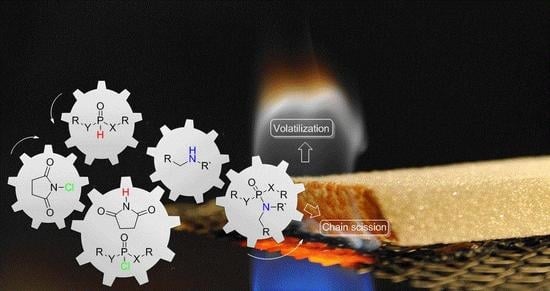
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Thermal and Fire Characterization, ICP-OES and NMR
2.3. Synthesis
2.3.1. Synthesis of Phosphonamidates (General Procedure)
2.3.2. Synthesis of Phosphoramidates (General Procedure)
2.4. Preparation of FPUF
3. Results and Discussion
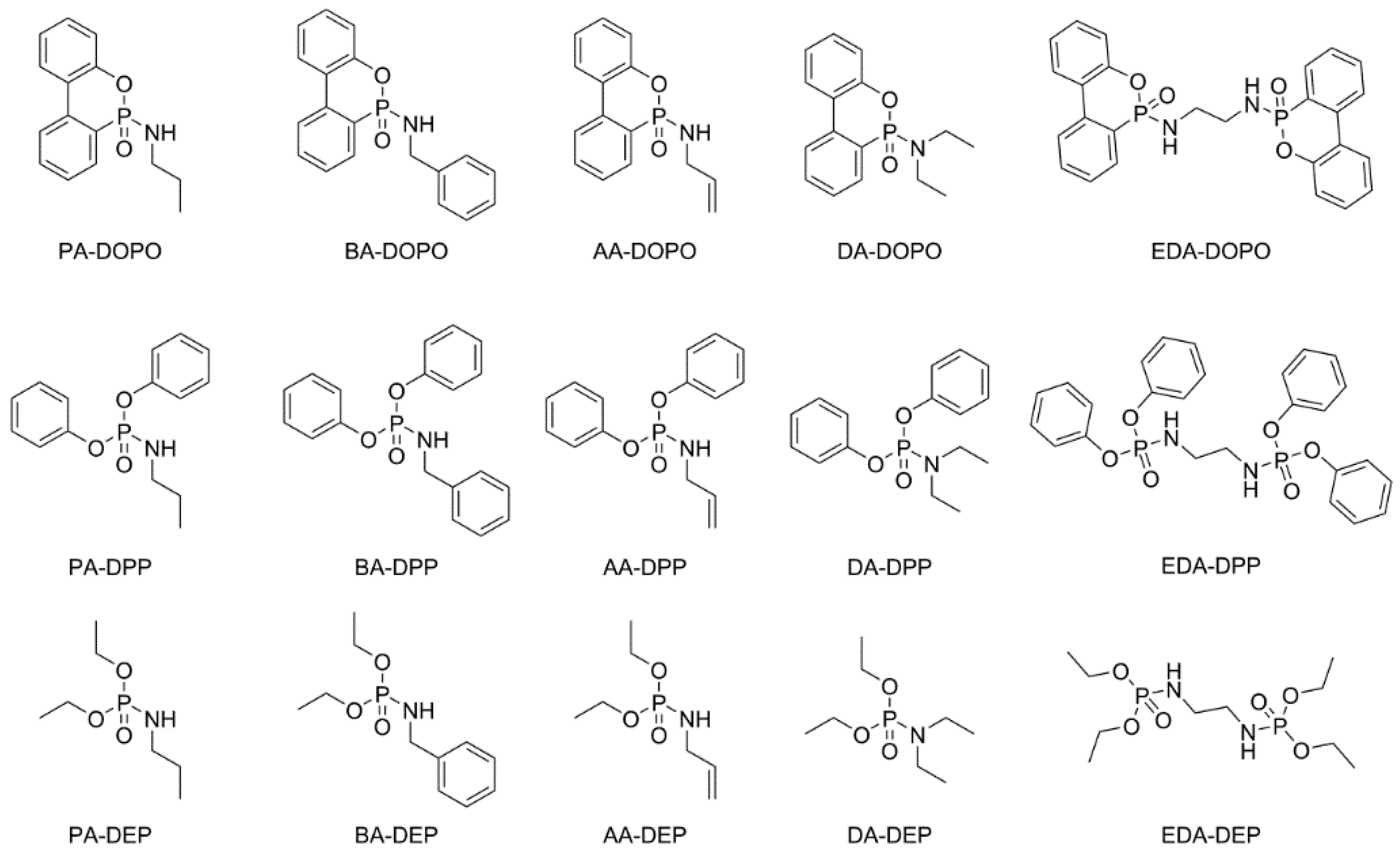
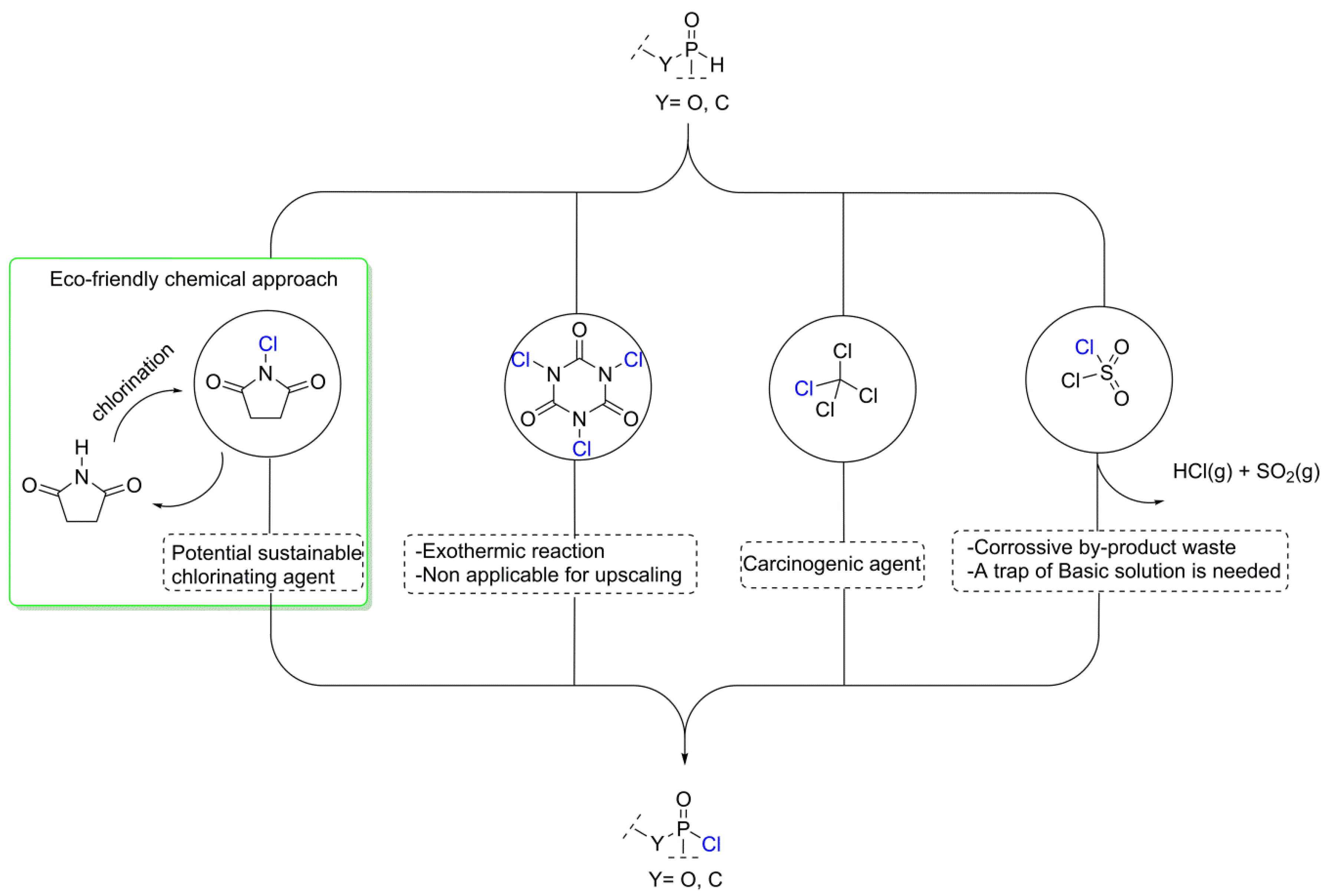
3.1. Synthesis of the P(O)-N Compounds
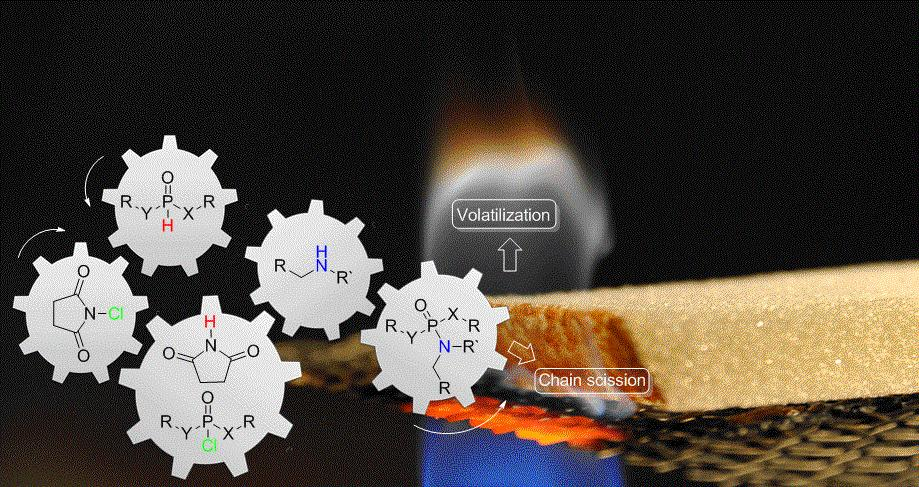
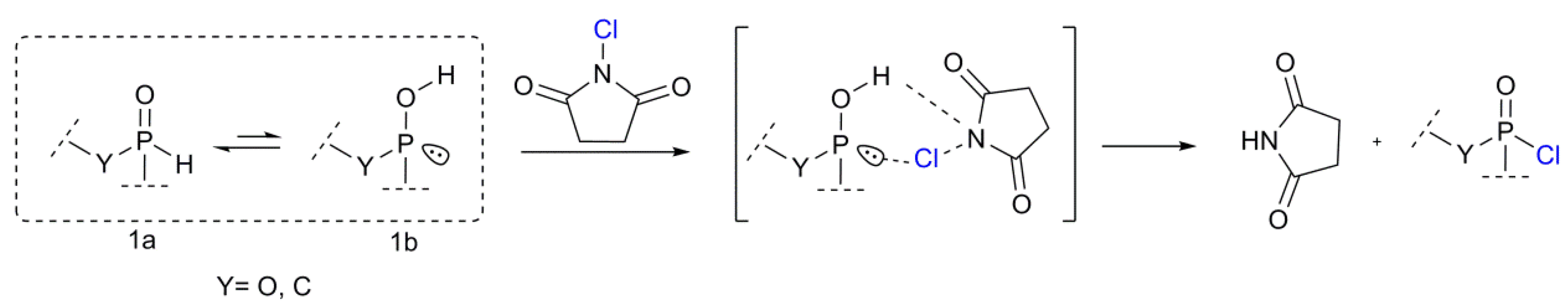
3.2. Proposed Chlorination Mechanism
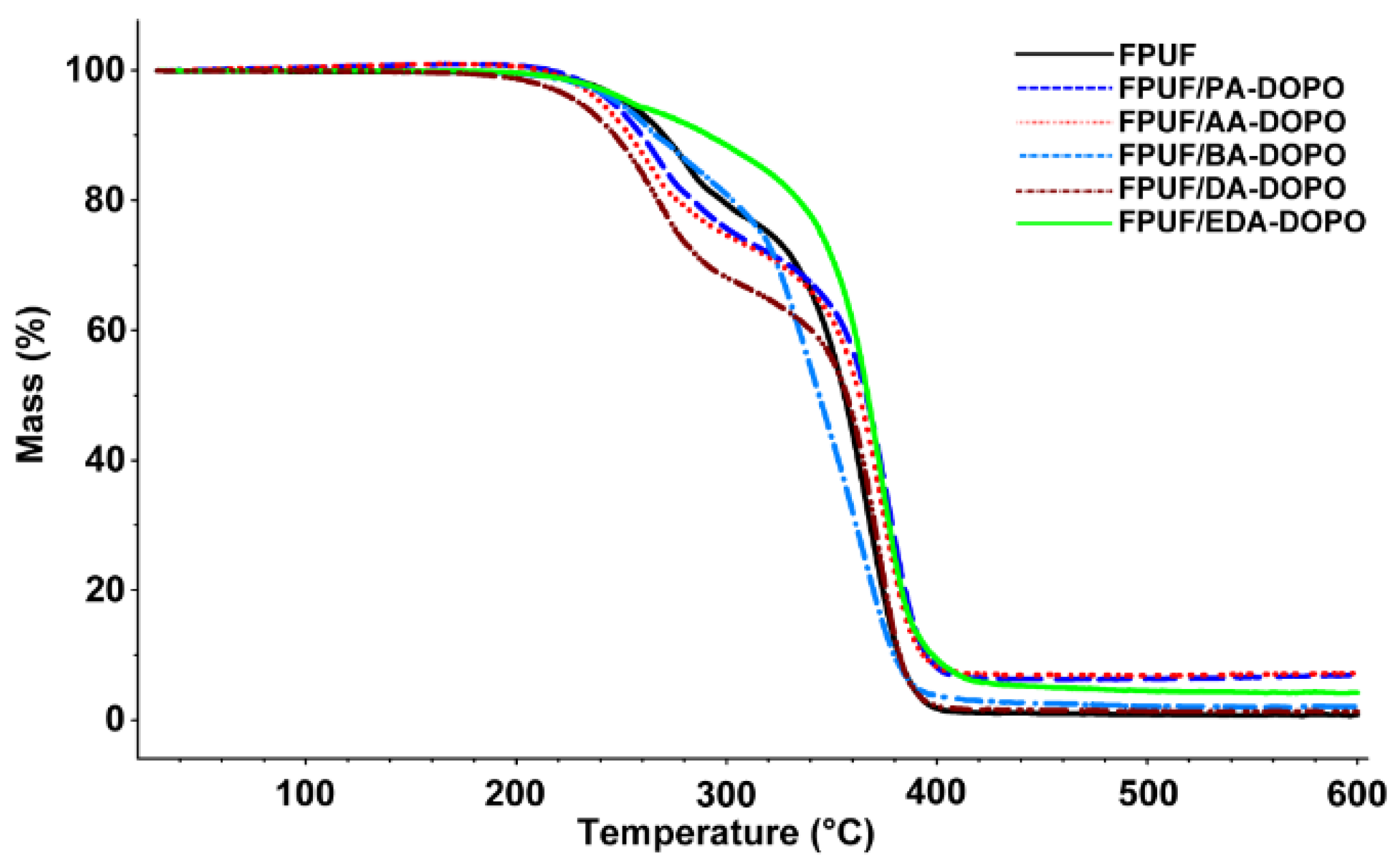
3.3. Thermal Analysis
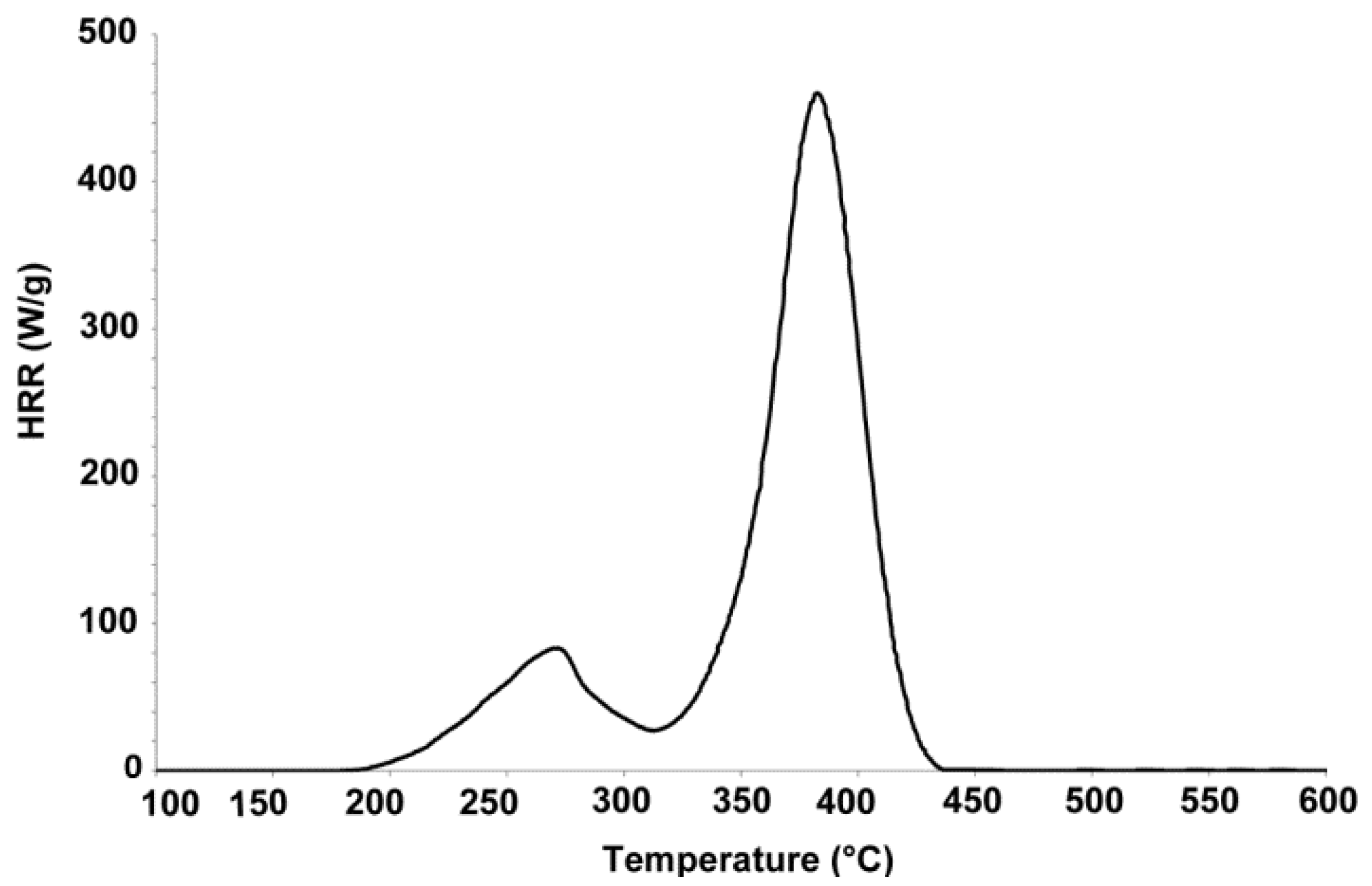
3.4. Small Scale Fire Test with Micro Scale Combustion Calorimeter
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CDCl3 | Deuterated chloroform |
| DOPO | 9,10-Dihydro-9-oxa-10-phosphaphenanthrene 10-oxide |
| FPUF | Flexible polyurethane foam |
| HRC | Heat release capacity |
| HRR | Heat release rate |
| MCC | Microscale combustion calorimeter |
| NMR | Nuclear magnetic resonance |
| TGA | Thermogravimetric analysis |
| THF | Tetrahydrofuran |
| THR | Total heat release rate |
References
- Gilheany, D.G. Structure and bonding in organophosphorus (III) compounds. In Organophosphorus Compounds; Hartley, F.R., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1990; Volume 1, pp. 9–49. [Google Scholar]
- Stawinski, J.; Kraszewski, A. How to get the most out of two phosphorus chemistries. Studies on H-phosphonates. Acc. Chem. Res. 2002, 35, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Marinozzi, M.; Pertusati, F.; Serpi, M. λ5-phosphorus-containing α-diazo compounds: A valuable tool for accessing phosphorus-functionalized molecules. Chem. Rev. 2016, 116, 13991–14055. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Tang, G.; Zhao, Y. Recent progress toward organophosphorus compounds based on phosphorus-centered radical difunctionalizations. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 589–596. [Google Scholar] [CrossRef]
- Oliveira, F.M.; Barbosa, L.C.A.; Ismail, F.M.D. The diverse pharmacology and medicinal chemistry of phosphoramidates—A review. RSC Adv. 2014, 4, 18998–19012. [Google Scholar] [CrossRef]
- Di Rocco, D.A.; Ji, Y.; Sherer, E.C.; Klapars, A.; Reibarkh, M.; Dropinski, J.; Mathew, R.; Maligres, P.; Hyde, A.M.; Limanto, J.; et al. A multifunctional catalyst that stereoselectively assembles prodrugs. Science 2017, 356, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Neisius, M.; Mispreuve, H.; Naescher, R.; Gaan, S. Flame retardancy and thermal decomposition of flexible polyurethane foams: Structural influence of organophosphorus compounds. Polym. Degrad. Stab. 2012, 97, 2428–2440. [Google Scholar] [CrossRef]
- Neisius, M.; Liang, S.; Mispreuve, H.; Gaan, S. Phosphoramidate-containing flame-retardant flexible polyurethane foams. Ind. Eng. Chem. Res. 2013, 52, 9752–9762. [Google Scholar] [CrossRef]
- Gaan, S.; Liang, S.; Mispreuve, H.; Perler, H.; Naescher, R.; Neisius, M. Flame retardant flexible polyurethane foams from novel dopo-phosphonamidate additives. Polym. Degrad. Stab. 2015, 113, 180–188. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, Z.; Fan, H.; Tian, S.; Chen, Y.; Yan, J. Waterborne polyurethane conjugated with novel diol chain-extender bearing cyclic phosphoramidate lateral group: Synthesis, flammability and thermal degradation mechanism. RSC Adv. 2016, 6, 56610–56622. [Google Scholar] [CrossRef]
- Zhao, B.; Liu, D.-Y.; Liang, W.-J.; Li, F.; Wang, J.-S.; Liu, Y.-Q. Bi-phase flame-retardant actions of water-blown rigid polyurethane foam containing diethyl-N,N-bis(2-hydroxyethyl) phosphoramide and expandable graphite. J. Anal. Appl. Pyrolysis 2017, 124, 247–255. [Google Scholar] [CrossRef]
- Nguyen, C.; Kim, J. Synthesis of a novel nitrogen-phosphorus flame retardant based on phosphoramidate and its application to PC, PBT, EVA, and ABS. Macromol. Res. 2008, 16, 620–625. [Google Scholar] [CrossRef]
- Zhao, W.; Li, B.; Xu, M.; Yang, K.; Lin, L. Novel intumescent flame retardants: Synthesis and application in polycarbonate. Fire Mater. 2013, 37, 530–546. [Google Scholar] [CrossRef]
- Kundu, C.K.; Yu, B.; Gangireddy, C.S.R.; Mu, X.; Wang, B.; Wang, X.; Song, L.; Hu, Y. UV grafting of a dopo-based phosphoramidate monomer onto polyamide 66 fabrics for flame retardant treatment. Ind. Eng. Chem. Res. 2017, 56, 1376–1384. [Google Scholar] [CrossRef]
- Kim, M.; Sanda, F.; Endo, T. Phosphonamidates as thermally latent initiators in the polymerization of epoxides. Polym. Bull. 2001, 46, 277–283. [Google Scholar] [CrossRef]
- Ma, C.; Yu, B.; Hong, N.; Pan, Y.; Hu, W.; Hu, Y. Facile synthesis of a highly efficient, halogen-free, and intumescent flame retardant for epoxy resins: Thermal properties, combustion behaviors, and flame-retardant mechanisms. Ind. Eng. Chem. Res. 2016, 55, 10868–10879. [Google Scholar] [CrossRef]
- Guo, W.; Yu, B.; Yuan, Y.; Song, L.; Hu, Y. In situ preparation of reduced graphene oxide/dopo-based phosphonamidate hybrids towards high-performance epoxy nanocomposites. Compos. Part B 2017, 123, 154–164. [Google Scholar] [CrossRef]
- Salmeia, K.; Gaan, S.; Malucelli, G. Recent advances for flame retardancy of textiles based on phosphorus chemistry. Polymers 2016, 8, 319. [Google Scholar] [CrossRef]
- Salmeia, K.A.; Jovic, M.; Ragaisiene, A.; Rukuiziene, Z.; Milasius, R.; Mikucioniene, D.; Gaan, S. Flammability of cellulose-based fibers and the effect of structure of phosphorus compounds on their flame retardancy. Polymers 2016, 8, 293. [Google Scholar] [CrossRef]
- Zhao, B.; Liu, Y.-T.; Zhang, C.-Y.; Liu, D.-Y.; Li, F.; Liu, Y.-Q. A novel phosphoramidate and its application on cotton fabrics: Synthesis, flammability and thermal degradation. J. Anal. Appl. Pyrolysis 2017, 125, 109–116. [Google Scholar] [CrossRef]
- Recknagel, R.O. Carbon tetrachloride hepatotoxicity: Status quo and future prospects. Trends Pharmacol. Sci. 1983, 4, 129–131. [Google Scholar] [CrossRef]
- Simeonova, P.P.; Gallucci, R.M.; Hulderman, T.; Wilson, R.; Kommineni, C.; Rao, M.; Luster, M.I. The role of tumor necrosis factor-α in liver toxicity, inflammation, and fibrosis induced by carbon tetrachloride. Toxicol. Appl. Pharmacol. 2001, 177, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Dahlstrom, D.; Snawder, J. Solvents and industrial hygiene. In Hayes’ Principles and Methods of Toxicology, 6th ed.; Hayes, A.W., Kruger, C.L., Eds.; CRC Press: Boca Raton, FL, USA, 2014; pp. 677–710. [Google Scholar]
- Mlodnosky, K.L.; Holmes, H.M.; Lam, V.Q.; Berkman, C.E. A convenient two-step one-pot synthesis of phosphonamidates. Tetrahedron Lett. 1997, 38, 8803–8806. [Google Scholar] [CrossRef]
- Yangt, G.; Zhao, K.; Landry, D.W. Tetrazole-catalyzed synthesis of phosphonamidate esters. Tetrahedron Lett. 1998, 39, 2449–2450. [Google Scholar] [CrossRef]
- Subramanyam, C.; Ramana, K.V.; Rasheed, S.; Adam, S.; Raju, C.N. Synthesis and biological activity of novel diphenyl N-substituted carbamimidoylphosphoramidate derivatives. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 1228–1235. [Google Scholar] [CrossRef]
- Panmand, D.S.; Tiwari, A.D.; Panda, S.S.; Monbaliu, J.-C.M.; Beagle, L.K.; Asiri, A.M.; Stevens, C.V.; Steel, P.J.; Hall, C.D.; Katritzky, A.R. New benzotriazole-based reagents for the phosphonylation of various N-, O-, and S-nucleophiles. Tetrahedron Lett. 2014, 55, 5898–5901. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, G.; Saga, Y.; Shen, R.; Goto, M.; Zhao, Y.; Han, L.-B. Stereospecific halogenation of P(O)-H bonds with copper(II) chloride affording optically active Z1Z2P(O)Cl. J. Org. Chem. 2010, 75, 7924–7927. [Google Scholar] [CrossRef] [PubMed]
- Neisius, N.M.; Lutz, M.; Rentsch, D.; Hemberger, P.; Gaan, S. Synthesis of dopo-based phosphonamidates and their thermal properties. Ind. Eng. Chem. Res. 2014, 53, 2889–2896. [Google Scholar] [CrossRef]
- Goldwhite, H.; Saunders, B.C. Esters containing phosphorus. Part XIII. Dialkyl phosphorobromidates. J. Chem. Soc. 1955, 3549–3564. [Google Scholar] [CrossRef]
- Au-Yeung, T.-L.; Chan, K.-Y.; Chan, W.-K.; Haynes, R.K.; Williams, I.D.; Yeung, L.L. Reactions of (RP)- and (SP)-tert-butylphenylphosphinobromidates and tert-butylphenylthionophosphinochloridates with heteroatom nucleophiles; preparation of P-chiral binol phosphinates and related compounds. Tetrahedron Lett. 2001, 42, 453–456. [Google Scholar] [CrossRef]
- Atherton, F.R.; Openshaw, H.T.; Todd, A.R. Studies on phosphorylation. Part I. Dibenzyl chlorophosphonate as a phosphorylating agent. J. Chem. Soc. 1945, 382–385. [Google Scholar] [CrossRef]
- McCombie, I.H.; Saunders, B.C.; Stacey, G.J. Esters containing phosphorus. Part I. J. Chem. Soc. 1945, 380–382. [Google Scholar] [CrossRef]
- Pretula, J.; Kaluzynski, K.; Szymanski, R.; Penczek, S. Preparation of poly(alkylene h-phosphonate)s and their derivatives by polycondensation of diphenyl H-phosphonate with diols and subsequent transformations. Macromolecules 1997, 30, 8172–8176. [Google Scholar] [CrossRef]
- Skowronska, A.; Mikolajczak, J.; Michalski, J. Pentacovalent intermediate in the arbuzov reaction. J. Chem. Soc. Chem. Commun. 1975, 791–792. [Google Scholar] [CrossRef]
- Fraser, J.; Wilson, L.J.; Blundell, R.K.; Hayes, C.J. Phosphoramidate synthesis via copper-catalysed aerobic oxidative coupling of amines and h-phosphonates. Chem. Commun. 2013, 49, 8919–8921. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Yamaguchi, K.; Mizuno, N. Copper-catalyzed oxidative cross-coupling of H-phosphonates and amides to N-acylphosphoramidates. Org. Lett. 2013, 15, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Dhineshkumar, J.; Prabhu, K.R. Cross-hetero-dehydrogenative coupling reaction of phosphites: A catalytic metal-free phosphorylation of amines and alcohols. Org. Lett. 2013, 15, 6062–6065. [Google Scholar] [CrossRef] [PubMed]
- Dar, B.A.; Dangroo, N.A.; Gupta, A.; Wali, A.; Khuroo, M.A.; Vishwakarma, R.A.; Singh, B. Iodine catalyzed solvent-free cross-dehydrogenative coupling of arylamines and H-phosphonates for the synthesis of N-arylphosphoramidates under atmospheric conditions. Tetrahedron Lett. 2014, 55, 1544–1548. [Google Scholar] [CrossRef]
- Wilkening, I.; del Signore, G.; Hackenberger, C.P.R. Synthesis of N,N-disubstituted phosphoramidates via a lewis acid-catalyzed phosphorimidate rearrangement. Chem. Commun. 2008, 2932–2934. [Google Scholar] [CrossRef] [PubMed]
- Bohrsch, V.; Serwa, R.; Majkut, P.; Krause, E.; Hackenberger, C.P.R. Site-specific functionalisation of proteins by a staudinger-type reaction using unsymmetrical phosphites. Chem. Commun. 2010, 46, 3176–3178. [Google Scholar] [CrossRef] [PubMed]
- Serwa, R.; Majkut, P.; Horstmann, B.; Swiecicki, J.-M.; Gerrits, M.; Krause, E.; Hackenberger, C.P.R. Site-specific PEGylation of proteins by a Staudinger-phosphite reaction. Chem. Sci. 2010, 1, 596–602. [Google Scholar] [CrossRef]
- Nischan, N.; Chakrabarti, A.; Serwa, R.A.; Bovee-Geurts, P.H.M.; Brock, R.; Hackenberger, C.P.R. Stabilization of peptides for intracellular applications by phosphoramidate-linked polyethylene glycol chains. Angew. Chem. Int. Ed. 2013, 52, 11920–11924. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Tao, J.; Jones, J.E.; Wojtas, L.; Zhang, X.P. Cobalt(II)-catalyzed intramolecular C−H amination with phosphoryl azides: Formation of 6- and 7-membered cyclophosphoramidates. Org. Lett. 2010, 12, 1248–1251. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Zhou, C.-Y.; Che, C.-M. Ruthenium(IV) porphyrin catalyzed phosphoramidation of aldehydes with phosphoryl azides as a nitrene source. Chem. Commun. 2012, 48, 5871–5873. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Park, J.; Kim, J.G.; Chang, S. Synthesis of phosphoramidates: A facile approach based on the C-N bond formation via ir-catalyzed direct C-H amidation. Org. Lett. 2014, 16, 5466–5469. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Jin, N.; Zhang, H.; Han, J.; Zhu, C. Iridium-catalyzed phosphoramidation of arene C-H bonds with phosphoryl azide. J. Org. Chem. 2014, 79, 9427–9432. [Google Scholar] [CrossRef] [PubMed]
- Meazza, M.; Kowalczuk, A.; Shirley, L.; Yang, J.W.; Guo, H.; Rios, R. Organophotocatalytic synthesis of phosphoramidates. Adv. Synth. Catal. 2016, 358, 719–723. [Google Scholar] [CrossRef]
- Kenner, G.W.; Todd, A.R.; Weymouth, F.J. Nucleotides. Part XVII. N-chloroamides as reagents for the chlorination of diesters of phosphorous acid. New synthesis of uridine-5′-pyrophosphate. J. Chem. Soc. 1952, 3675–3681. [Google Scholar] [CrossRef]
- Goldwhite, H.; Saunders, B.C. Esters containing phosphorus. Part XII. Esters of phosphofluoridic acid. J. Chem. Soc. 1955, 2038–2056. [Google Scholar] [CrossRef]
- Skrzypczynski, Z.; Michalski, J. Stereoselective synthesis and stereochemistry of optically active tert-butylphenylphosphine sulfide. J. Org. Chem. 1988, 53, 4549–4551. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C. Chapter 4—Purification of organic chemicals. In Purification of Laboratory Chemicals, 7th ed.; Butterworth-Heinemann: Boston, MA, USA, 2013; pp. 103–554. [Google Scholar]
- Keglevich, G.; Szelke, H.; Kerenyi, A.; Kudar, V.; Hanusz, M.; Simon, K.; Imre, T.; Ludanyi, K. New chiral P-ligands: P-amino- and P-cycloalkoxy dibenzo[c.e][1,2]oxaphosphorines. Tetrahedron Asymmetry 2005, 16, 4015–4021. [Google Scholar] [CrossRef]
- Stock, J.A.; Hopwood, W.J.; Regan, P.D. Amide as a protecting group in phosphate ester synthesis. Part I. The acid hydrolysis of some phosphoramidic diesters. J. Chem. Soc. C 1966, 637–639. [Google Scholar] [CrossRef]
- Li, B.-J.; Simard, R.D.; Beauchemin, A.M. O-phthalaldehyde catalyzed hydrolysis of organophosphinic amides and other P(=O)-NH containing compounds. Chem. Commun. 2017, 53, 8667–8670. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.; Kim, J. Thermal stabilities and flame retardancies of nitrogen–phosphorus flame retardants based on bisphosphoramidates. Polym. Degrad. Stab. 2008, 93, 1037–1043. [Google Scholar] [CrossRef]
- Shiri, A.; Khoramabadi-zad, A. Preparation of several active N-chloro compounds from trichloroisocyanuric acid. Synthesis 2009, 2797–2801. [Google Scholar] [CrossRef]
- Yang, Z.; Xu, J. Convenient and environment-friendly synthesis of sulfonyl chlorides from S-alkylisothiourea salts via N-chlorosuccinimide chlorosulfonation. Synthesis 2013, 45, 1675–1682. [Google Scholar] [CrossRef]
- Gloede, J.; Pieper, U.; Costisella, B.; Krueger, R.-P. A stable trichloro-oxyphosphorane with an oxaphosphorin ring. Z. Anorg. Allg. Chem. 2003, 629, 998–1000. [Google Scholar] [CrossRef]
- Acharya, J.; Gupta, A.K.; Shakya, P.D.; Kaushik, M.P. Trichloroisocyanuric acid: An efficient reagent for the synthesis of dialkyl chlorophosphates from dialkyl phosphites. Tetrahedron Lett. 2005, 46, 5293–5295. [Google Scholar] [CrossRef]
- Cink, R.D.; Chambournier, G.; Surjono, H.; Xiao, Z.; Richter, S.; Naris, M.; Bhatia, A.V. Investigation of the stability of the Corey−Kim intermediate. Org. Process Res. Dev. 2007, 11, 270–274. [Google Scholar] [CrossRef]
- Higuchi, T.; Hasegawa, J. Rate of exchange of chlorine between dimethylchloramine and succinimide1. J. Phys. Chem. 1965, 69, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Antelo, J.M.; Arce, F.; Franco, J.; Garcia Lopez, M.C.; Sanchez, M.; Varela, A. Kinetics of the chlorination of secondary amines by N-chlorosuccinimide. Int. J. Chem. Kinet. 1988, 20, 397–409. [Google Scholar] [CrossRef]
- Pastoriza, C.; Antelo, J.M.; Crugeiras, J.; Pena-Gallego, A. Kinetic study of the formation of N-chloro compounds using N-chlorosuccinimide. J. Phys. Org. Chem. 2014, 27, 407–418. [Google Scholar] [CrossRef]
- Salmeia, K.; Fage, J.; Liang, S.; Gaan, S. An overview of mode of action and analytical methods for evaluation of gas phase activities of flame retardants. Polymers 2015, 7, 504–526. [Google Scholar] [CrossRef]
- Allan, D.; Daly, J.; Liggat, J.J. Thermal volatilisation analysis of TDI-based flexible polyurethane foam. Polym. Degrad. Stab. 2013, 98, 535–541. [Google Scholar] [CrossRef]
- Chambers, J.; Jiricny, J.; Reese, C.B. The thermal decomposition of polyurethanes and polyisocyanurates. Fire Mater. 1981, 5, 133–141. [Google Scholar] [CrossRef]
- Benbow, A.W.; Cullis, C.F. The combustion of flexible polyurethane foams: Mechanisms and evaluation of flame retardance. Combust. Flame 1975, 24, 217–230. [Google Scholar] [CrossRef]
- Troev, K.; Tsekova, A.; Tsevi, R. Chemical degradation of polyurethanes: Degradation of flexible polyester polyurethane foam by phosphonic acid dialkyl esters. J. Appl. Polym. Sci. 2000, 78, 2565–2573. [Google Scholar] [CrossRef]



 ) and derivative thermogravimetry (DTG) (
) and derivative thermogravimetry (DTG) ( ) curves for pure FPUF under N2 atmosphere.
) curves for pure FPUF under N2 atmosphere.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Starting Material | Product (a) | Yield (%) (b) | 31P NMR (ppm) (c) |
|---|---|---|---|---|
| 1 |  |  | 100 | 4.1 |
| 2 (d) |  |  | 95 | −5.1 |
| 3 (e) |  |  | 98 | 19.7 |

| Entry | Solvent | Base | Yield (%) |
|---|---|---|---|
| 1 | chloroform | triethylamine | 62 |
| 2 | toluene | triethylamine | 48 |
| 3 | acetonitrile | triethylamine | 55 |
| 4 | dichloromethane | triethylamine | 90 |
| 5 | dichloromethane | K2CO3 | 60 |
| Entry | Substances | Appearance | mp (a) (°C)/bp (b) (°C) | Yield (%) |
|---|---|---|---|---|
| PA-DOPO | White solid | 131 (a) | 90 | |
| AA-DOPO | White solid | 103 (a) | 91 | |
| 1 | BA-DOPO | Off-white solid | 159 (a) | 98 |
| DA-DOPO | Red solid | 114 (a) | 59 | |
| EDA-DOPO | White solid | 266 (a) | 75 | |
| PA-DPP | Off-white solid | 55 (a) | 68 | |
| AA-DPP | White solid | 55 (a) | 83 | |
| 2 | BA-DPP | Off-white solid | 106 (a) | 87 |
| DA-DPP | White solid | 55 (a) | 63 | |
| EDA-DPP | White solid | 134 (a) | 85 | |
| PA-DEP | Colorless oil | 82–85 (b) at 0.13 mbar | 81 | |
| AA-DEP | Pale yellow oil | ND | 68 | |
| 3 | BA-DEP | Pale Yellow oil | ND | 94 |
| DA-DEP | Pale yellow oil | 55–56 (b) at 0.29 mbar | 74 | |
| EDA-DEP | Off-white solid | 83 | 95 |
| Sampless | Stage 1 | Stage 2 | Stage 3 | Char Residue at 600 °C (wt %) ± 0.5 | |||
|---|---|---|---|---|---|---|---|
| Tonset (°C) | Tmax(a) (°C) | Tonset (°C) | Tmax(a) (°C) | Tonset (°C) | Tmax(a) (°C) | ||
| FPUF | 262 | 282 | 346 | 367 | --- | --- | 1 |
| FPUF/PA-DOPO | 249 | 267 | 354 | 371 | --- | --- | 7 |
| FPUF/AA-DOPO | 238 | 266 | 357 | 374 | --- | --- | 7 |
| FPUF/BA-DOPO | 251 | 320 | 262 | 365 | --- | --- | 2 |
| FPUF/DA-DOPO | 258 | 267 | 356 | 372 | --- | --- | 1 |
| FPUF/EDA-DOPO | 259 | 279 | 364 | 380 | --- | --- | 4 |
| FPUF/PA-DPP | 219 | 246 | 357 | 373 | --- | --- | 0 |
| FPUF/AA-DPP | 222 | 247 | 357 | 372 | --- | --- | 2 |
| FPUF/BA-DPP | 267 | 280 | 352 | 368 | --- | --- | 2 |
| FPUF/DA-DPP | 208 | 232 | 348 | 364 | --- | --- | 0 |
| FPUF/EDA-DPP | 257 | 271 | 334 | 353 | --- | --- | 6 |
| FPUF/PA-DEP | 125 | 155 | 272 | 285 | 354 | 370 | 0 |
| FPUF/AA-DEP | 128 | 151 | 270 | 290 | 357 | 374 | 1 |
| FPUF/BA-DEP | 193 | 213 | 267 | 280 | 346 | 367 | 0 |
| FPUF/DA-DEP | 122 | 130 | 265 | 292 | 353 | 368 | 6 |
| FPUF/EDA-DEP | 241 | 268 | 333 | 360 | --- | --- | 3 |
| Samples | P-Content (wt %) | THR(kJ/g) (a) | HRC (J/g K) (b) | PHRR(W/g) (c) | Char Residue (wt %) |
|---|---|---|---|---|---|
| FPUF | --- | 26.0 ± 0.9 | 489.0 ± 7.0 | 484.8 ± 12.6 | 1.1 ± 0.4 |
| FPUF/PA-DOPO | 2.12 ± 0.09 | 28.1 ± 0.9 | 479.0 ± 13.1 | 479.4 ± 11.7 | 1.8 ± 1.0 |
| FPUF/AA-DOPO | 2.10 ± 0.04 | 26.7 ± 0.8 | 487.7 ± 72.1 | 461.3 ± 21.4 | 1.8 ± 0.7 |
| FPUF/BA-DOPO | 1.93 ± 0.03 | 27.9 ± 1.5 | 326.3 ± 85.5 | 461.3 ± 21.4 | 0.2 ± 0.2 |
| FPUF/DA-DOPO | 2.04 ± 0.11 | 25.5 ± 0.8 | 561.7 ± 24.9 | 565.5 ± 20.5 | 0.7 ± 0.6 |
| FPUF/EDA-DOPO | 2.09 ± 0.08 | 23.6 ± 0.9 | 516. 7 ± 2.9 | 516.3 ± 3.0 | 6.5 ± 0.6 |
| FPUF/PA-DPP | 2.00 ± 0.08 | 26.9 ± 1.1 | 593.7 ± 18.3 | 593.7 ± 18.9 | 2.3 ± 2.3 |
| FPUF/AA-DPP | 2.11 ± 0.04 | 26.2 ± 0.8 | 556.7 ± 19.1 | 550.0 ± 13.9 | 0.6 ± 0.5 |
| FPUF/BA-DPP | 2.13 ± 0.01 | 26.8 ± 0.3 | 530.0 ± 4.0 | 529.8 ± 7.4 | 0.3 ± 0.2 |
| FPUF/DA-DPP | 2.08 ± 0.06 | 26.2 ± 2.4 | 581.0 ± 30.5 | 572.1 ± 27.7 | 0.8 ± 0.4 |
| FPUF/EDA-DPP | 1.98 ± 0.11 | 26.4 ± 0.1 | 387.3 ± 23.9 | 394.2 ± 21.7 | 4.7 ± 1.2 |
| FPUF/PA-DEP | 2.07 ± 0.00 | 26.1 ± 3.3 | 575.7 ± 5.7 | 564.2 ± 3.4 | 0.9 ± 0.2 |
| FPUF/AA-DEP | 2.15 ± 0.01 | 23.1 ± 0.8 | 600.0 ± 11.3 | 616.8 ± 9.2 | 0.6 ± 0.6 |
| FPUF/BA-DEP | 2.13 ± 0.00 | 25.8 ± 1.2 | 558.0 ± 14.0 | 546.9 ± 10.2 | 0.2 ± 0.2 |
| FPUF/DA-DEP | 2.00 ± 0.08 | 23.6 ± 1.0 | 659.0 ± 4.4 | 667.7 ± 14.3 | 1.8 ± 1.3 |
| FPUF/EDA-DEP | 2.07 ± 0.07 | 25.5 ± 0.8 | 490.3 ± 29.4 | 486.0 ± 31.9 | 2.1 ± 1.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salmeia, K.A.; Flaig, F.; Rentsch, D.; Gaan, S. One-Pot Synthesis of P(O)-N Containing Compounds Using N-Chlorosuccinimide and Their Influence in Thermal Decomposition of PU Foams. Polymers 2018, 10, 740. https://doi.org/10.3390/polym10070740
Salmeia KA, Flaig F, Rentsch D, Gaan S. One-Pot Synthesis of P(O)-N Containing Compounds Using N-Chlorosuccinimide and Their Influence in Thermal Decomposition of PU Foams. Polymers. 2018; 10(7):740. https://doi.org/10.3390/polym10070740
Chicago/Turabian StyleSalmeia, Khalifah A., Florence Flaig, Daniel Rentsch, and Sabyasachi Gaan. 2018. "One-Pot Synthesis of P(O)-N Containing Compounds Using N-Chlorosuccinimide and Their Influence in Thermal Decomposition of PU Foams" Polymers 10, no. 7: 740. https://doi.org/10.3390/polym10070740
APA StyleSalmeia, K. A., Flaig, F., Rentsch, D., & Gaan, S. (2018). One-Pot Synthesis of P(O)-N Containing Compounds Using N-Chlorosuccinimide and Their Influence in Thermal Decomposition of PU Foams. Polymers, 10(7), 740. https://doi.org/10.3390/polym10070740