Synthesis and Aqueous Solution Properties of an Amino Bisphosphonate Methacrylate Homopolymer via RAFT Polymerization


Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of MAC2NP2 Monomer
2.3. Synthesis of PMAC2NP2 via RAFT Polymerization
2.4. Hydrolysis of the Phosphonated Ester Groups of PMAC2NP2
2.5. Characterization
2.5.1. Gel Permeation Chromatography (GPC)
2.5.2. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.5.3. Potentiometric Titration
2.5.4. Electrokinetic measurements
3. Results and Discussion
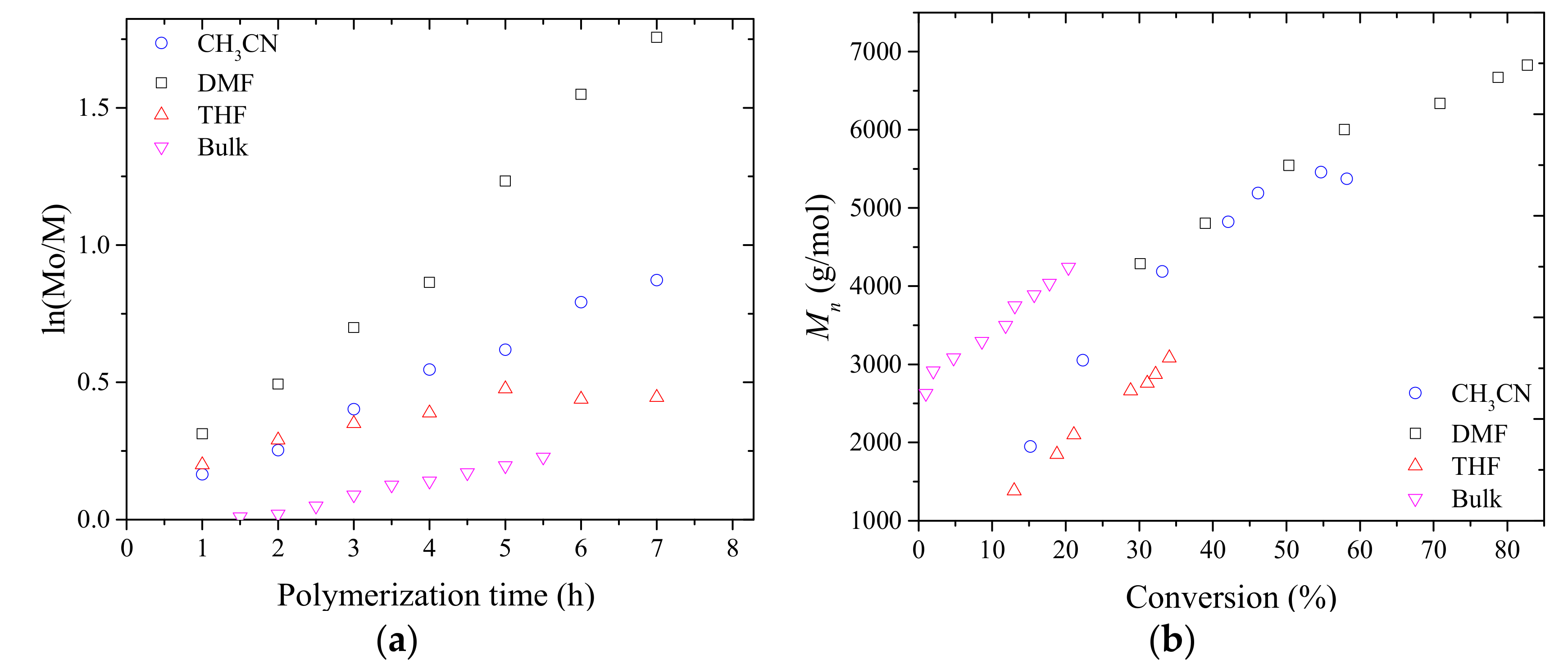
3.1. Synthesis of PMAC2NP2 Homopolymers via RAFT Polymerization
3.2. Hydrolysis of PMAC2NP2 Polymers
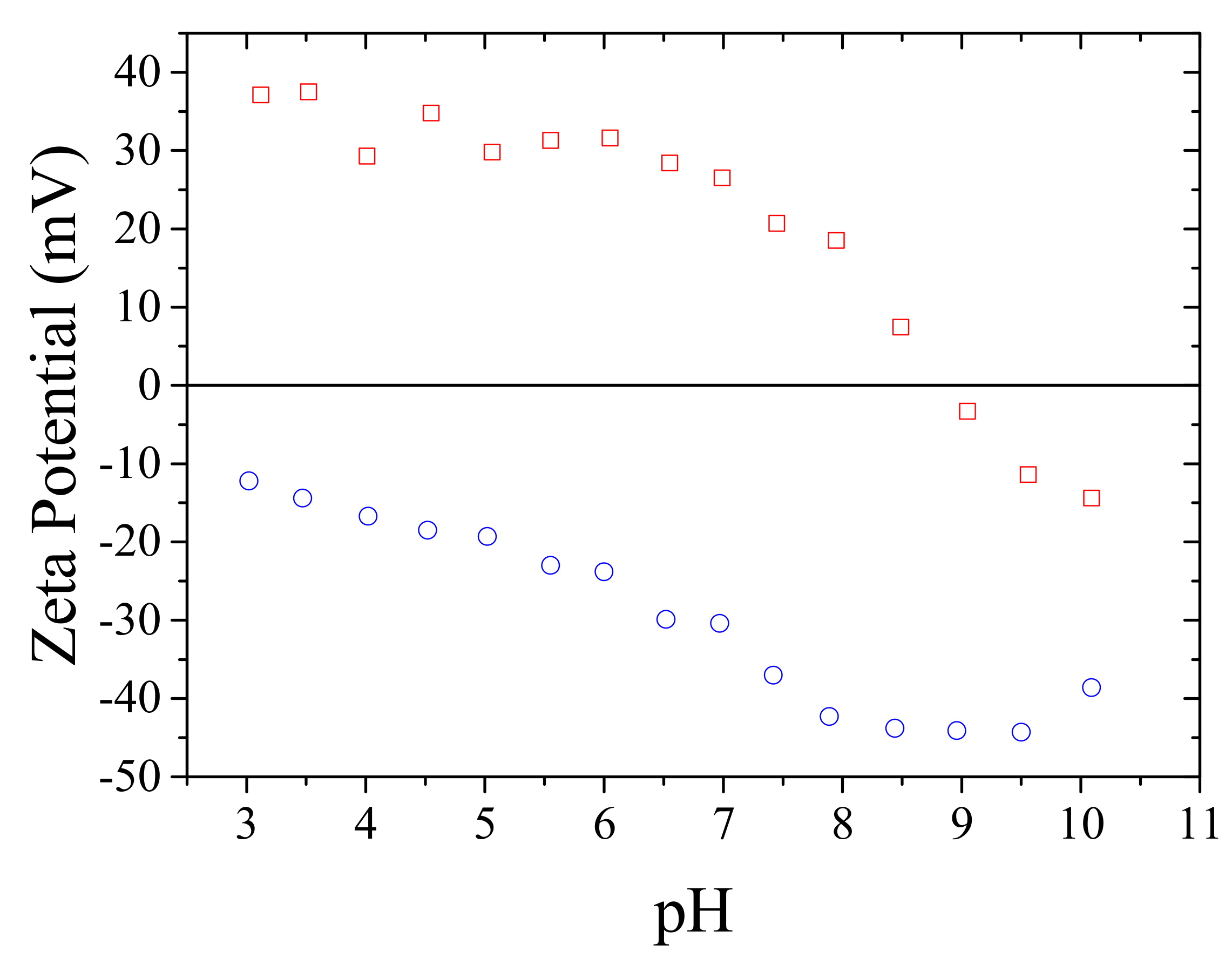
3.3. Aqueous Solution Properties of HPMAC2NP2 Homopolymer
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mou, L.; Singh, G.; Nicholson, J.W. Synthesis of a hydrophilic phosphonic acid monomer for dental materials. Chem. Commun. 2000, 5, 345–346. [Google Scholar] [CrossRef]
- Tripathi, B.P.; Shahi, V.K. Organic–inorganic nanocomposite polymer electrolyte membranes for fuel cell applications. Prog. Polym. Sci. 2011, 36, 945–979. [Google Scholar] [CrossRef]
- Tamura, Y.; Sheng, L.; Nakazawa, S.; Higashihara, T.; Ueda, M. Polymer electrolyte membranes based on polystyrenes with phosphonic acid via long alkyl side chains. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 4334–4340. [Google Scholar] [CrossRef]
- Levchik, S.V.; Weil, E.D. Overview of recent developments in the flame retardancy of polycarbonates. Polym. Int. 2005, 54, 981–998. [Google Scholar] [CrossRef]
- Levchik, S.V.; Weil, E.D. A review of recent progress in phosphorus-based flame retardants. J. Fire Sci. 2006, 24, 345–364. [Google Scholar] [CrossRef]
- Kaiser, J.; Schied, B.; Trautmann, N.; Vogt, W. Cation exchange resins based on phosphonomethyl-substituted phenols. Die Makromol. Chem. 1992, 193, 799–810. [Google Scholar] [CrossRef]
- Essahli, M.; Colomines, G.; Monge, S.; Robin, J.-J.; Collet, A.; Boutevin, B. Synthesis and characterization of ionomers based on telechelic phosphonic polyether or aromatic polyesters. Polymer 2008, 49, 4510–4518. [Google Scholar] [CrossRef]
- Monge, S.; Canniccioni, B.; Graillot, A.; Robin, J.-J. Phosphorus-containing polymers: A great opportunity for the biomedical field. Biomacromolecules 2011, 12, 1973–1982. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Nakagawa, C.; Ohtomi, M.; Ishihara, K.; Akiyoshi, K. Novel biodegradable polyphosphate cross-linker for making biocompatible hydrogel. Biomacromolecules 2004, 5, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Mao, H.-Q.; Leong, K.W. A novel biodegradable gene carrier based on polyphosphoester. J. Am. Chem. Soc. 2001, 123, 9480–9481. [Google Scholar] [CrossRef] [PubMed]
- David, G.; Asri, Z.E.; Rich, S.; Castignolles, P.; Guillaneuf, Y.; Lacroix-Desmazes, P.; Boutevin, B. Peculiar behavior of degenerative chain transfer polymerization of a phosphonated methacrylate. Macromol. Chem. Phys. 2009, 210, 631–639. [Google Scholar] [CrossRef]
- David, G.; Negrell, C.; Manseri, A.; Boutevin, B. Poly(mma)-b-poly(monophosphonic acrylate) diblock copolymers obtained by atrp and used as additives for anticorrosive coatings. J. Appl. Polym. Sci. 2009, 114, 2213–2220. [Google Scholar] [CrossRef]
- Banks, M.; Ebdon, J.R.; Johnson, M. Influence of covalently bound phosphorus-containing groups on the flammability of poly(vinyl alcohol), poly(ethylene-co-vinyl alcohol) and low-density polyethylene. Polymer 1993, 34, 4547–4556. [Google Scholar] [CrossRef]
- Kohler, J.; Keul, H.; Moller, M. Post-polymerization functionalization of linear polyglycidol with diethyl vinylphosphonate. Chem. Commun. 2011, 47, 8148–8150. [Google Scholar] [CrossRef] [PubMed]
- Tayouo, R.; David, G.; Améduri, B.; Rozière, J.; Roualdès, S. New fluorinated polymers bearing pendant phosphonic acid groups. Proton conducting membranes for fuel cell. Macromolecules 2010, 43, 5269–5276. [Google Scholar] [CrossRef]
- Suzuki, S.; Whittaker, M.R.; Grøndahl, L.; Monteiro, M.J.; Wentrup-Byrne, E. Synthesis of soluble phosphate polymers by raft and their in vitro mineralization. Biomacromolecules 2006, 7, 3178–3187. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Matyjaszewski, K. Atom transfer radical polymerization of dimethyl(1-ethoxycarbonyl)vinyl phosphate and corresponding block copolymers. Macromolecules 2005, 38, 3577–3583. [Google Scholar] [CrossRef]
- Zhou, F.; Huck, W.T.S. Three-stage switching of surface wetting using phosphate-bearing polymer brushes. Chem. Commun. 2005, 5999–6001. [Google Scholar] [CrossRef] [PubMed]
- Hua, D.; Tang, J.; Jiang, J.; Zhu, X. Synthesis of phenylphosphinic acid-containing amphiphilic homopolymers by reversible addition-fragmentation transfer (raft) polymerization and its aggregation in water. Polymer 2009, 50, 5701–5707. [Google Scholar] [CrossRef]
- Hua, D.; Tang, J.; Jiang, J.; Zhu, X.; Bai, R. A facile approach for preparation of phenylphosphinic acid-functionalized pst microspheres by emulsion polymerization using amphiphilic macro-raft agent as emulsifier. Macromolecules 2009, 42, 8697–8701. [Google Scholar] [CrossRef]
- Blidi, I.; Geagea, R.; Coutelier, O.; Mazieres, S.; Violleau, F.; Destarac, M. Aqueous raft/madix polymerisation of vinylphosphonic acid. Polym. Chem. 2012, 3, 609–612. [Google Scholar] [CrossRef]
- Mukumoto, K.; Zhong, M.; Matyjaszewski, K. Atom transfer radical polymerization of dimethyl(methacryloyloxymethyl) phosphonate. Eur. Polym. J. 2014, 56, 11–16. [Google Scholar] [CrossRef]
- Kumar, A.; Pisula, W.; Markova, D.; Klapper, M.; Müllen, K. Proton-conducting poly(phenylene oxide)–poly(vinyl benzyl phosphonic acid) block copolymers via atom transfer radical polymerization. Macromol. Chem. Phys. 2012, 213, 489–499. [Google Scholar] [CrossRef]
- Graillot, A.; Monge, S.; Faur, C.; Bouyer, D.; Robin, J.-J. Synthesis by raft of innovative well-defined (co)polymers from a novel phosphorus-based acrylamide monomer. Polym. Chem. 2013, 4, 795–803. [Google Scholar] [CrossRef]
- Canniccioni, B.; Monge, S.; David, G.; Robin, J.-J. Raft polymerization of dimethyl(methacryloyloxy)methyl phosphonate and its phosphonic acid derivative: A new opportunity for phosphorus-based materials. Polym. Chem. 2013, 4, 3676–3685. [Google Scholar] [CrossRef]
- Markova, D.; Kumar, A.; Klapper, M.; Müllen, K. Phosphonic acid-containing homo-, ab and bab block copolymers via atrp designed for fuel cell applications. Polymer 2009, 50, 3411–3421. [Google Scholar] [CrossRef]
- Keddie, D.J.; Moad, G.; Rizzardo, E.; Thang, S.H. Raft agent design and synthesis. Macromolecules 2012, 45, 5321–5342. [Google Scholar] [CrossRef]
- Gegenhuber, T.; De Keer, L.; Goldmann, A.S.; Van Steenberge, P.H.M.; Mueller, J.O.; Reyniers, M.-F.; Menzel, J.P.; D’hooge, D.R.; Barner-Kowollik, C. Fusing light-induced step-growth processes with raft chemistry for segmented copolymer synthesis: A synergetic experimental and kinetic modeling study. Macromolecules 2017, 50, 6451–6467. [Google Scholar] [CrossRef]
- Derboven, P.; Van Steenberge, P.H.; Vandenbergh, J.; Reyniers, M.F.; Junkers, T.; D’hooge, D.R.; Marin, G.B. Improved livingness and control over branching in raft polymerization of acrylates: Could microflow synthesis make the difference? Macromol. Rapid Commun. 2015, 36, 2149–2155. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H. Bisphosphonates: A review of their pharmacokinetic properties. Bone 1996, 18, 75–85. [Google Scholar] [CrossRef]
- Dyba, M.; Jezowska-Bojczuk, M.; Kiss, E.; Kiss, T.; Kozlowski, H.; Leroux, Y.; El Manouni, D. 1-hydroxyalkane-1,1-diyldiphosphonates as potent chelating agents for metal ions. Potentiometric and spectroscopic studies of copper(ii) co-ordination. J. Chem. Soc. Dalton Trans. 1996, 6, 1119–1123. [Google Scholar] [CrossRef]
- Gumienna-Kontecka, E.; Jezierska, J.; Lecouvey, M.; Leroux, Y.; Kozlowski, H. Bisphosphonate chelating agents: Coordination ability of 1-phenyl-1-hydroxymethylene bisphosphonate towards Cu2+ ions. J. Inorg. Biochem. 2002, 89, 13–17. [Google Scholar] [CrossRef]
- Mady, M.F.; Kelland, M.A. Overview of the synthesis of salts of organophosphonic acids and their application to the management of oilfield scale. Energy Fuels 2017, 31, 4603–4615. [Google Scholar] [CrossRef]
- Chougrani, K.; Boutevin, B.; David, G.; Seabrook, S.; Loubat, C. Acrylate based anticorrosion films using novel bis-phosphonic methacrylates. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 7972–7984. [Google Scholar] [CrossRef]
- Cui, J.; Zhu, C.; He, M.; Ke, Z.; Liu, Y.; Tai, Q.; Xiao, X.; Hu, Y. Preparation and thermal properties of a novel core-shell structure flame-retardant copolymer. Polym. Adv. Technol. 2018, 29, 541–550. [Google Scholar] [CrossRef]
- Vahabi, H.; Longuet, C.; Ferry, L.; David, G.; Robin, J.-J.; Lopez-Cuesta, J.-M. Effect of aminobisphosphonated copolymer on the thermal stability and flammability of poly(methyl methacrylate). Polym. Int. 2012, 61, 129–134. [Google Scholar] [CrossRef]
- Bachler, P.R.; Schulz, M.D.; Sparks, C.A.; Wagener, K.B.; Sumerlin, B.S. Aminobisphosphonate polymers via raft and a multicomponent kabachnik–fields reaction. Macromol. Rapid Commun. 2015, 36, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Chougrani, K.; Boutevin, B.; David, G.; Boutevin, G. New n,n-amino-diphosphonate-containing methacrylic derivatives, their syntheses and radical copolymerizations with mma. Eur. Polym. J. 2008, 44, 1771–1781. [Google Scholar] [CrossRef]
- Chougrani, K.; Niel, G.; Boutevin, B.; David, G. Regioselective ester cleavage during the preparation of bisphosphonate methacrylate monomers. Beilstein J. Organ. Chem. 2011, 7, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Chong, Y.K.; Krstina, J.; Le, T.P.T.; Moad, G.; Postma, A.; Rizzardo, E.; Thang, S.H. Thiocarbonylthio compounds [sc(ph)s−r] in free radical polymerization with reversible addition-fragmentation chain transfer (raft polymerization). Role of the free-radical leaving group (r). Macromolecules 2003, 36, 2256–2272. [Google Scholar] [CrossRef]
- Chong, Y.K.; Moad, G.; Rizzardo, E.; Skidmore, M.A.; Thang, S.H. Reversible addition fragmentation chain transfer polymerization of methyl methacrylate in the presence of lewis acids: An approach to stereocontrolled living radical polymerization. Macromolecules 2007, 40, 9262–9271. [Google Scholar] [CrossRef]
- Mellon, V.; Rinaldi, D.; Bourgeat-Lami, E.; D’Agosto, F. Block copolymers of γ-methacryloxypropyltrimethoxysilane and methyl methacrylate by raft polymerization. A new class of polymeric precursors for the sol−gel process. Macromolecules 2005, 38, 1591–1598. [Google Scholar] [CrossRef]
- Derboven, P.; Van Steenberge, P.H.M.; Reyniers, M.-F.; Barner-Kowollik, C.; D’Hooge, D.R.; Marin, G.B. A novel method for the measurement of degenerative chain transfer coefficients: Proof of concept and experimental validation. Polym. Chem. 2016, 7, 3334–3349. [Google Scholar] [CrossRef]
- Reichardt, C.; Welton, T. Empirical parameters of solvent polarity. In Solvents and Solvent Effects in Organic Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 425–508. [Google Scholar]
- Wood, M.R.; Duncalf, D.J.; Findlay, P.; Rannard, S.P.; Perrier, S. Investigation of the experimental factors affecting the trithiocarbonate-mediated raft polymerization of methyl acrylate. Aust. J. Chem. 2007, 60, 772–778. [Google Scholar] [CrossRef]
- Van Wazer, J.R. Phosphorus and Its Compounds; Interscience Publishers: New York, NY, USA, 1958. [Google Scholar]
- Alfrey, T.; Pinner, S.H. Preparation and titration of amphoteric polyelectrolytes. J. Polym. Sci. 1957, 23, 533–547. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Conv. (%) a | Mn b (g/mol) | Mn,th. c(g/mol) | Mw/Mn |
|---|---|---|---|---|---|
| 1 | DMF | 82 | 6800 | 9500 | 1.24 |
| 2 | CH3CN | 58 | 5300 | 6700 | 1.21 |
| 3 | THF | 34 | 3000 | 4800 | 1.14 |
| 4 | Bulk | 47 d | 4600 | 5400 | 1.21 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falireas, P.G.; Negrell, C.; David, G. Synthesis and Aqueous Solution Properties of an Amino Bisphosphonate Methacrylate Homopolymer via RAFT Polymerization. Polymers 2018, 10, 711. https://doi.org/10.3390/polym10070711
Falireas PG, Negrell C, David G. Synthesis and Aqueous Solution Properties of an Amino Bisphosphonate Methacrylate Homopolymer via RAFT Polymerization. Polymers. 2018; 10(7):711. https://doi.org/10.3390/polym10070711
Chicago/Turabian StyleFalireas, Panagiotis G., Claire Negrell, and Ghislain David. 2018. "Synthesis and Aqueous Solution Properties of an Amino Bisphosphonate Methacrylate Homopolymer via RAFT Polymerization" Polymers 10, no. 7: 711. https://doi.org/10.3390/polym10070711
APA StyleFalireas, P. G., Negrell, C., & David, G. (2018). Synthesis and Aqueous Solution Properties of an Amino Bisphosphonate Methacrylate Homopolymer via RAFT Polymerization. Polymers, 10(7), 711. https://doi.org/10.3390/polym10070711