Acetalised Galactarate Polyesters: Interplay between Chemical Structure and Polymerisation Kinetics




Abstract

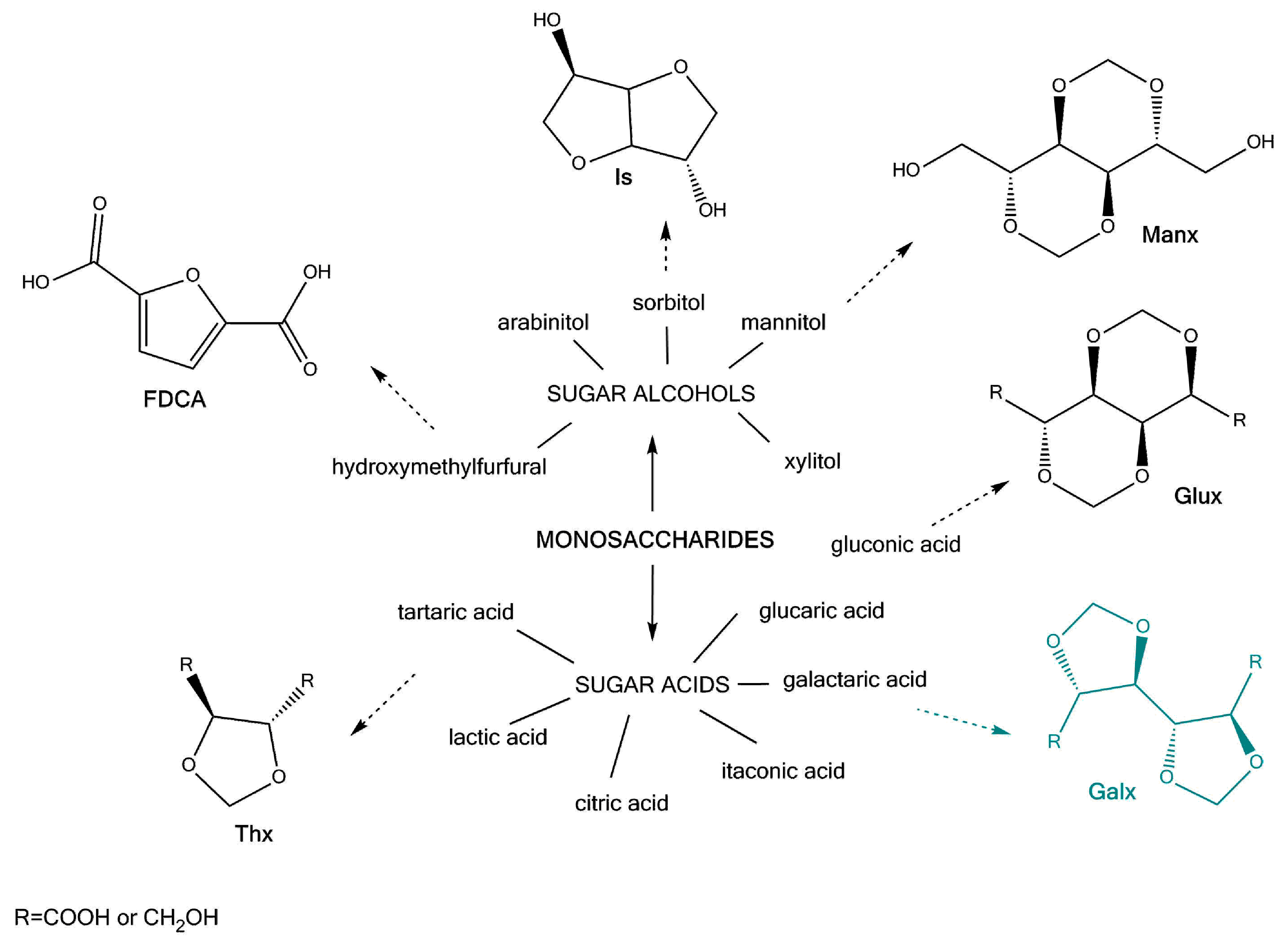
1. Introduction
2. Materials and Methods
2.1. Materials
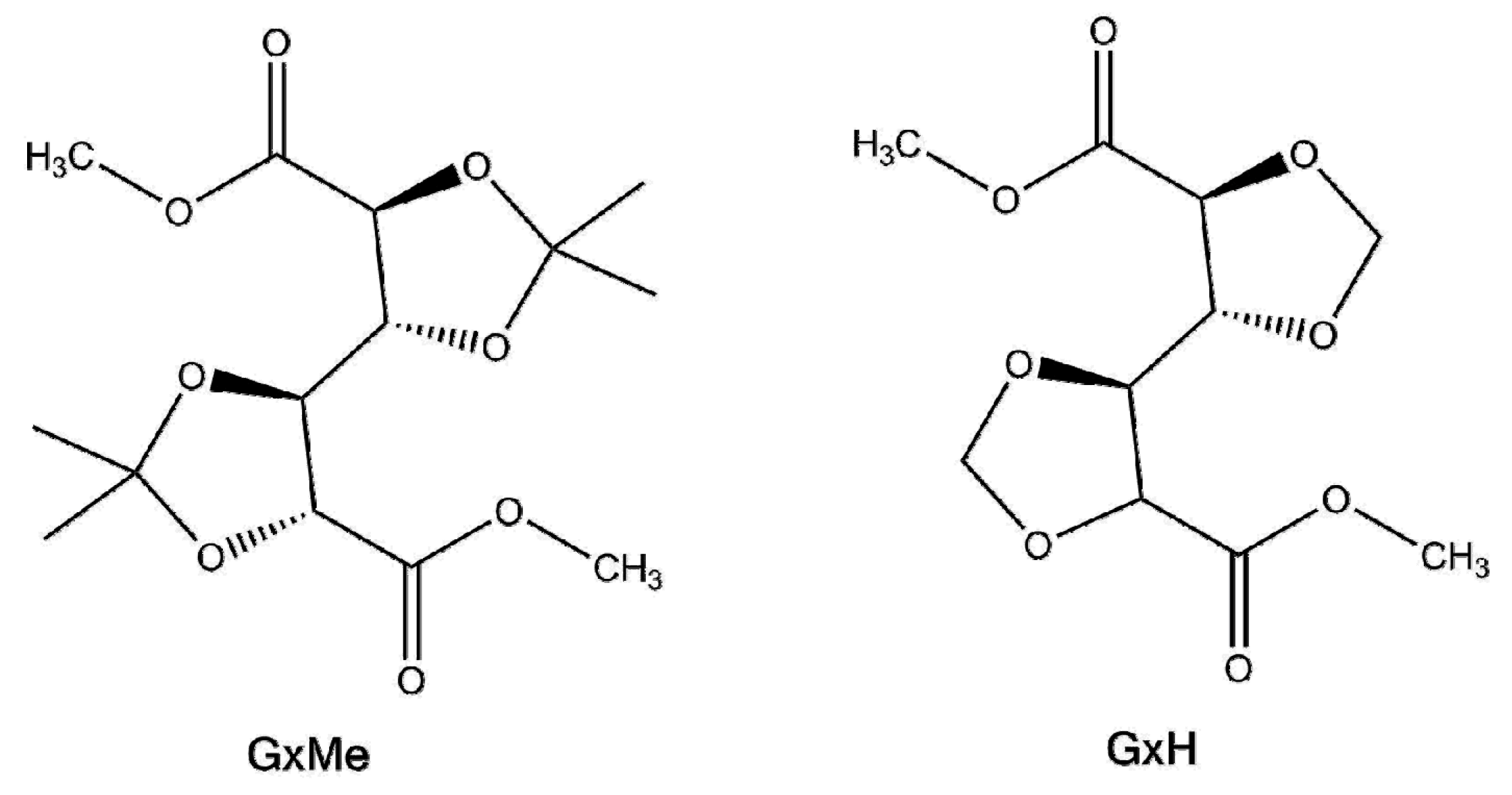
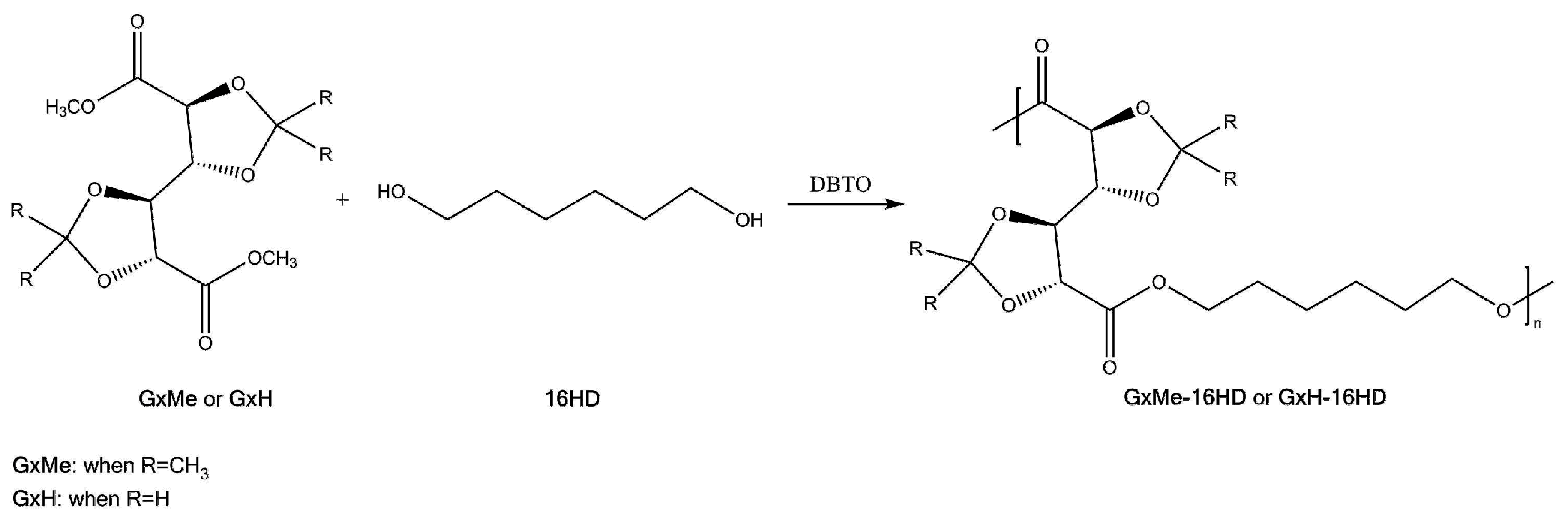
2.2. Synthesis Procedure
2.3. Equipment and Technique
3. Results and Discussion
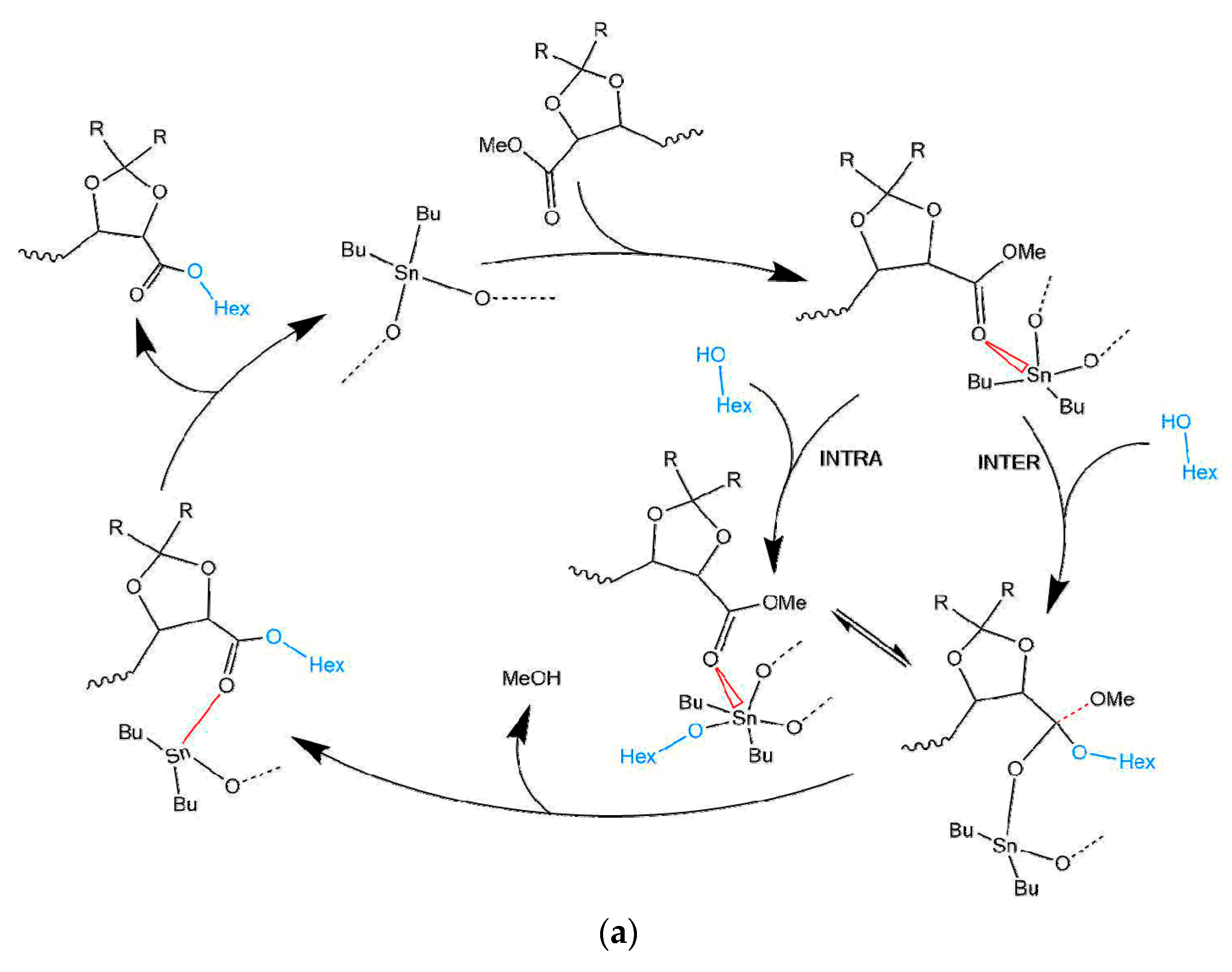
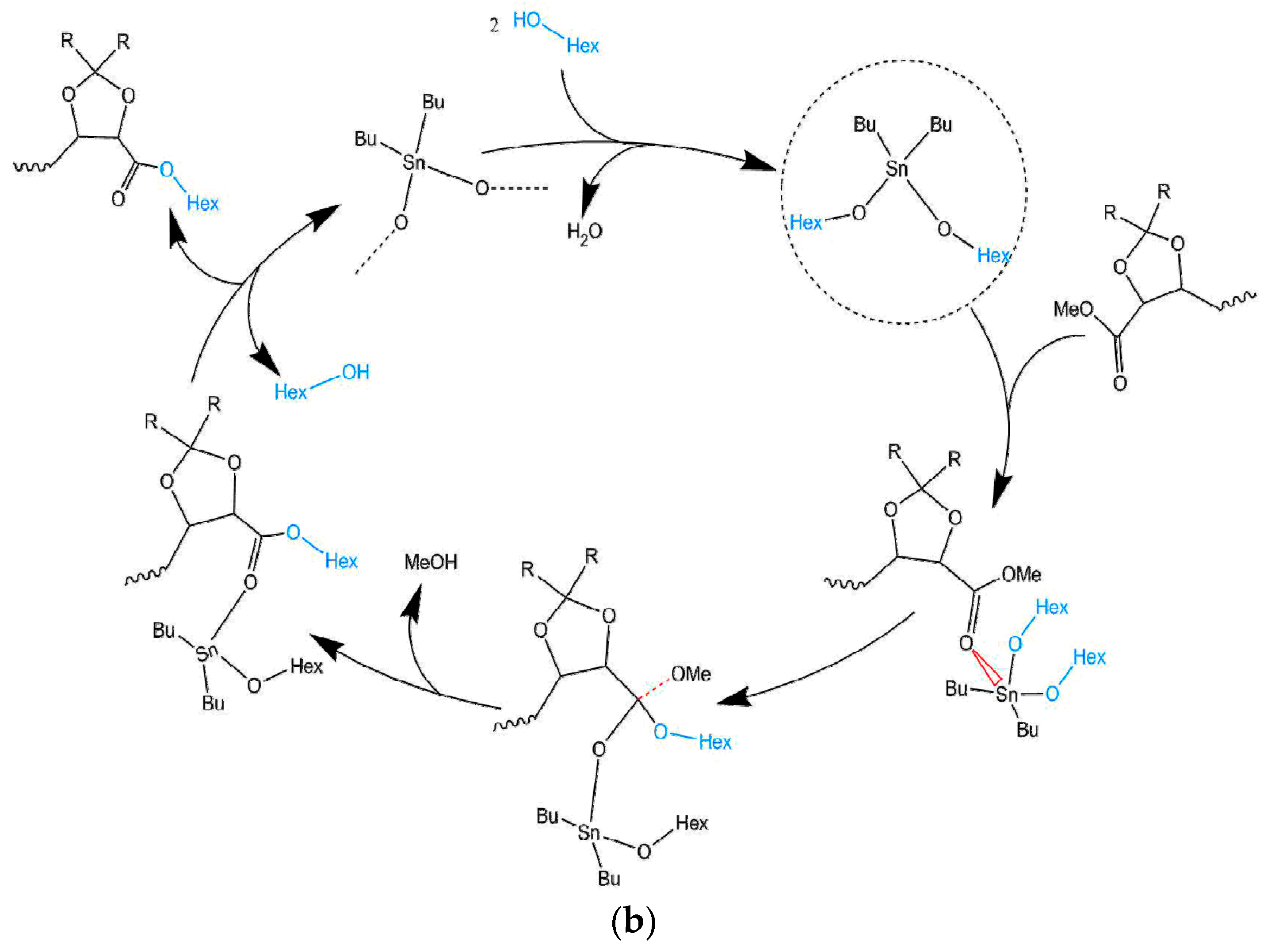
3.1. Mechanism of the DBTO Catalysed Transesterification
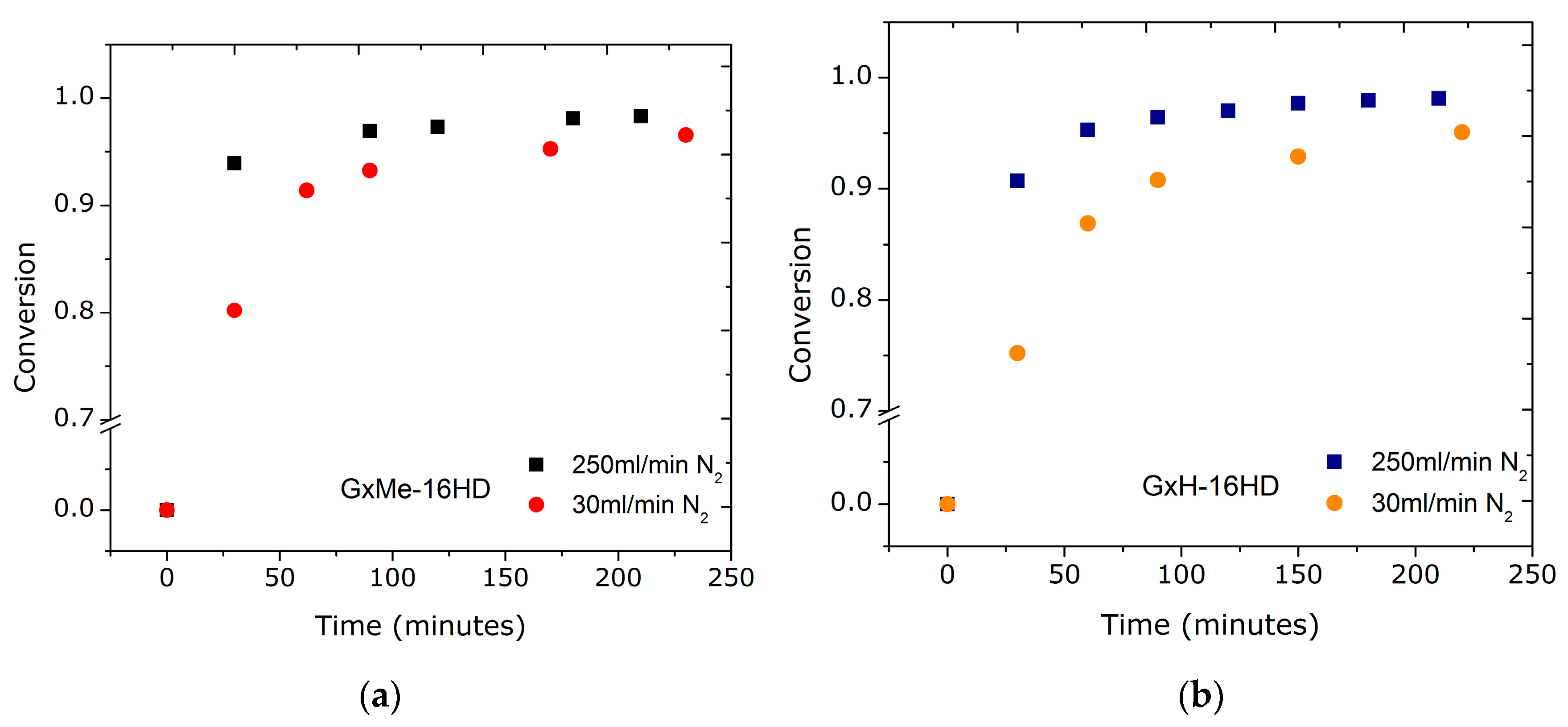
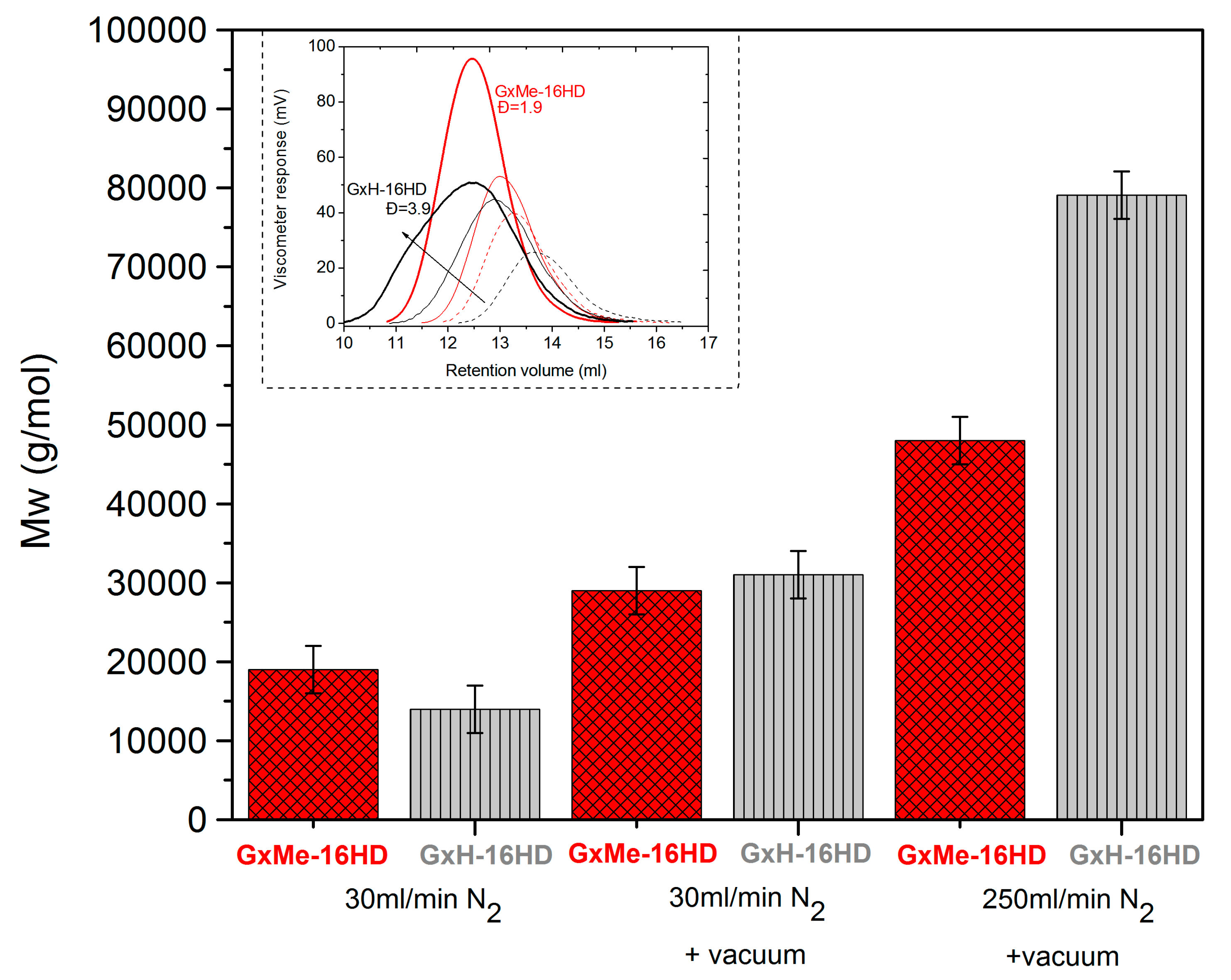
3.1.1. Effect of the N2 Flow Rate
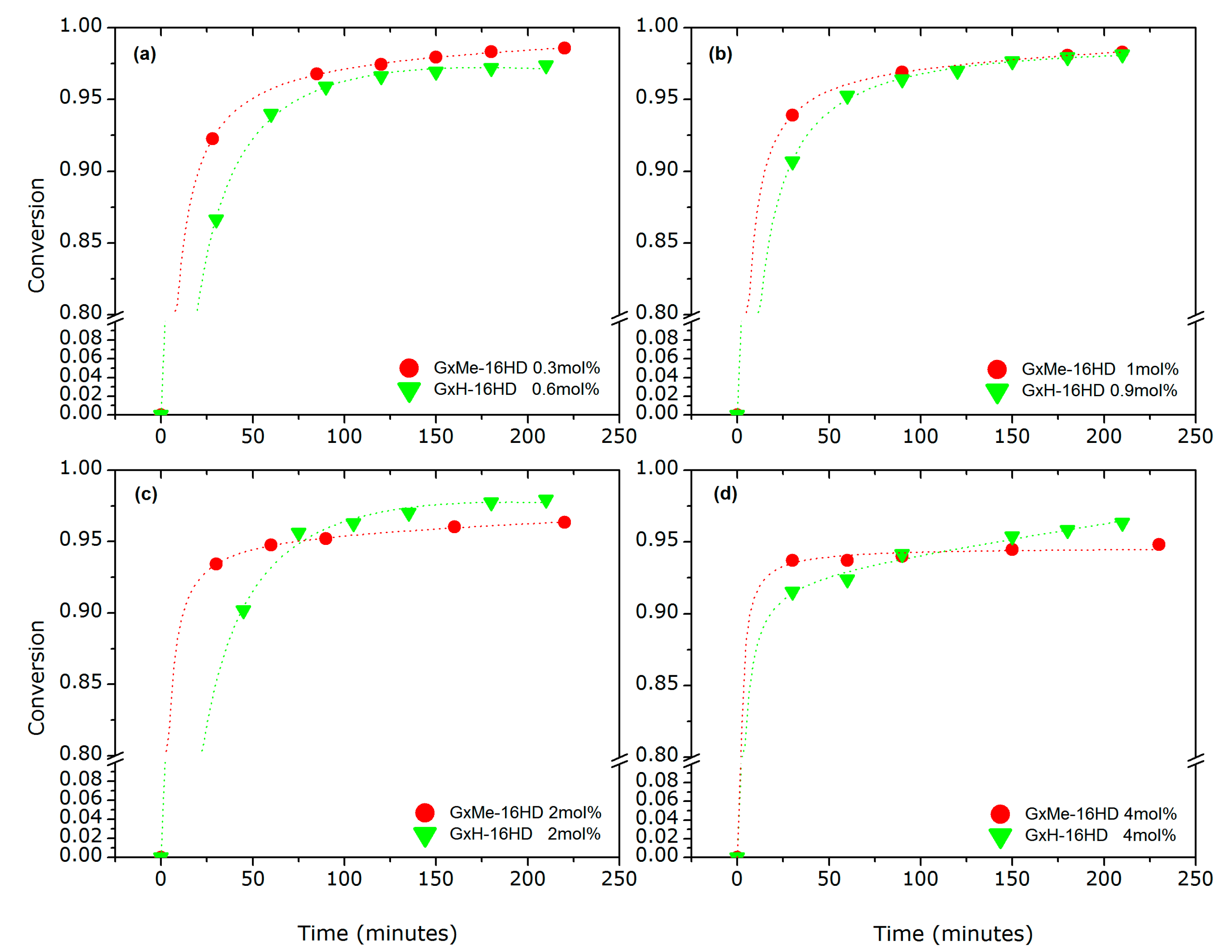
3.1.2. Variation of the Catalyst Concentration
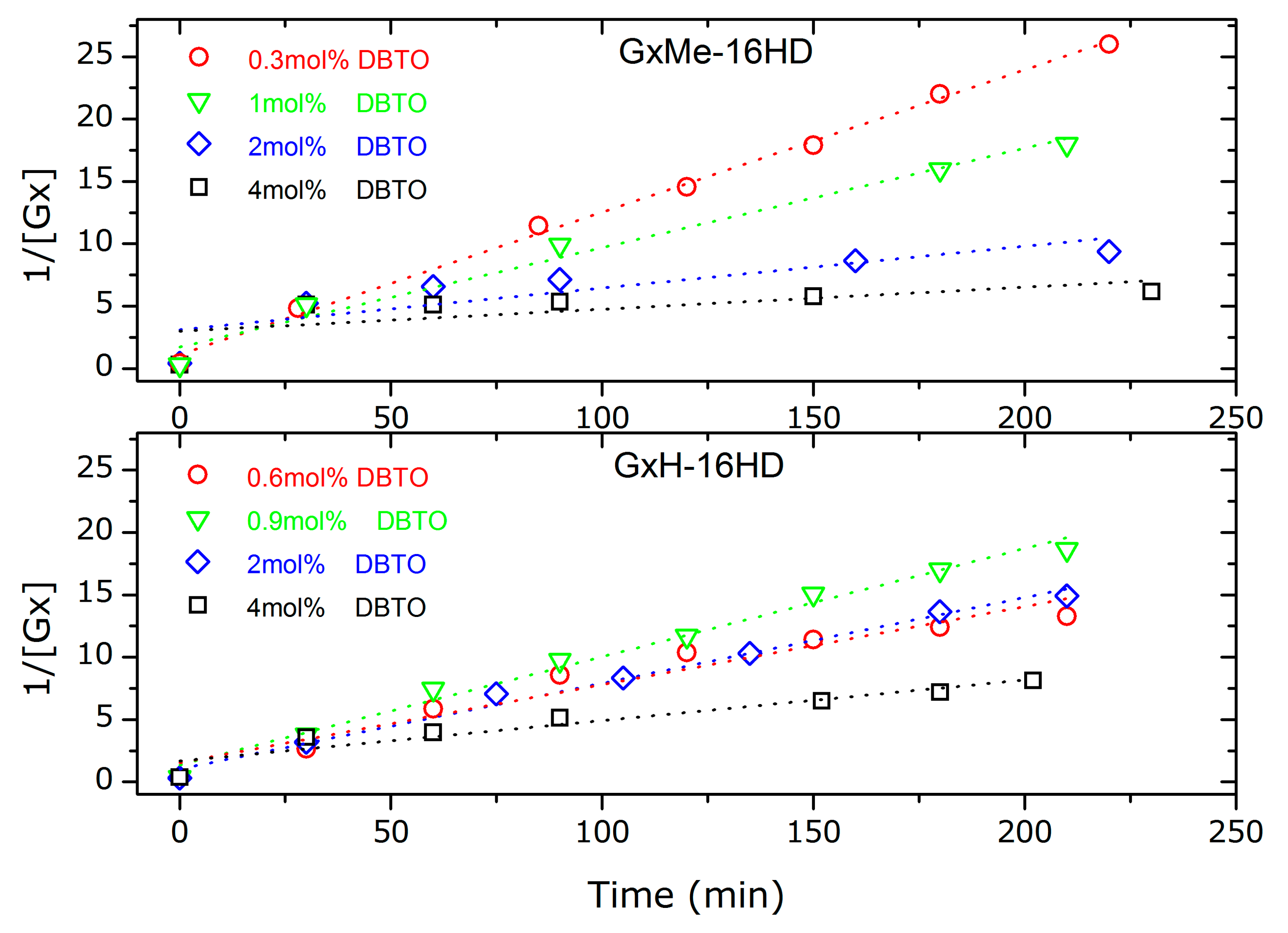
3.1.3. Analysis of the Kinetics of the Transesterification Reactions
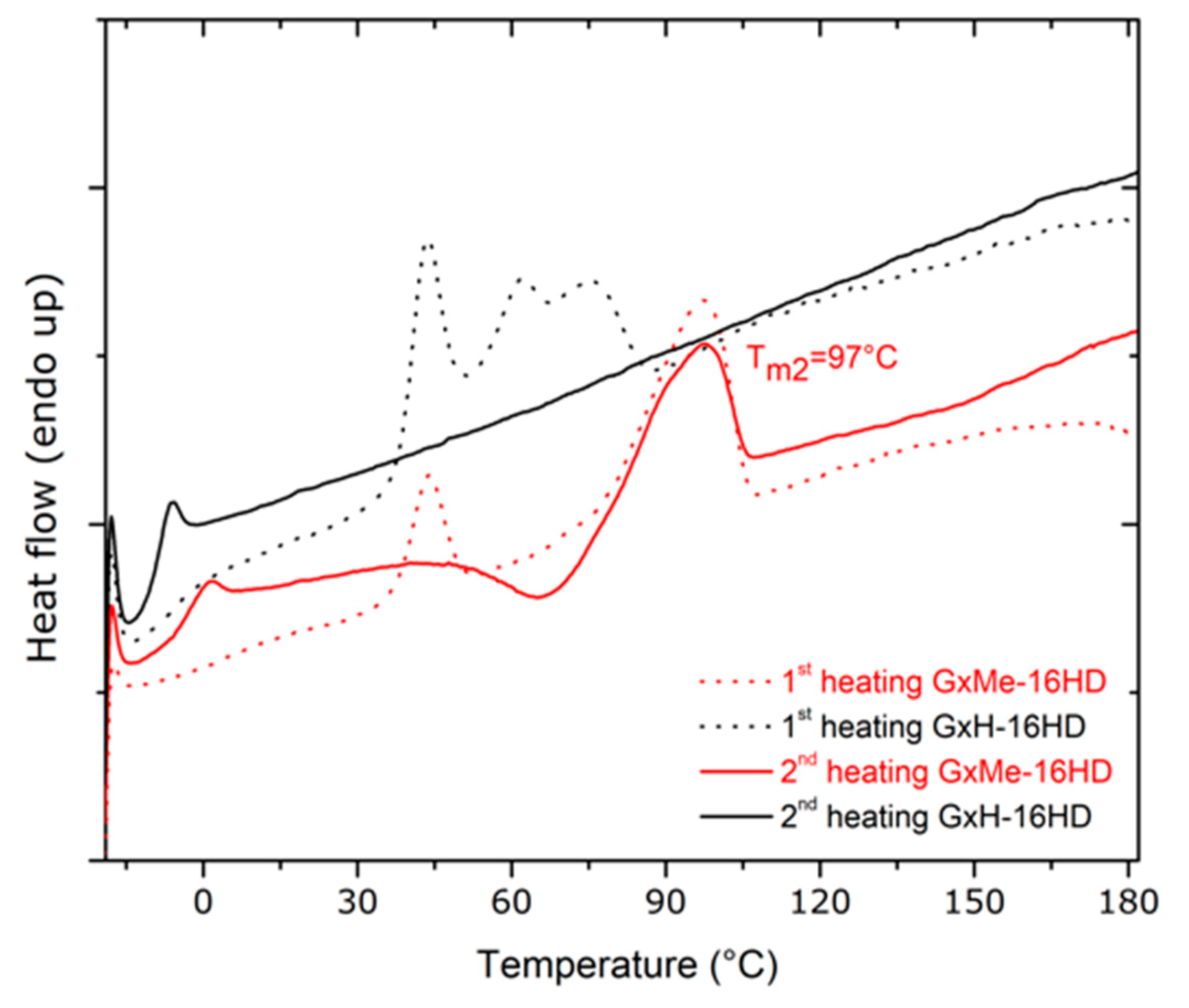
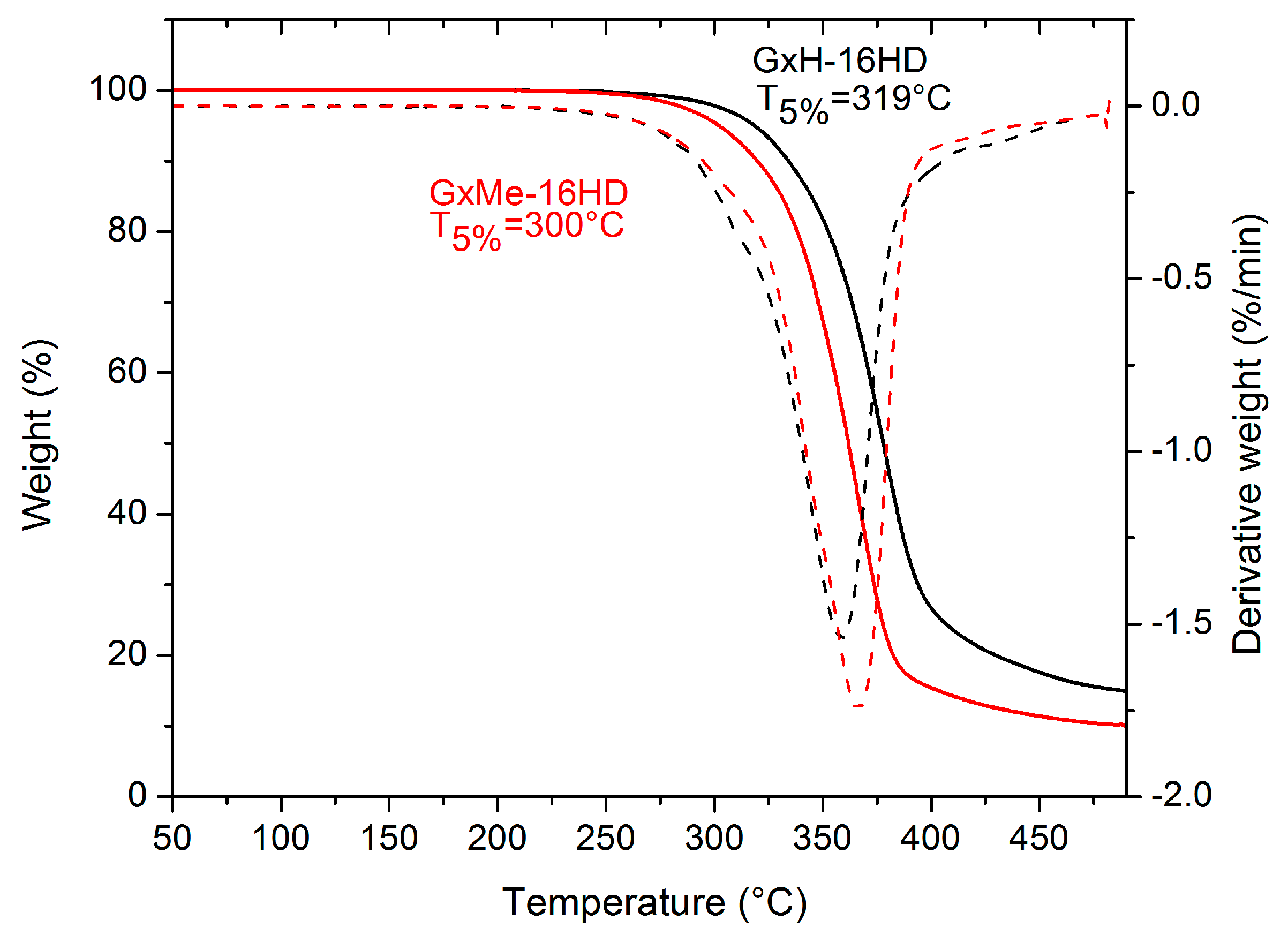
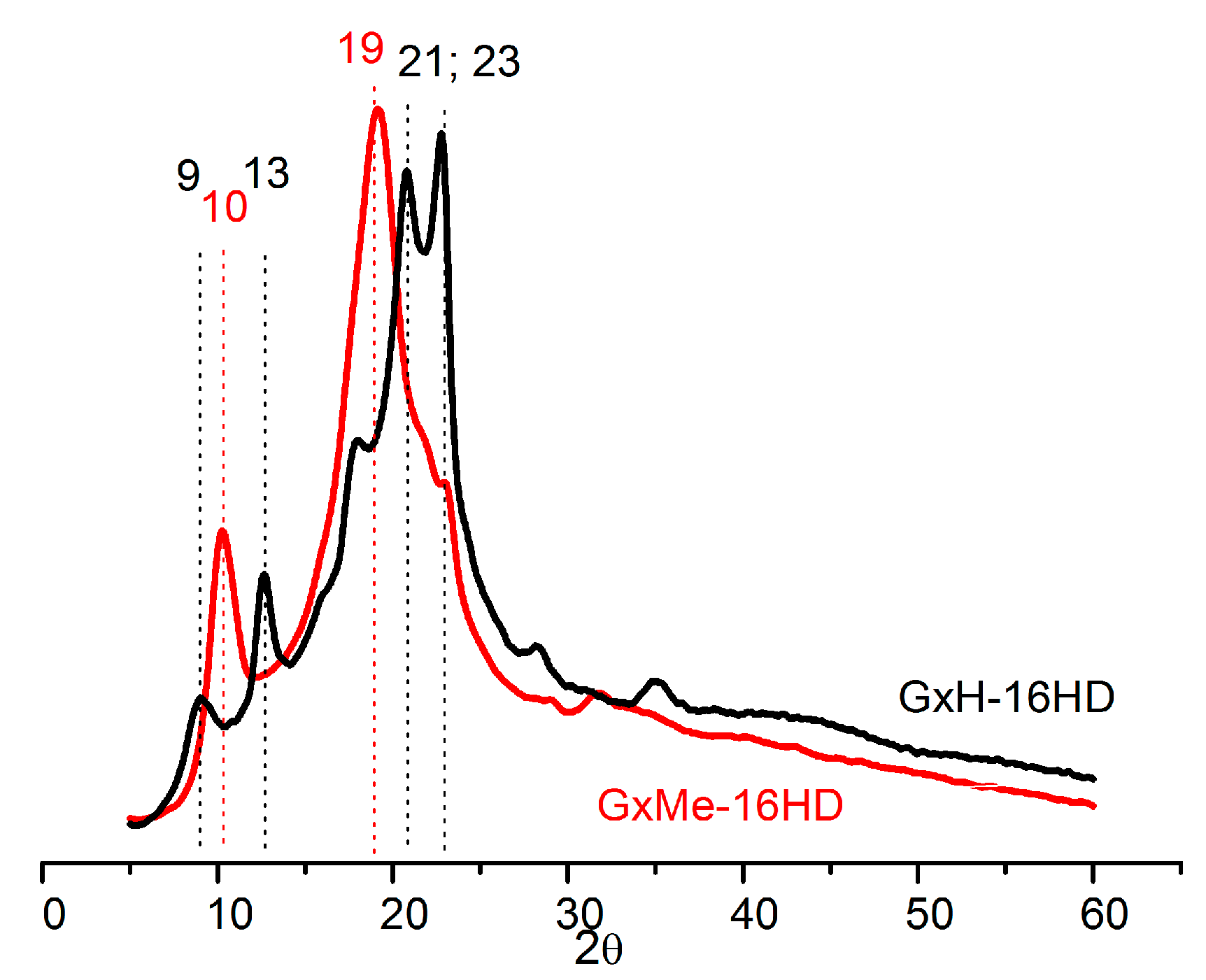
3.2. Thermal and Mechanical Characterisation
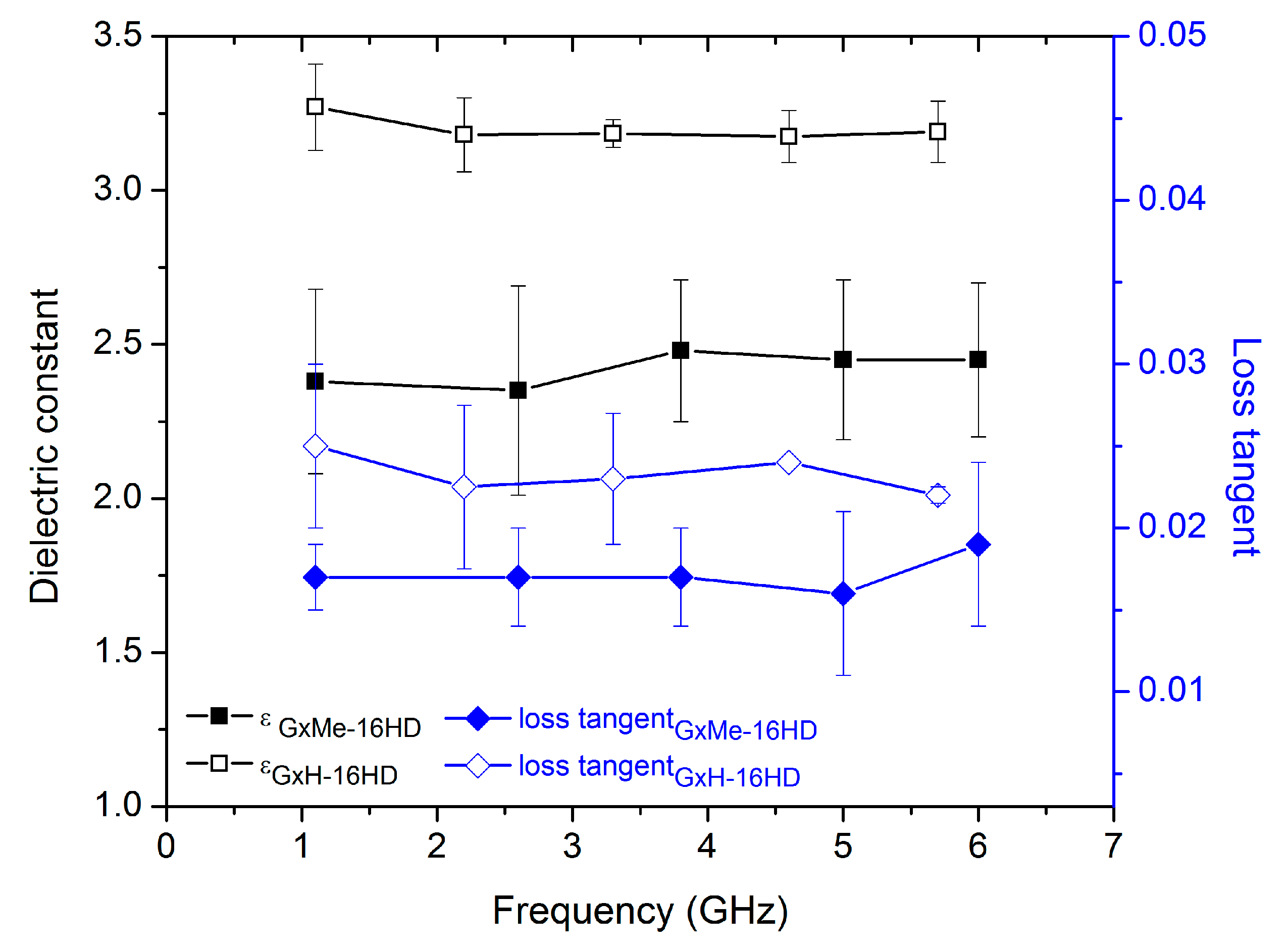
3.3. Dielectric Constant Characterisation
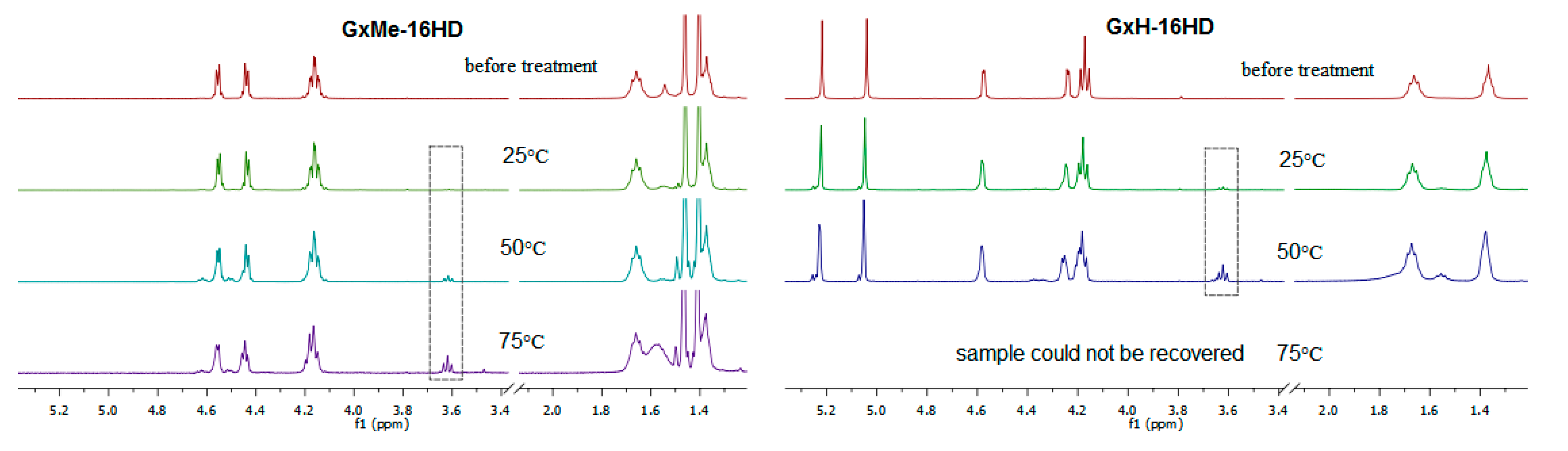
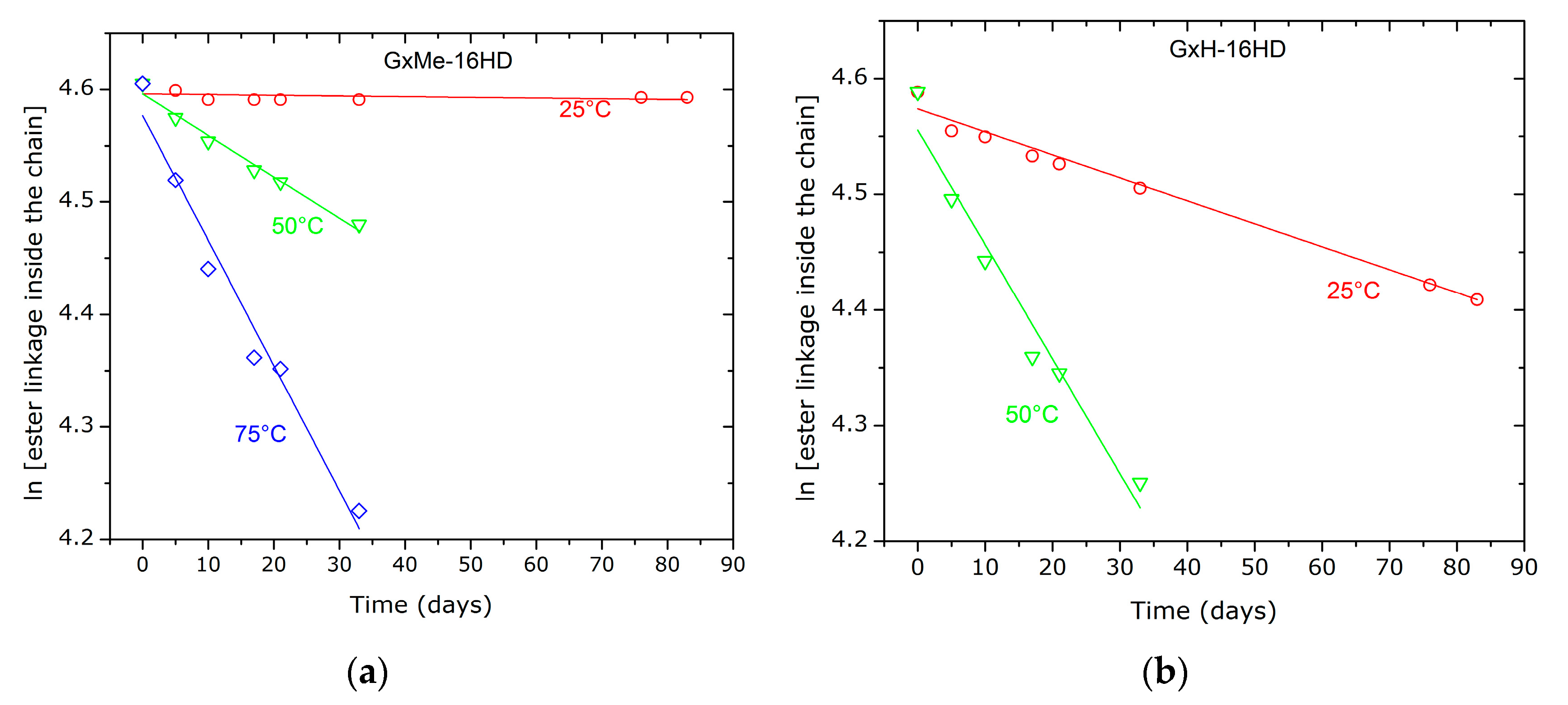
3.4. Hydrolytic Stability
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zamora, F.; Hakkou, K.; Alla, A.; Rivas, M.; Roffé, I.; Mancera, M.; Muñoz-Guerra, S.; Galbis, J.A. Aromatic polyesters from naturally occurring monosaccharides: Poly(ethylene terephthalate) and poly(ethylene isophthalate) analogs derived from D-mannitol and galactitol. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 4570–4577. [Google Scholar] [CrossRef]
- Yokoe, M.; Aoi, K.; Okada, M. Biodegradable polymers based on renewable resources. IX. Synthesis and degradation behavior of polycarbonates based on 1,4:3,6-dianhydrohexitols and tartaric acid derivatives with pendant functional groups. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 3909–3919. [Google Scholar] [CrossRef]
- Prompers, G.; Keul, H.; Hocker, H. Polyurethanes with pendant hydroxy groups: polycondensation of 1,6-bis-O-phenoxycarbonyl-2,3:4,5-di-O-isopropylidenegalactitol and 1,6-di-O-phenoxycarbonylgalactitol with diamines. Green Chem. 2006, 8, 467. [Google Scholar] [CrossRef]
- Alla, A.; Hakkou, K.; Zamora, F.; Martınez de Ilarduya, A.; Galbis, J.A.; Munoz-Guerra, S. Poly (butylene terephthalate) copolyesters derived from l-arabinitol and xylitol. Macromolecules 2006, 39, 1410–1416. [Google Scholar] [CrossRef]
- Muñoz-Guerra, S.; Lavilla, C.; Japu, C.; Martínez de Ilarduya, A. Renewable terephthalate polyesters from carbohydrate-based bicyclic monomers. Green Chem. 2014, 16, 1716. [Google Scholar] [CrossRef]
- Gandini, A. Furans as offspring of sugars and polysaccharides and progenitors of a family of remarkable polymers: A review of recent progress. Polym. Chem. 2010, 1, 245. [Google Scholar] [CrossRef]
- Fenouillot, F.; Rousseau, A.; Colomines, G.; Saint-Loup, R.; Pascault, J.-P. Polymers from renewable 1,4:3,6-dianhydrohexitols (isosorbide, isomannide and isoidide): A review. Prog. Polym. Sci. 2010, 35, 578–622. [Google Scholar] [CrossRef]
- De Jong, E.; Higson, A.; Walsh, P.; Wellisch, M. Bio-based Chemicals Value Added Products from Biorefineries; IEA BioEnergy: Paris, France, 2011. [Google Scholar]
- Aeschelmann, F.; Carus, M. Bio-based Building Blocks and Polymers in the World—Capacities, Production and Applications: Status Quo and Trends toward 2020; Nova-Institute GmbH: Hurth, Germany, 2015. [Google Scholar]
- De Jong, E.; Dam, M.A.; Sipos, L.; Gruter, G.-J.M. Furandicarboxylic Acid (FDCA), a Versatile Building Block for a Very Interesting Class of Polyesters. In Biobased Monomers, Polymers, and Materials; American Chemical Society: Washington, DC, USA, 2012; pp. 1–13. [Google Scholar]
- Weinberger, S.; Canadell, J.; Quartinello, F.; Yeniad, B.; Arias, A.; Pellis, A.; Guebitz, G. Enzymatic Degradation of Poly(ethylene 2,5-furanoate) Powders and Amorphous Films. Catalysts 2017, 7, 318. [Google Scholar] [CrossRef]
- Van Es, D.S.; van Der, K.F.; Knoop, R.J.I.; Molenveld, K.; Sijtsma, L.; Van Haveren, J. Other Polyesters from Biomass Derived Monomers. In Bio-Based Plastics—Materials and Applications; Kabasci, S., Ed.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2014; pp. 241–274. [Google Scholar]
- Lavilla, C.; Gubbels, E.; Mart, A.; Noordover, B.A.J.; Koning, C.E. Solid-State Modification of PBT with Cyclic Acetalized Galactitol and D-Mannitol: Influence of Composition and Chemical Microstructure on Thermal Properties. Macromolecules 2013, 46, 4335–4345. [Google Scholar] [CrossRef]
- Fradet, A.; Tessier, M. Polyesters. In Synthetic Methods in Step-Growth Polymers; Rogers, M.E., Long, T.E., Eds.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2003; ISBN 3175723993. [Google Scholar]
- Hu, G.; Sun, Y.; Lambla, M. Catalysis of the transesterification of poly[ethylene-co-(vinyl acetate)] by organometallic compounds in the melt. Makromol. Chem. 1993, 194, 665–675. [Google Scholar] [CrossRef]
- Lavilla, C.; Alla, A.; de Ilarduya, A.M.; Benito, E.; García-Martín, M.G.; Galbis, J.A.; Muñoz-Guerra, S. Carbohydrate-based polyesters made from bicyclic acetalized galactaric acid. Biomacromolecules 2011, 12, 2642–2652. [Google Scholar] [CrossRef] [PubMed]
- Meneghetti, M.R.; Meneghetti, S.M.P. Sn(IV)-based organometallics as catalysts for the production of fatty acid alkyl esters. Catal. Sci. Technol. 2015, 5, 765–771. [Google Scholar] [CrossRef]
- Otera, J.; Dan-oh, N.; Nozaki, H.; Catalyst, D. Novel Template Effects of Distannoxane Catalysts in Highly Efficient Transesterification and Esterification. J. Org. Chem. 1991, 56, 5307–5311. [Google Scholar] [CrossRef]
- Poller, R.C.; Retout, S.P. Organotin compounds as transesterification catalysts. J. Organomet. Chem. 1979, 173. [Google Scholar] [CrossRef]
- Lavilla, C.; Alla, A.; Martínez de Ilarduya, A.; Muñoz-Guerra, S. High T(g) bio-based aliphatic polyesters from bicyclic D-mannitol. Biomacromolecules 2013, 14, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Pell, T.M.; Davis, T.G. Diffusion and Reaction in Polyester Melts. J. Polym. Sci. Polym. Phys. 1973, 11, 1671–1682. [Google Scholar] [CrossRef]
- Shah, T.H.; Bhatty, J.I.; Gamlen, G.A.; Dollimore, D. Aspects of the chemistry of poly(ethylene terephthalate): 5. Polymerization of bis(hydroxyethyl)terephthalate by various metallic catalysts. Polymer (Guildf) 1984, 25, 1333–1336. [Google Scholar] [CrossRef]
- Lu, T.; Sun, Y.; Wang, C. Novel Copolyesters Containing Naphthalene Structure, II. Copolyesters Prepared from 2,6-Dimethyl Naphthalate, 1,4-Dimethyl Terephthalate, and Ethylene Glycol. J. Polym. Sci. Part A Polym. Chem. 1995, 33, 2841–2850. [Google Scholar] [CrossRef]
- Park, S.S.; Im, S.S.; Kim, D.K. Kinetics for the catalyzed fromation of Poly(ethylene 2,6-naphtalate) (PEN) by various metal compounds. J. Polym. Sci. Part A Polym. Chem. 1994, 32, 2873–2881. [Google Scholar] [CrossRef]
- Jacquel, N.; Freyermouth, F.; Fenouillot, F.; Rousseau, A.; Pascault, J.P.; Fuertes, P.; Saint-Loup, R. Synthesis and properties of poly(butylene succinate): Efficiency of different transesterification catalysts. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 5301–5312. [Google Scholar] [CrossRef]
- Ferreira, A.B.; Lemos Cardoso, A.; da Silva, M.J. Tin-Catalyzed Esterification and Transesterification Reactions: A Review. ISRN Renew. Energy 2012, 2012, 1–13. [Google Scholar] [CrossRef]
- Wang, C.; Sun, Y. Studies on the Formation of PEN. II. Polymerization of Bis (hydroxyethyl ) naphthalate by Various Metallic Catalysts. J. Polym. Sci. Part A Polym. Chem. 1994, 32, 1305–1315. [Google Scholar] [CrossRef]
- Ratner, B.D. Comprehensive Polymer Science: The synthesis, Characterization, Reactions and Applications of Polymers; Pergamon Press: Oxford, UK, 1989; Volume 7, pp. 202–203. [Google Scholar]
- Pilati, F. Polyesters. In Comprehensive Polymer Science and Supplements; Pergamon Press: Oxford, UK, 1989; pp. 275–315. ISBN 9780080967011. [Google Scholar]
- Duda, A.; Penczek, S. Mechanisms of Aliphatic Polyester Formation. In Biopolymers Online; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2005; pp. 371–383. ISBN 9783527600038. [Google Scholar]
- Lavilla, C.; Alla, A.; Martínez de Ilarduya, A.; Benito, E.; García-Martín, M.G.; Galbis, J.A.; Muñoz-Guerra, S. Biodegradable aromatic copolyesters made from bicyclic acetalized galactaric acid. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 3393–3406. [Google Scholar] [CrossRef]
- Allcock, H.R. Introduction to Materials Chemistry; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2008; ISBN 978-0-470-29333-1. [Google Scholar]
- Ryan, A.J.; Bras, W.; Mant, G.R.; Derbyshire, G.E. A direct method to determine the degree of crystallinity and lamellar thickness of polymers: application to polyethylene. Polymer 1994, 35, 4537–4544. [Google Scholar] [CrossRef]
- Van Krevelen, D.W. Properties of Polymers, 4th ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2009; ISBN 9780080548197. [Google Scholar]
- Ahmad, Z. Polymeric Dielectric Materials. In Dielectric Material; InTech Open Access Publisher: Rijeka, Croatia, 2012. [Google Scholar]
- Ramani, R.; Ramachandran, R.; Amarendra, G.; Alam, S. Direct correlation between free volume and dielectric constant in a fluorine-containing polyimide blend. J. Phys. Conf. Ser. 2015, 618, 12025. [Google Scholar] [CrossRef]
- Giants, T.W. Crystallinity and Dielectric Properties of PEEK, a Thermoplastic Poly(ether ether ketone). IEEE Trans. Dielectr. Electr. Insul. 1994, 1. [Google Scholar] [CrossRef]
- Deslongchamps, P.; Dory, Y.L.; Li, S. The relative rate of hydrolysis of a series of acyclic and six-membered cyclic acetals, ketals, orthoesters, and orthocarbonates. Tetrahedron 2000, 56, 3533–3537. [Google Scholar] [CrossRef]
- Arias, V.; Olsen, P.; Odelius, K.; Hoglund, A.; Albertsson, A.C. Forecasting linear aliphatic copolyester degradation through modular block design. Polym. Degrad. Stab. 2016, 130, 58–67. [Google Scholar] [CrossRef]
- Díaz, A.; Katsarava, R.; Puiggalí, J. Synthesis, properties and applications of biodegradable polymers derived from diols and dicarboxylic acids: from polyesters to poly(ester amide)s. Int. J. Mol. Sci. 2014, 15, 7064–7123. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Noordover, B.A.J.; Sablong, R.J.; Koning, C.E. Thermoplastic poly(urethane urea)s from novel, bio-based amorphous polyester diols. Macromol. Chem. Phys. 2012, 213, 2541–2549. [Google Scholar] [CrossRef]
- Ignatov, V.N.; Pilati, F.; Berti, C.; Tartari, V.; Nadali, G.; Fiorini, M.; Toselli, M. PET Synthesis in the Presence of Lanthanide Catalysts. J. Appl. Polym. Sci. 1995, 58, 771–777. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | [DBTO] | ||||||
|---|---|---|---|---|---|---|---|
| 0.3 mol % | 0.6 mol % | 0.9 mol % | 1 mol % | 2 mol % | 4 mol % | ||
| GxMe-16HD | ka′ (L/mol s) R2 fit | 0.110 0.996 | - - | - - | 0.071 0.974 | 0.021 0.688 | 0.006 0.344 |
| GxH-16HD | ka′ (L/mol s) R2 fit | - - | 0.063 0.938 | 0.087 0.988 | - | 0.069 0.986 | 0.033 0.910 |
| Sample | Tg, * °C | Tm *, °C | Melting Enthalpy ΔH *, J/g | Crystallinity **, % | Contact Angle, deg. | Elastic Modulus, GPa *** |
|---|---|---|---|---|---|---|
| GxMe-16HD | −3 | 97 | 19.5 | 44 | 86 | 0.15 ± 0.025 |
| GxH-16HD | −8 | - | - | 35 | 75 | 0.1 ± 0.02 |
| Entry | Dielectric Constant | |
|---|---|---|
| Amorphous | Semicrystalline | |
| GxMe-16HD | 2.74 | 2.67 |
| GxH-16HD | 3.24 | 3.15 |
| Temperature | khydrolysis, 1/s | |
|---|---|---|
| GxMe-16HD | GxH-16HD | |
| 25 °C | 6.28 × 10−5 | 1.98 × 10−3 |
| 50 °C | 3.69 × 10−3 | 9.9 × 10−3 |
| 75 °C | 1.11 × 10−2 | n.a. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gavrila, I.; Raffa, P.; Picchioni, F. Acetalised Galactarate Polyesters: Interplay between Chemical Structure and Polymerisation Kinetics. Polymers 2018, 10, 248. https://doi.org/10.3390/polym10030248
Gavrila I, Raffa P, Picchioni F. Acetalised Galactarate Polyesters: Interplay between Chemical Structure and Polymerisation Kinetics. Polymers. 2018; 10(3):248. https://doi.org/10.3390/polym10030248
Chicago/Turabian StyleGavrila, Ionela, Patrizio Raffa, and Francesco Picchioni. 2018. "Acetalised Galactarate Polyesters: Interplay between Chemical Structure and Polymerisation Kinetics" Polymers 10, no. 3: 248. https://doi.org/10.3390/polym10030248
APA StyleGavrila, I., Raffa, P., & Picchioni, F. (2018). Acetalised Galactarate Polyesters: Interplay between Chemical Structure and Polymerisation Kinetics. Polymers, 10(3), 248. https://doi.org/10.3390/polym10030248