State of the Art in Dual-Curing Acrylate Systems



Abstract
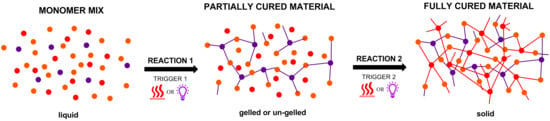
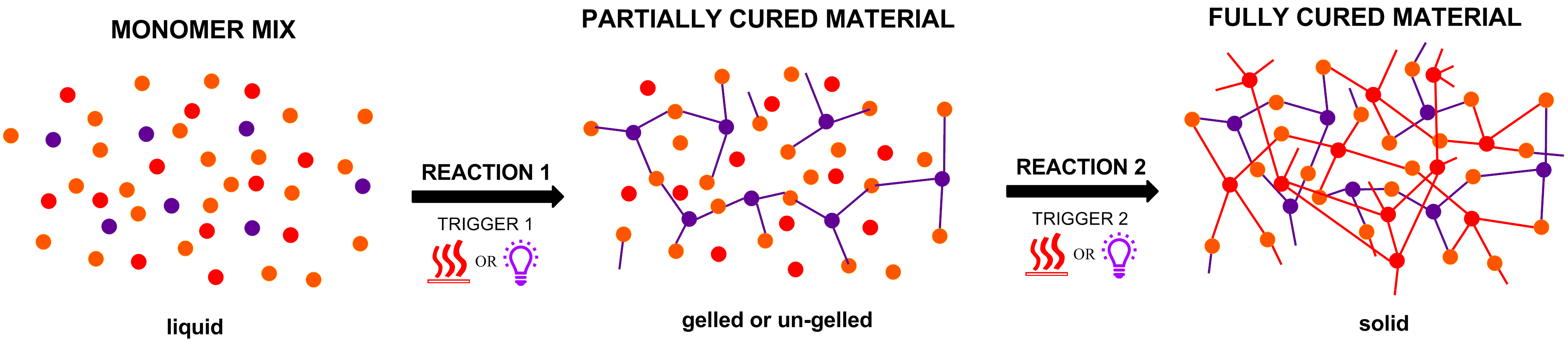{kind=link}
{kind=link}
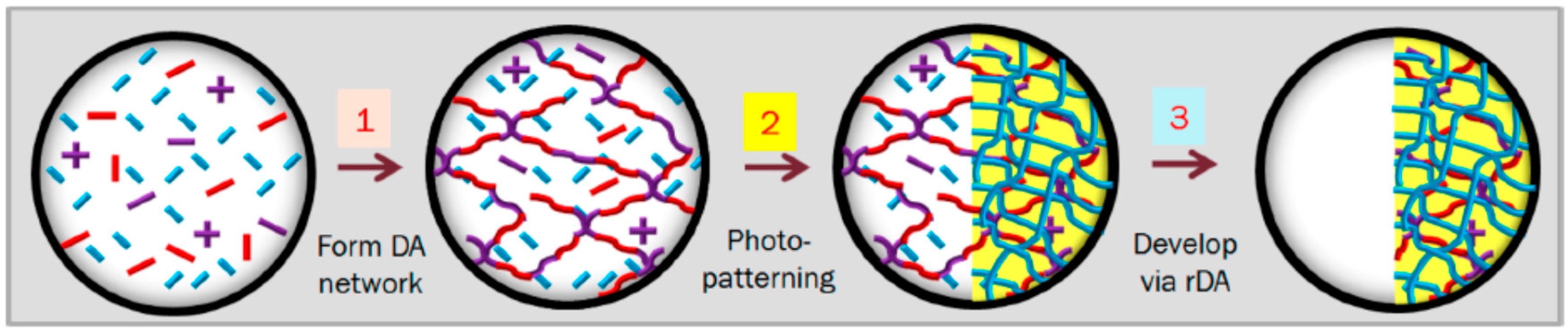{kind=link}
{kind=link}
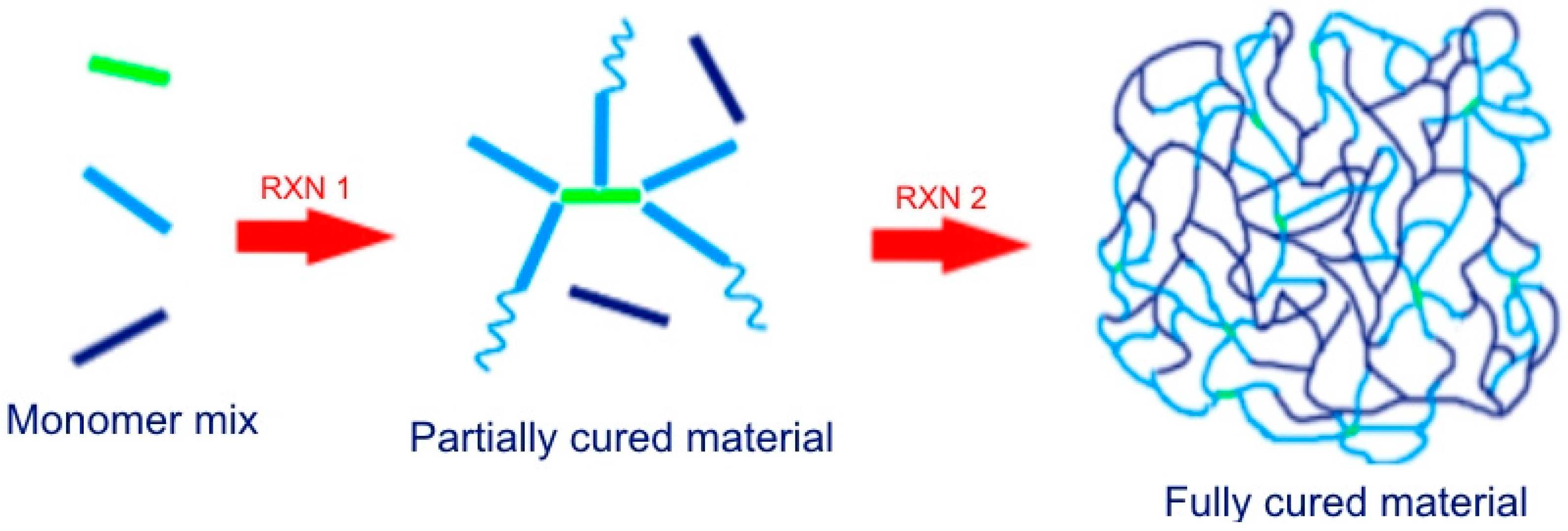{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
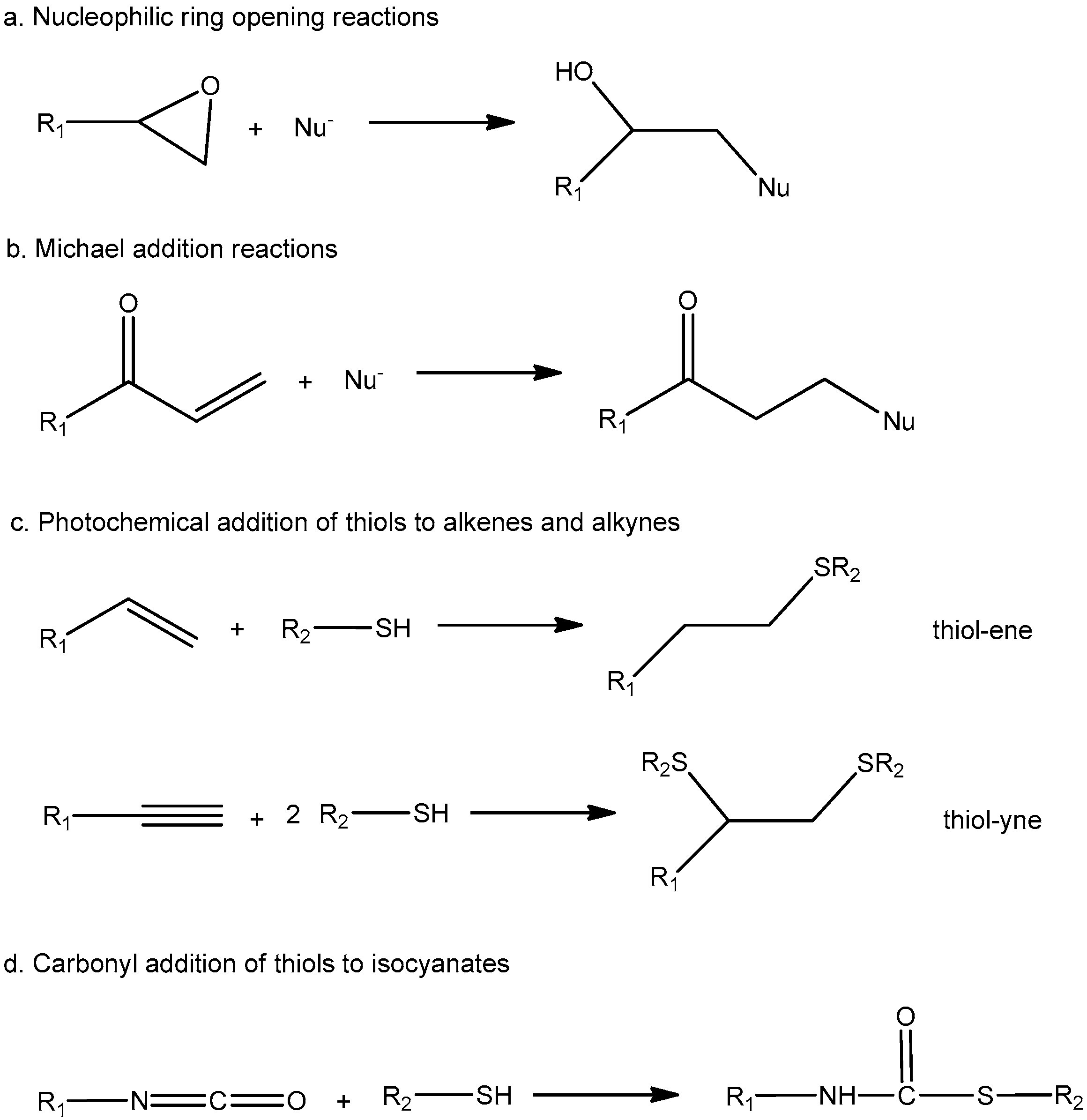
2. Acrylate Reactions in Dual-Curing Systems
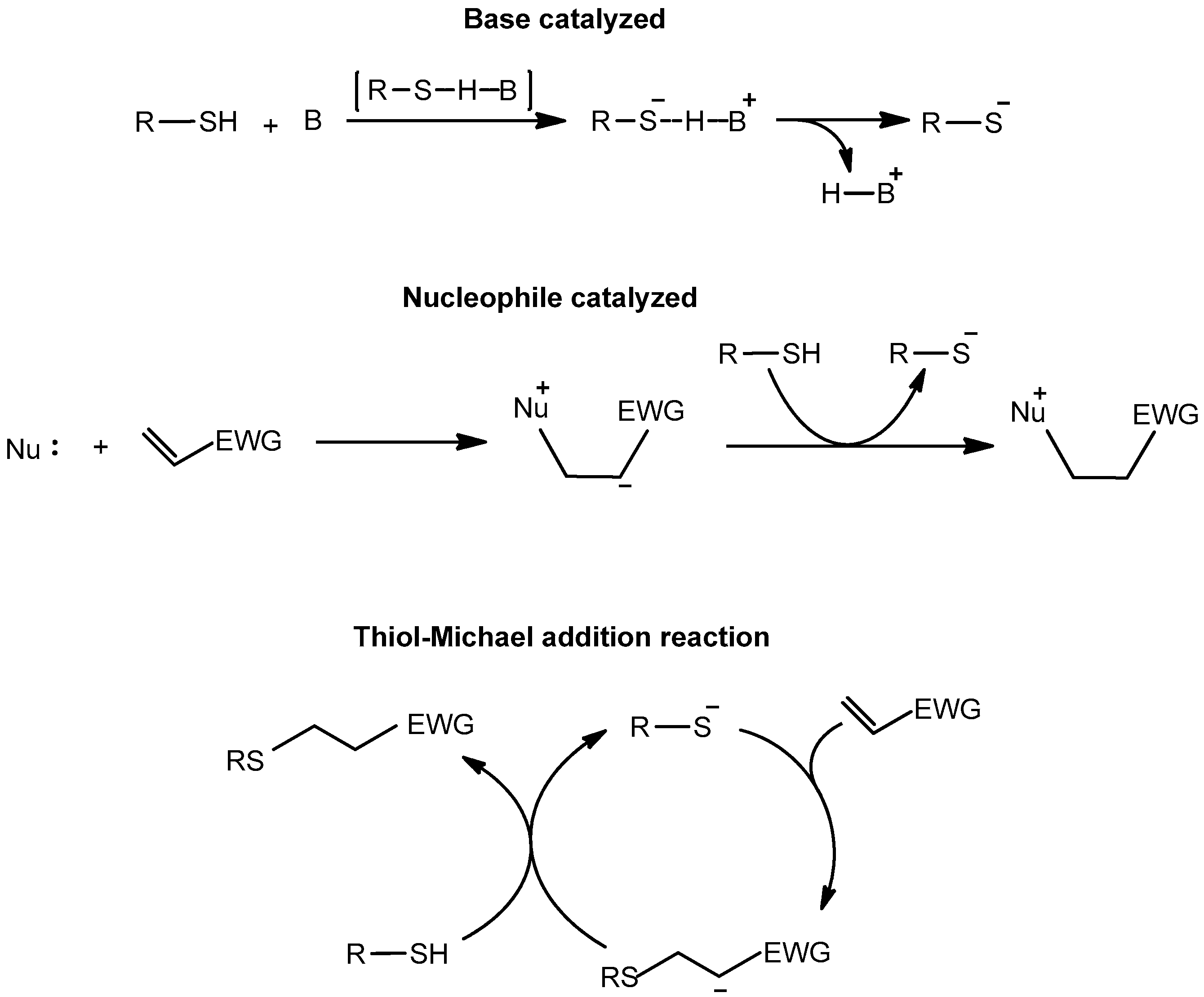
2.1. Thiol–Acrylate Reaction
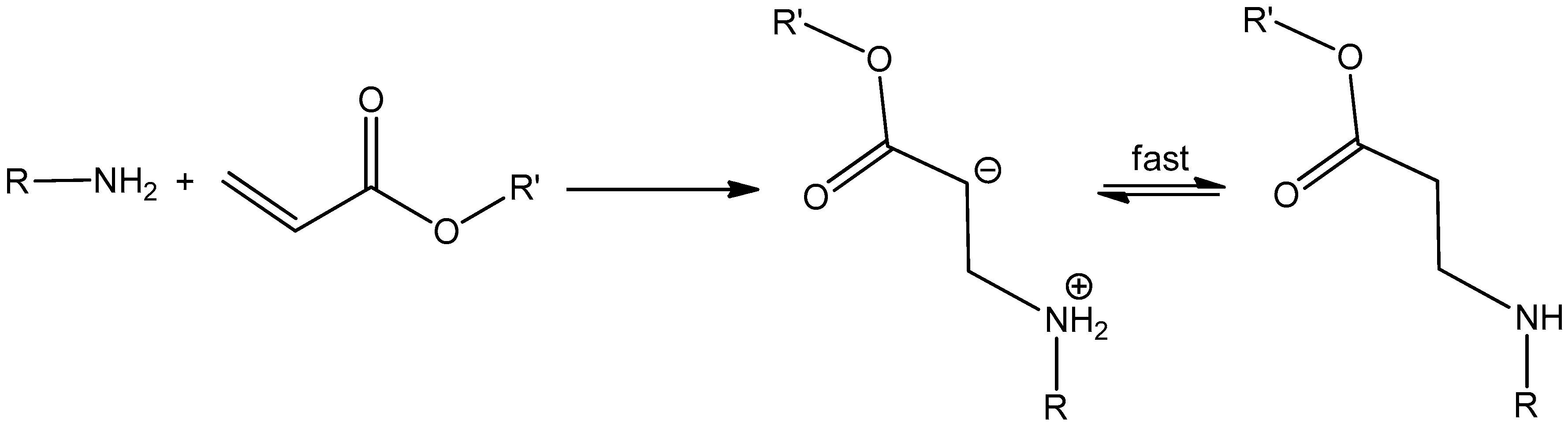
2.2. Aza-Michael Addition
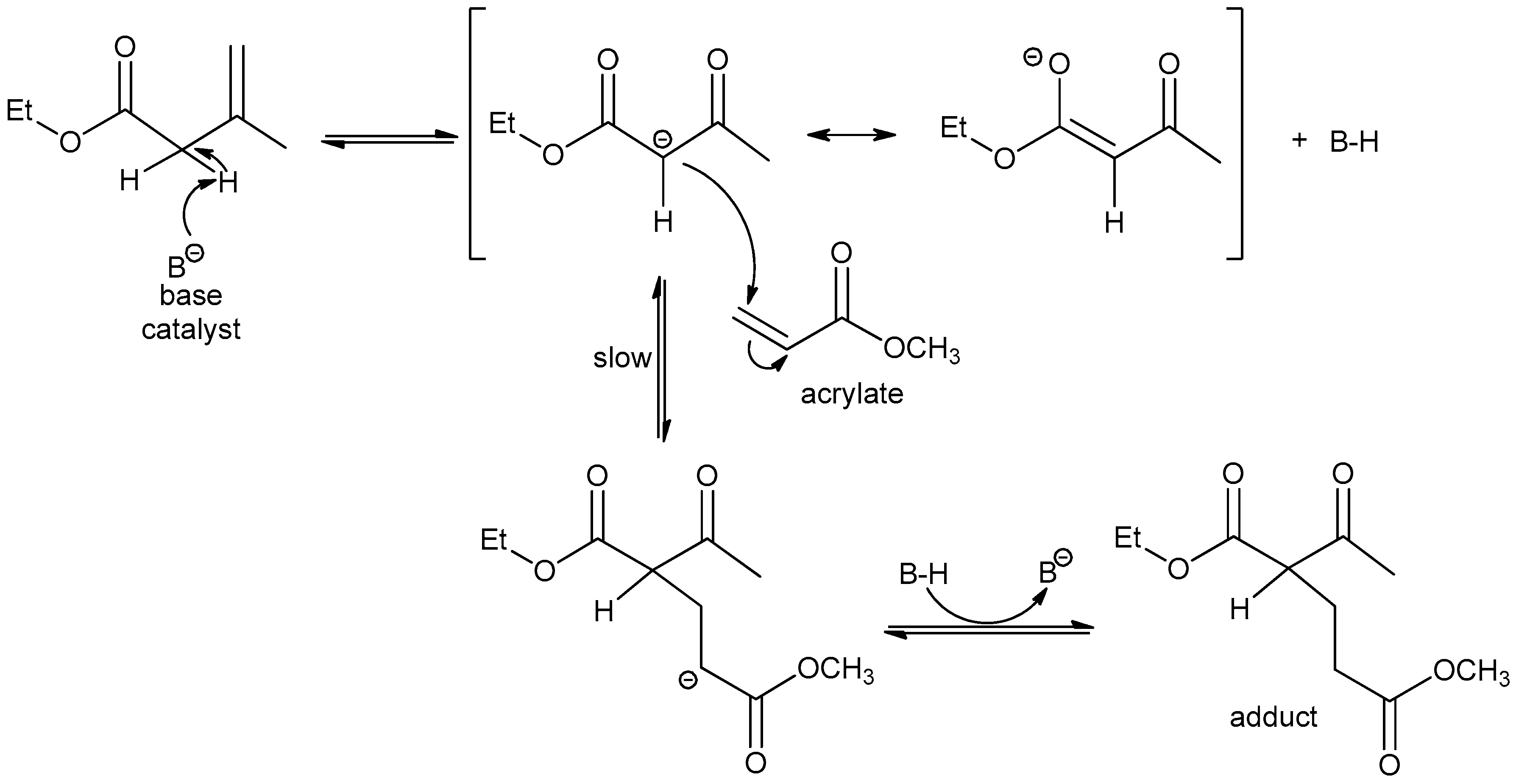
2.3. Acetoacetate–Acrylate Reaction
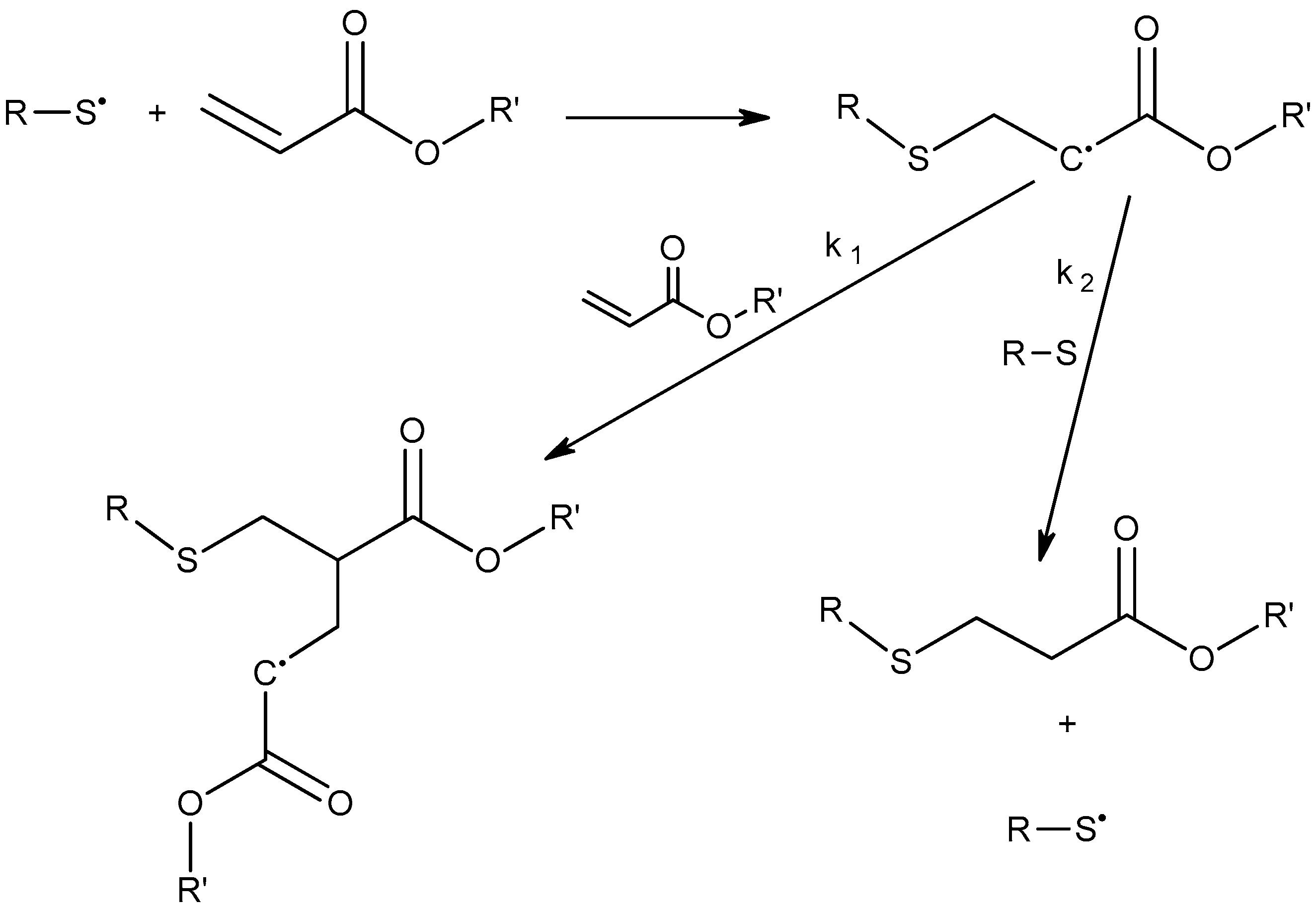
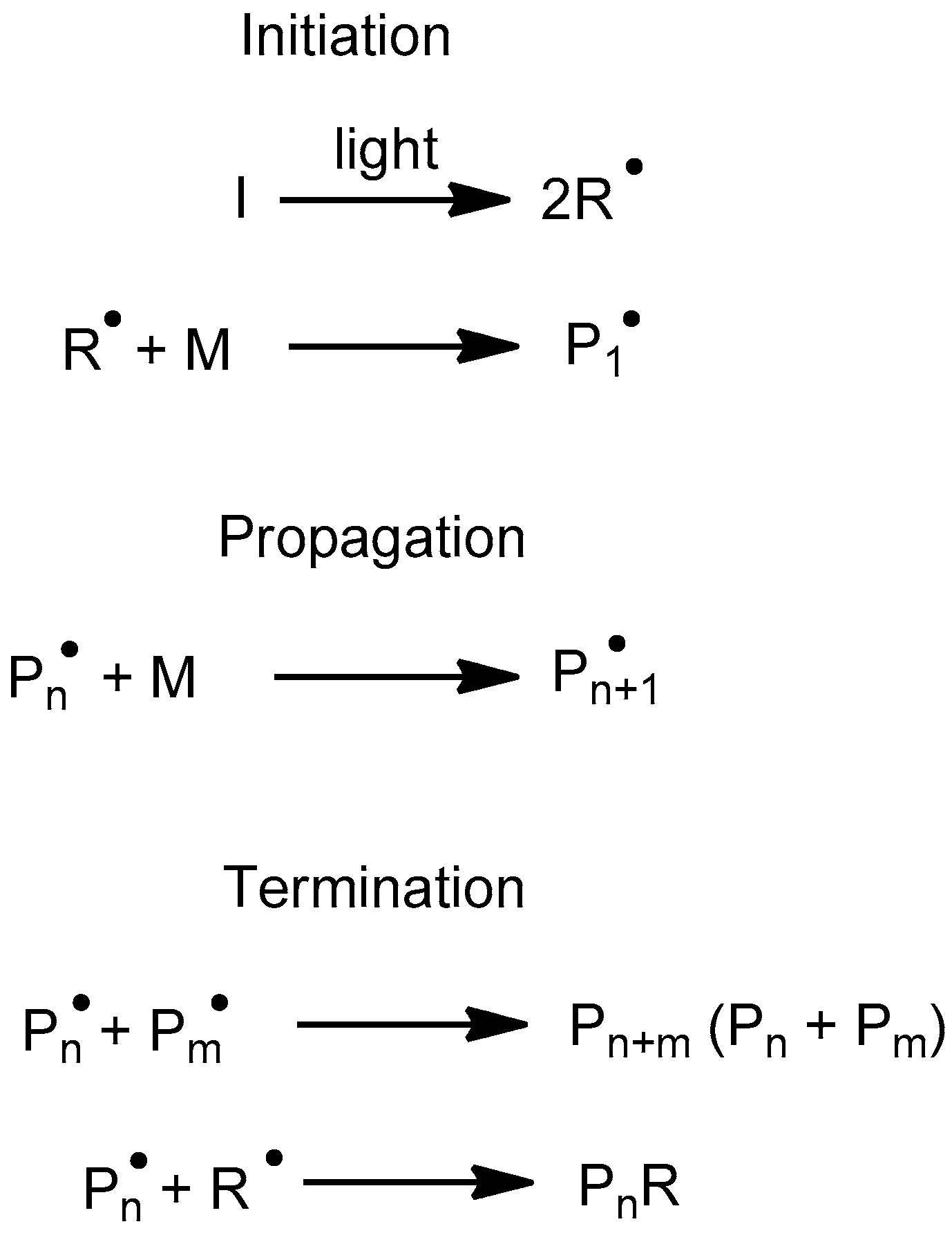
2.4. Radical Homopolymerization of Acrylates
3. Acrylate-Based Dual-Curing Systems
3.1. Thiol–Acrylate and Thiol–Epoxy Reactions
3.2. Thiol–Acrylate and Thiol–Ene (or Thiol–Yne) Reactions
3.3. Thiol–Acrylate and Acetoacetate–Acrylate Reactions
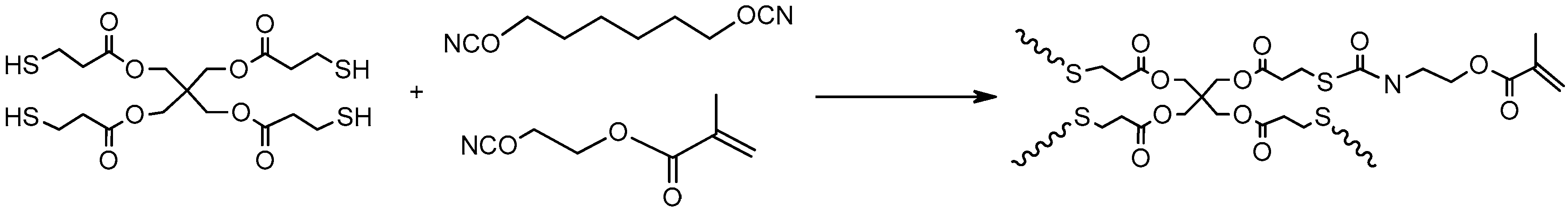
3.4. Thiol–Acrylate and Thiol–Isocyanate Reactions
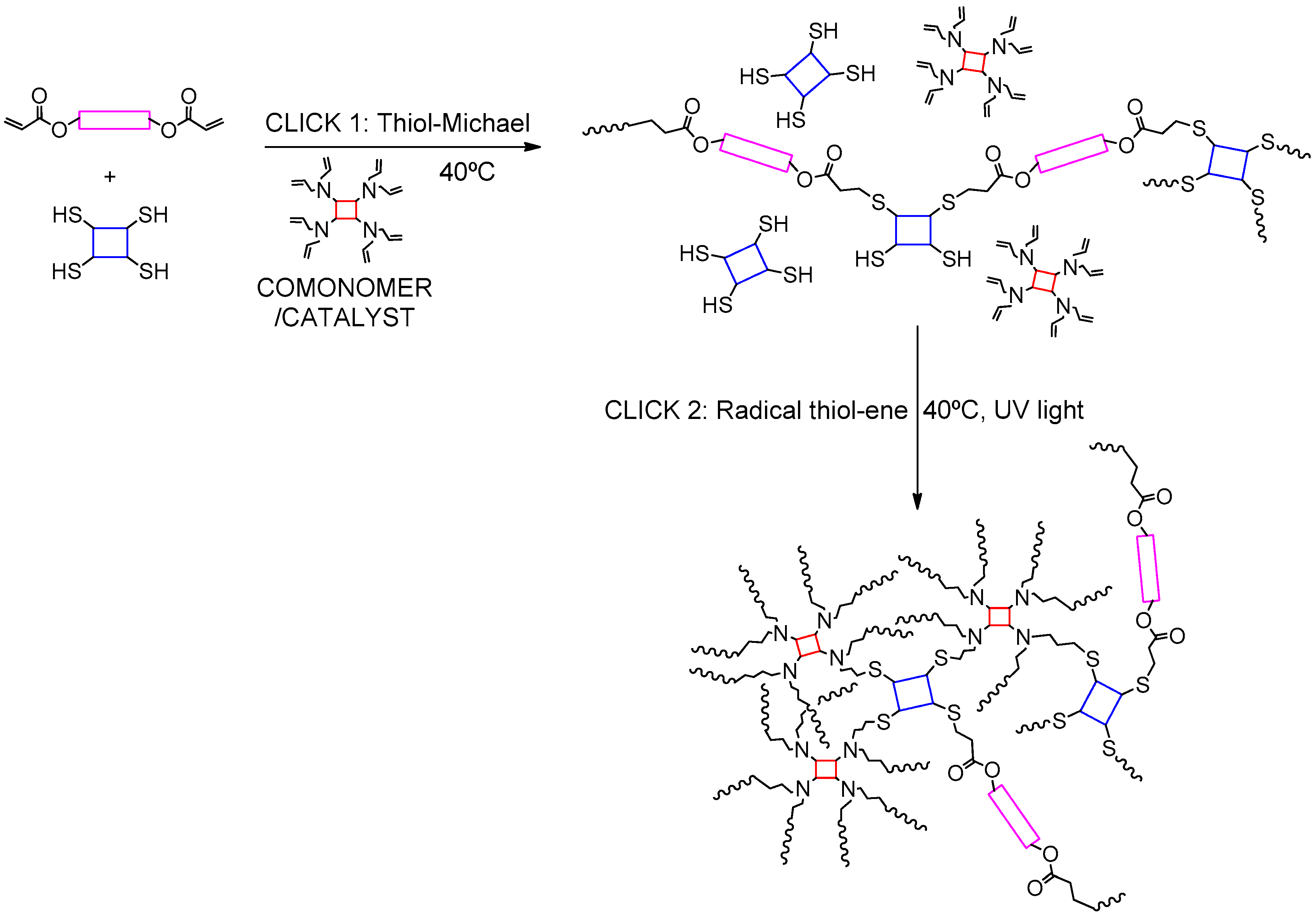
3.5. Combination of Two Thiol–Michael Additions on Different Acceptors
3.6. Michael Addition Followed by Homopolymerization
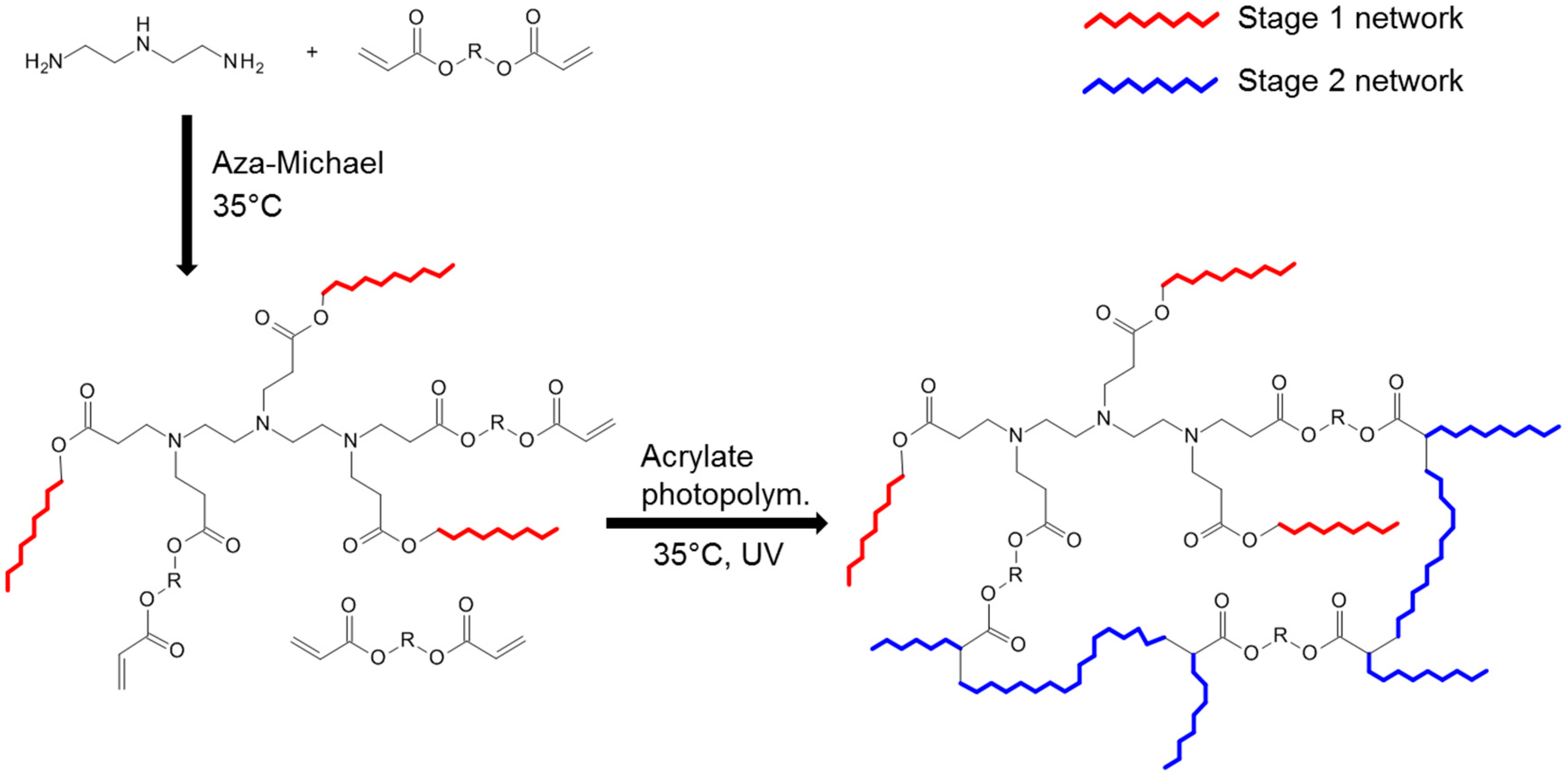
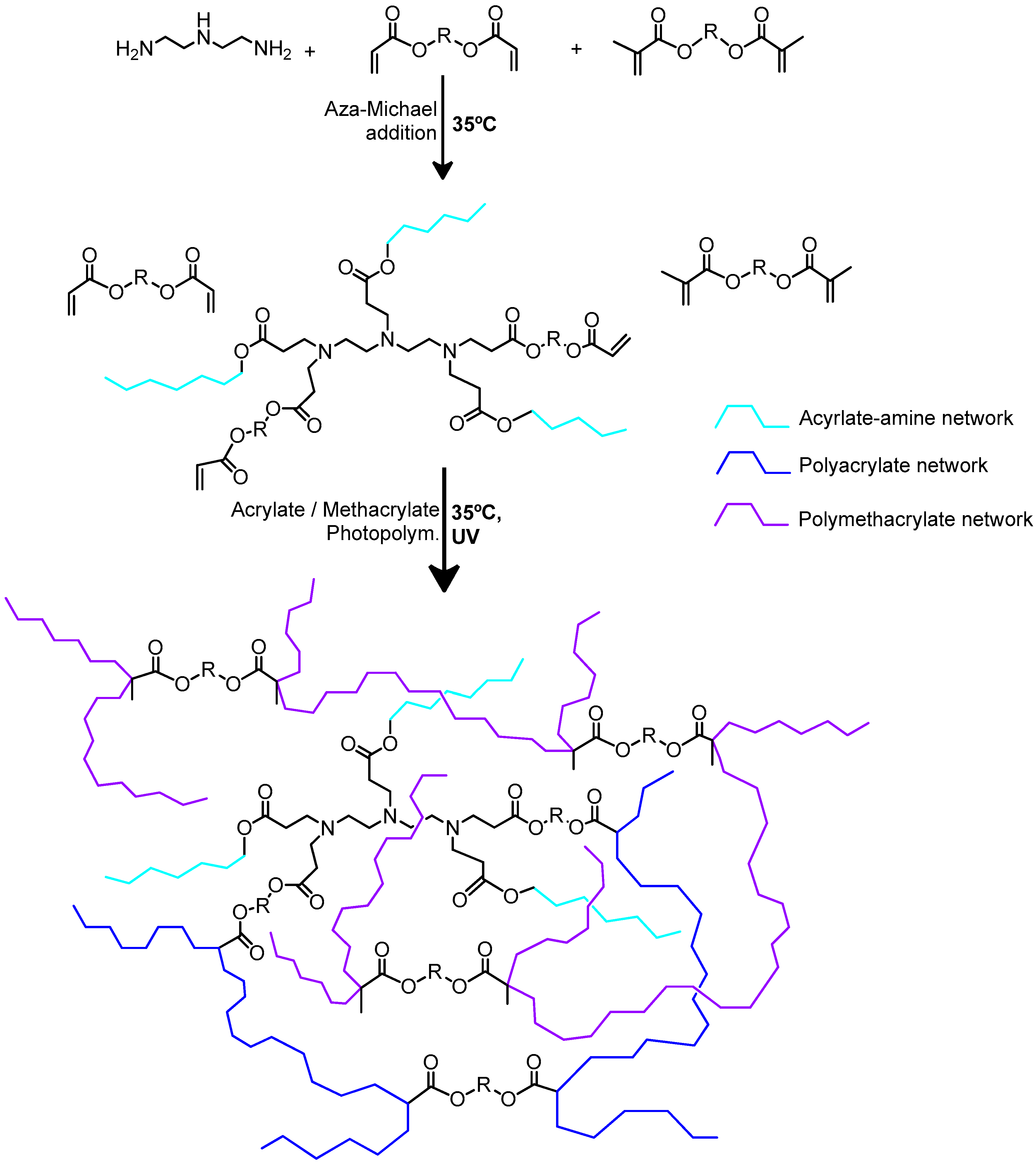
3.7. Michael Addition Followed by Methacrylate Homopolymerization
3.8. Isocyanate–Thiol Followed by Acrylate Homopolymerization
3.9. Diels–Alder Reaction Followed by Acrylate Homopolymerization
3.10. Combination of Acrylate Homopolymerization and Other Reactions
4. The Comonomer Approach
5. Polymer Modification/Functionalization
6. Organic–Inorganic Hybrid Dual-Curing Systems
7. Applications
7.1. Shape-Memory Systems
7.2. Optical Materials
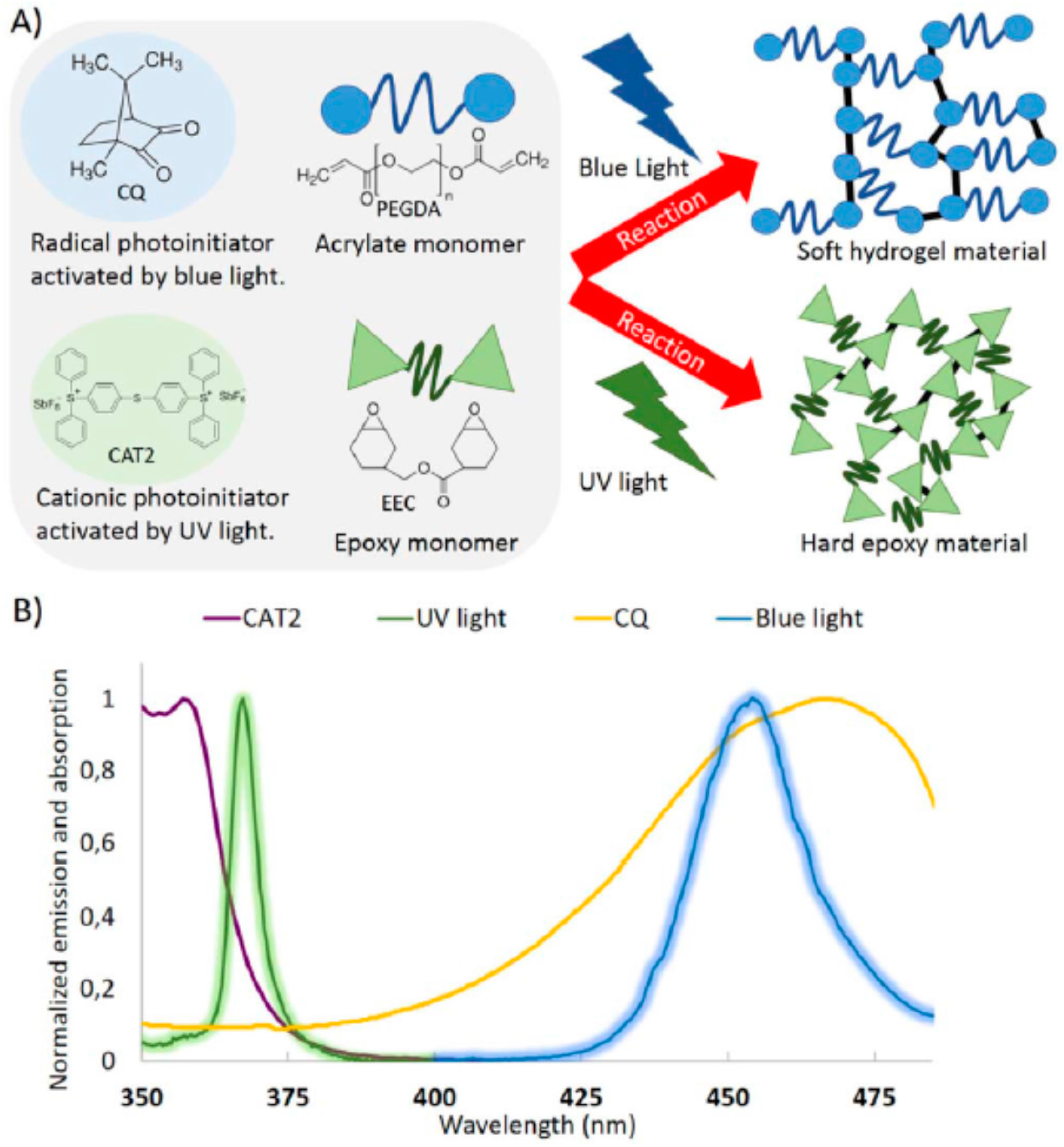
7.3. Lithography and Photopatterning
7.4. Creating Surface Topology
7.5. Holography
8. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ramis, X.; Fernández-Francos, X.; de la Flor, S.; Ferrando, F.; Serra, À. Click-based dual-curing thermosets and their applications. In Thermosets Structure, Properties, and Applications, 2nd ed.; Guo, Q., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; Chapter 16. [Google Scholar]
- Studer, J.; Dransfeld, C.; Masania, K. An analytical model for B-stage joining and co-curing of carbon fibre epoxy composites. Compos. Part A Appl. Sci. Manuf. 2016, 87, 282–289. [Google Scholar] [CrossRef]
- White, S.R.; Kim, Y.K. Staged curing of composite materials. Compos. Part A Appl. Sci. Manuf. 1996, 27, 219–227. [Google Scholar] [CrossRef]
- Penczek, P.; Czub, P.; Pielichowski, J. Unsaturated Polyester Resins: Chemistry and Technology. In Crosslinking in Materials Science; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar] [CrossRef]
- Tasdelen, M.A.; Kahveci, M.U.; Yagci, Y. Telechelic polymers by living and controlled/living polymerization methods. Prog. Polym. Sci. 2011, 36, 455–567. [Google Scholar] [CrossRef]
- Van Caeter, P.; Goethals, E.J. Telechelic polymers-new developments. Trends Polym. Sci. 1995, 3, 227–233. [Google Scholar]
- Sherman, C.L.; Zeigler, R.C.; Verghese, N.E.; Marks, M.J. Structure-property relationships of controlled epoxy networks with quantified levels of excess epoxy etherification. Polymer 2008, 49, 1164–1172. [Google Scholar] [CrossRef]
- Fernández-Francos, X.; Santiago, D.; Ferrando, F.; Ramis, X.; Salla, J.M.; Serra, À.; Sangermano, M. Network structure and thermomechanical properties of hybrid DGEBA networks cured with 1-methylimidazole and hyperbranched poly(ethyleneimine)s. J. Polym. Sci. Part B Polym. Phys. 2012, 50, 1489–1503. [Google Scholar] [CrossRef]
- Van Landuyt, K.L.; Snauwaert, J.; de Munck, J.; Peumans, M.; Yoshida, Y.; Poitevin, A.; Coutinho, E.; Suzuki, K.; Lambrechts, P.; van Meerbeek, B. Systematic review of the chemical composition of contemporary dental adhesives. Biomaterials 2007, 28, 3757–3785. [Google Scholar] [CrossRef] [PubMed]
- Studer, K.; Decker, C.; Beck, E.; Schwalm, R. Thermal and photochemical curing of isocyanate and acrylate functionalized oligomers. Eur. Polym. J. 2005, 41, 157–167. [Google Scholar] [CrossRef]
- Nowers, J.R.; Costanzo, J.A.; Narasimhan, B. Structure–Property Relationships in Acrylate/Epoxy Interpenetrating Polymer Networks: Effects of the Reaction Sequence and Composition. J. Appl. Polym. Sci. 2007, 104, 891–901. [Google Scholar] [CrossRef]
- Chen, F.; Cook, W.D. Curing kinetics and morphology of IPNs from a flexible dimethacrylate and a rigid epoxy via sequential photo and thermal polymerization. Eur. Polym. J. 2008, 44, 1796–1813. [Google Scholar] [CrossRef]
- Decker, C.; Viet, T.N.T.; Decker, D. UV-radiation curing of acrylate/epoxide systems. Polymer 2001, 42, 5531–5541. [Google Scholar] [CrossRef]
- Sperling, L.H.; Mishra, V. The Current Status of Interpenetrating Polymer Networks. Polym. Adv. Technol. 1996, 7, 197–208. [Google Scholar] [CrossRef]
- Ramis, X.; Cadenato, A.; Morancho, J.M.; Salla, J.M. Polyurethane—Unsaturated polyester interpenetrating polymer networks: Thermal and dynamic mechanical thermal behavior. Polymer 2001, 42, 9469–9479. [Google Scholar] [CrossRef]
- Decker, C. Kinetic Study and New Applications of UV Radiation Curing. Macromol. Rapid Commun. 2002, 23, 1067–1093. [Google Scholar] [CrossRef]
- Lee, S.-B.; Takata, T.; Endo, T. Quaternary ammonium salts as useful cationic initiators. 6. Synthesis, activity, and thermal latency of N-benzylpyridinium salts and the role of the pyridine moiety. Macromolecules 1991, 24, 2689–2693. [Google Scholar] [CrossRef]
- Dietliker, K.; Jung, T.; Benkhoff, J.; Kura, H.; Matsumoto, A.; Oka, H.; Hristova, D.; Gescheidt, G.; Rist, G. New developments in photoinitiators. Macromol. Symp. 2004, 217, 77–97. [Google Scholar] [CrossRef]
- Guzmán, D.; Ramis, X.; Fernández-Francos, X.; Serra, A. New catalysts for diglycidyl ether of bisphenol A curing based on thiol–epoxy click reaction. Eur. Polym. J. 2014, 59, 377–386. [Google Scholar] [CrossRef]
- Decker, C.; Masson, F.; Schwalm, R. Dual-curing of waterborne urethane-acrylate coatings by UV and thermal processing. Macromol. Mater. Eng. 2003, 288, 17–28. [Google Scholar] [CrossRef]
- Ruiz, E.; Trochu, F. Numerical analysis of cure temperature and internal stresses in thin and thick RTM parts. Compos. Part A Appl. Sci. Manuf. 2005, 36, 806–826. [Google Scholar] [CrossRef]
- Guzmán, D.; Ramis, X.; Fernández-Francos, X.; Serra, A. Preparation of click thiol-ene/thiol-epoxy thermosets by controlled photo/thermal dual curing sequence. RSC Adv. 2015, 5, 101623–101633. [Google Scholar] [CrossRef]
- Fernández-Francos, X.; Konuray, A.O.; Belmonte, A.; de la Flor, S.; Serra, À.; Ramis, X. Sequential curing of off-stoichiometric thiol–epoxy thermosets with a custom-tailored structure. Polym. Chem. 2016, 7, 2280–2290. [Google Scholar] [CrossRef]
- Chatani, S.; Nair, D.P.; Bowman, C.N. Relative reactivity and selectivity of vinyl sulfones and acrylates towards the thiol–Michael addition reaction and polymerization. Polym. Chem. 2013, 4, 1048–1055. [Google Scholar] [CrossRef]
- Belmonte, A.; Fernández-Francos, X.; Serra, À.; De la Flor, S. Phenomenological characterization of sequential dual-curing of off-stoichiometric “thiol-epoxy” systems: Towards applicability. Mater. Des. 2017, 113, 116–127. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Binder, W.H.; Sachsenhofer, R. “Click” chemistry in polymer and material science: An Update. Macromol. Rapid Commun. 2008, 29, 952–981. [Google Scholar] [CrossRef]
- Lahann, J. Click Chemistry: A Universal Ligation Strategy for Biotechnology and Materials Science. In Click Chemistry for Biotechnology Materials Science; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2009. [Google Scholar] [CrossRef]
- Lowe, A.; Bowman, C. (Eds.) Thiol-X Chemistries in Polymer and Materials Science; The Royal Society of Chemistry: London, UK, 2013. [Google Scholar] [CrossRef]
- Moses, J.E.; Moorhouse, A.D. The growing applications of click chemistry. Chem. Soc. Rev. 2007, 36, 1249–1262. [Google Scholar] [CrossRef] [PubMed]
- Azagarsamy, M.A.; Anseth, K.S. Bioorthogonal click chemistry: An indispensable tool to create multifaceted cell culture scaffolds. ACS Macro Lett. 2013, 2, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Tunca, U. Orthogonal multiple click reactions in synthetic polymer chemistry. J. Polym. Sci. Part A Polym. Chem. 2014, 52, 3147–3165. [Google Scholar] [CrossRef]
- Schwalm, R. UV Coatings: Basics, Recent Developments and New Applications; Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar] [CrossRef]
- Yao, B.; Zhao, H.; Wang, L.; Liu, Y.; Zheng, C.; Li, H. Synthesis of acrylate-based UV/thermal dual-cure coatings for antifogging. J. Coat. Technol. Res. 2018, 15, 149–158. [Google Scholar] [CrossRef]
- Yildiz, Z.; Onen, H.A. Dual-curable PVB based adhesive formulations for cord/rubber composites: The influence of reactive diluents. Int. J. Adhes. Adhes. 2017, 78, 38–44. [Google Scholar] [CrossRef]
- Jang, S.C.; Yi, S.C.; Kim, Y.B.; Hong, J.W. Dual-curable fluorinated poly(methacrylate) copolymers for optical adhesives. Polym. Adv. Technol. 2005, 16, 484–488. [Google Scholar] [CrossRef]
- Narayan-Sarathy, S.; Gould, M.; Ananthachar, S.; Zaranec, A.; Hahn, L. Radiation-curable inks and coatings from novel multifunctional acrylate oligomers. Tech. In Proceedings of the RadTech 2004 Conference, Charlotte, NC, USA, 2–5 May, 2004; pp. 47–50. [Google Scholar]
- Moszner, N.; Salz, U. New developments of polymeric dental composites. Prog. Polym. Sci. 2001, 26, 535–576. [Google Scholar] [CrossRef]
- Mather, B.D.; Viswanathan, K.; Miller, K.M.; Long, T.E. Michael addition reactions in macromolecular design for emerging technologies. Prog. Polym. Sci. 2006, 31, 487–531. [Google Scholar] [CrossRef]
- Retailleau, M.; Ibrahim, A.; Croutxé-Barghorn, C.; Allonas, X.; Ley, C.; Le Nouen, D. One-Pot Three-Step Polymerization System Using Double Click Michael Addition and Radical Photopolymerization. ACS Macro Lett. 2015, 4, 1327–1331. [Google Scholar] [CrossRef]
- Nair, D.P.; Cramer, N.B.; Gaipa, J.C.; McBride, M.K.; Matherly, E.M.; McLeod, R.R.; Shandas, R.; Bowman, C.N. Two-Stage Reactive Polymer Network Forming Systems. Adv. Funct. Mater. 2012, 22, 1502–1510. [Google Scholar] [CrossRef]
- González, G.; Fernández-Francos, X.; Serra, À.; Sangermano, M.; Ramis, X. Environmentally-friendly processing of thermosets by two-stage sequential aza-Michael addition and free-radical polymerization of amine–acrylate mixtures. Polym. Chem. 2015, 6, 6987–6997. [Google Scholar] [CrossRef]
- Konuray, A.O.; Liendo, F.; Fernández-Francos, X.; Serra, À.; Sangermano, M.; Ramis, X. Sequential curing of thiol-acetoacetate-acrylate thermosets by latent Michael addition reactions. Polymer 2017, 113, 193–199. [Google Scholar] [CrossRef]
- Konuray, A.O.; Fernández-Francos, X.; Serra, À.; Ramis, X. Sequential curing of amine-acrylate-methacrylate mixtures based on selective aza-Michael addition followed by radical photopolymerization. Eur. Polym. J. 2016, 84, 256–267. [Google Scholar] [CrossRef]
- Xi, W.; Peng, H.; Aguirre-Soto, A.; Kloxin, C.J.; Stansbury, J.W.; Bowman, C.N. Spatial and temporal control of thiol-michael addition via photocaged superbase in photopatterning and two-stage polymer networks formation. Macromolecules 2014, 47, 6159–6165. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.W.; Hoyle, C.E.; Lowe, A.B.; Bowman, M. Nucleophile-Initiated Thiol-Michael Reactions: Effect of Organocatalyst, Thiol, and Ene. Macromolecules 2010, 43, 6381–6388. [Google Scholar] [CrossRef]
- Li, G.-Z.; Randev, R.; Soeriyadi, A.H.; Rees, G.J.; Boyer, C.; Tong, Z.; Becer, C.R.; Haddleton, D.M. Investigation into thiol-(meth)acrylate Michael addition reactions using amine and phosphine catalysts. Polym. Chem. 2010, 1, 1196–1204. [Google Scholar] [CrossRef]
- Chatani, S.; Sheridan, R.J.; Podgórski, M.; Nair, D.P.; Bowman, C.N. Temporal control of thiol-click chemistry. Chem. Mater. 2013, 25, 3897–3901. [Google Scholar] [CrossRef]
- Chatani, S.; Gong, T.; Earle, B.A.; Podgorski, M.; Bowman, C.N. Visible-light initiated thiol-Michael addition photopolymerization reactions. ACS Macro Lett. 2014, 3, 315–318. [Google Scholar] [CrossRef]
- Jian, Y.; He, Y.; Sun, Y.; Yang, H.; Yang, W.; Nie, J. Thiol-epoxy/thiol-acrylate hybrid materials synthesized by photopolymerization. J. Mater. Chem. C 2013, 1, 4481–4489. [Google Scholar] [CrossRef]
- Konuray, A.O.; Fernández-Francos, X.; Ramis, X. Latent curing of epoxy-thiol thermosets. Polymer 2017, 116, 191–203. [Google Scholar] [CrossRef]
- Uygun, M.; Tasdelen, M.A.; Yagci, Y. Influence of type of initiation on thiol-ene “click” Chemistry. Macromol. Chem. Phys. 2010, 211, 103–110. [Google Scholar] [CrossRef]
- Flores, M.; Tomuta, A.M.; Fernández-Francos, X.; Ramis, X.; Sangermano, M.; Serra, A. A new two-stage curing system: Thiol-ene/epoxy homopolymerization using an allyl terminated hyperbranched polyester as reactive modifier. Polymer 2013, 54, 5473–5481. [Google Scholar] [CrossRef]
- Ma, S.J.; Mannino, S.J.; Wagner, N.J.; Kloxin, C.J. Photodirected Formation and Control of Wrinkles on a Thiol−ene Elastomer. ACS Macro Lett. 2013, 2, 474–477. [Google Scholar] [CrossRef]
- Acebo, C.; Fernández-Francos, X.; Ramis, X.; Serra, À. Multifunctional allyl-terminated hyperbranched poly(ethyleneimine) as component of new thiol-ene/thiol-epoxy materials. React. Funct. Polym. 2016, 99, 17–25. [Google Scholar] [CrossRef]
- Durmaz, H.; Butun, M.; Hizal, G.; Tunca, U. Postfunctionalization of polyoxanorbornene via sequential Michael addition and radical thiol-ene click reactions. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 3116–3125. [Google Scholar] [CrossRef]
- Shin, J.; Matsushima, H.; Comer, C.M.; Bowman, C.N.; Hoyle, C.E. Thiol-isocyanate-ene ternary networks by sequential and simultaneous thiol click reactions. Chem. Mater. 2010, 22, 2616–2625. [Google Scholar] [CrossRef]
- Cramer, N.B.; Bowman, C.N. Kinetics of thiol-ene and thiol-acrylate photopolymerizations with real-time Fourier transform infrared. J. Polym. Sci. Part A Polym. Chem. 2001, 39, 3311–3319. [Google Scholar] [CrossRef]
- Nair, D.P.; Podgórski, M.; Chatani, S.; Gong, T.; Xi, W.; Fenoli, C.R.; Bowman, C.N. The Thiol-Michael addition click reaction: A powerful and widely used tool in materials chemistry. Chem. Mater. 2014, 26, 724–744. [Google Scholar] [CrossRef]
- Xi, W.; Wang, C.; Kloxin, C.J.; Bowman, C.N. Nitrogen-centered nucleophile catalyzed thiol-vinylsulfone addition, another thiol-ene “click” reaction. ACS Macro Lett. 2012, 1, 811–814. [Google Scholar] [CrossRef]
- Konuray, A.O.; Fernández-Francos, X.; Ramis, X. Curing kinetics and characterization of dual-curable thiol-acrylate-epoxy thermosets with latent reactivity. React. Funct. Polym. 2018, 122, 60–67. [Google Scholar] [CrossRef]
- Fouassier, J.-P. Photoinitiation, Photopolymerization and Photocuring—Fundamentals and Applications; Hanser Publishers: Munich, Germany, 1995. [Google Scholar] [CrossRef]
- Jin, K.; Wilmot, N.; Heath, W.H.; Torkelson, J.M. Phase-Separated Thiol-Epoxy-Acrylate Hybrid Polymer Networks with Controlled Cross-Link Density Synthesized by Simultaneous Thiol-Acrylate and Thiol-Epoxy Click Reactions. Macromolecules 2016, 49, 4115–4123. [Google Scholar] [CrossRef]
- Chatani, S.; Wang, C.; Podgórski, M.; Bowman, C.N. Triple Shape Memory Materials Incorporating Two Distinct Polymer Networks Formed by Selective Thiol–Michael Addition Reactions. Macromolecules 2014, 47, 4949–4954. [Google Scholar] [CrossRef]
- Chan, J.W.; Hoyle, C.E.; Lowe, A.B. Sequential phosphine-catalyzed, nucleophilic thiol ene/radical-mediated thiol-yne reactions and the facile orthogonal synthesis of polyfunctional materials. J. Am. Chem. Soc. 2009, 131, 5751–5753. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Nair, D.P.; Kowalski, B.A.; Xi, W.; Gong, T.; Wang, C.; Cole, M.; Cramer, N.B.; Xie, X.; McLeod, R.R.; et al. High performance graded rainbow holograms via two-stage sequential orthogonal thiol-click chemistry. Macromolecules 2014, 47, 2306–2315. [Google Scholar] [CrossRef]
- Peng, H.; Wang, C.; Xi, W.; Kowalski, B.A.; Gong, T.; Xie, X.; Wang, W.; Nair, D.P.; McLeod, R.R.; Bowman, C.N. Facile image patterning via sequential thiol-Michael/thiol-yne click reactions. Chem. Mater. 2014, 26, 6819–6826. [Google Scholar] [CrossRef]
- Konuray, A.O.; Ramis, X.; Fernández-Francos, X.; Serra, À. New allyl-functional catalytic comonomers for sequential thiol-acrylate Michael and radical thiol-ene reactions. Polymer 2018, 138C, 369–377. [Google Scholar] [CrossRef]
- Matsushima, H.; Shin, J.; Bowman, C.N.; Hoyle, C.E. Thiol-Isocyanate-Acrylate Ternary Networks by Selective Thiol-Click Chemistry. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 3255–3264. [Google Scholar] [CrossRef]
- Moszner, N.; Rheinberger, V. Reaction behaviour of monomeric β-ketoesters, 4. Polymer network formation by Michael reaction of multifunctional acetoacetates with multifunctional acrylates. Macromol. Rapid Commun. 1995, 16, 135–138. [Google Scholar] [CrossRef]
- Pavlinec, J.; Moszner, N. Photocured polymer networks based on multifunctional beta-ketoesters and acrylates. J. Appl. Polym. Sci. 1997, 65, 165–178. [Google Scholar] [CrossRef]
- Moy, T.M.; Dammann, L.; Loza, R. Liquid Oligomers Containing Unsaturation. US Patent No 5945489, 31 August 1999. [Google Scholar]
- Dammann, L.G.; Gould, M.L. Liquid Uncrosslinked Michael Addition Oligomers Prepared in the Presence of a Catalyst Having both an Epoxy Moiety and a Quaternary Salt. US Patent No US6706414 B1, 16 March 2004. [Google Scholar]
- Pietschmann, N.; Stengel, K.; Hoesselbarth, B. Investigations into vinylogic addition reactions of modified polyester resins. Prog. Org. Coat. 1999, 36, 64–69. [Google Scholar] [CrossRef]
- Nair, D.P.; Cramer, N.B.; McBride, M.K.; Gaipa, J.C.; Shandas, R.; Bowman, C.N. Enhanced two-stage reactive polymer network forming systems. Polymer 2012, 53, 2429–2434. [Google Scholar] [CrossRef] [PubMed]
- Nair, D.P.; Cramer, N.B.; McBride, M.K.; Gaipa, J.C.; Lee, N.C.; Shandas, R.; Bowman, C.N. Fabrication and characterization of novel high modulus, two-stage reactive thiol-acrylate composite polymer systems. Macromol. Symp. 2013, 329, 101–107. [Google Scholar] [CrossRef]
- Klee, J.E.; Neidhart, F.; Flammersheim, H.-J.; Mülhaupt, R. Monomers for low shrinking composites, 2. Synthesis of branched methacrylates and their application in dental composites. Macromol. Chem. Phys. 1999, 200, 517–523. [Google Scholar] [CrossRef]
- Konuray, A.O.; Ruiz, A.; Morancho, J.M.; Salla, J.M.; Fernández-Francos, X.; Serra, À.; Ramis, X. Sequential dual curing by selective Michael addition and free radical polymerization of acetoacetate-acrylate-methacrylate mixtures. Eur. Polym. J. 2018, 98, 39–46. [Google Scholar] [CrossRef]
- Podgórski, M.; Nair, D.P.; Chatani, S.; Berg, G.; Bowman, C.N. Programmable mechanically assisted geometric deformations of glassy two-stage reactive polymeric materials. ACS Appl. Mater. Interfaces 2014, 6, 6111–6119. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.J.; Gong, T.; Fenoli, C.R.; Bowman, C.N. A dual-cure, solid-state photoresist combining a thermoreversible diels-alder network and a chain growth acrylate network. Macromolecules 2014, 47, 3473–3482. [Google Scholar] [CrossRef]
- Li, W.; Noodeh, M.B.; Delpouve, N.; Saiter, J.M.; Tan, L.; Negahban, M. Printing continuously graded interpenetrating polymer networks of acrylate/epoxy by manipulating cationic network formation during stereolithography. Express Polym. Lett. 2016, 10, 1003–1015. [Google Scholar] [CrossRef]
- Larsen, E.K.U.; Larsen, N.B.; Almdal, K.; Larsen, E.K.U.; Larsen, N.B.; Almdal, K. Multimaterial hydrogel with widely tunable elasticity by selective photopolymerization of PEG diacrylate and epoxy monomers. J. Polym. Sci. Part B Polym. Phys. 2016, 54, 1195–1201. [Google Scholar] [CrossRef]
- Park, C.H.; Lee, S.W.; Park, J.W.; Kim, H.J. Preparation and characterization of dual curable adhesives containing epoxy and acrylate functionalities. React. Funct. Polym. 2013, 73, 641–646. [Google Scholar] [CrossRef]
- Bertoldo, M.; Bronco, S.; Narducci, P.; Rossetti, S.; Scoponi, M. Polymerization kinetics and characterization of dual cured polyurethane-acrylate nanocomposites for laminates. Macromol. Mater. Eng. 2005, 290, 475–484. [Google Scholar] [CrossRef]
- Bounds, C.O.; Upadhyay, J.; Totaro, N.; Thakuri, S.; Garber, L.; Vincent, M.; Huang, Z.; Hupert, M.; Pojman, J.A. Fabrication and characterization of stable hydrophilic microfluidic devices prepared via the in situ tertiary-amine catalyzed Michael addition of multifunctional thiols to multifunctional acrylates. ACS Appl. Mater. Interfaces 2013, 5, 1643–1655. [Google Scholar] [CrossRef] [PubMed]
- Malucelli, G. Hybrid Organic/Inorganic Coatings Through Dual-Cure Processes: State of the Art and Perspectives. Coatings 2016, 6, 10. [Google Scholar] [CrossRef]
- Kickelbick, G. Introduction to Hybrid Materials. In Hybrid Mater; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; Chapter 1. [Google Scholar] [CrossRef]
- Ciriminna, R.; Fidalgo, A.; Pandarus, V.; Béland, F.; Ilharco, L.M.; Pagliaro, M. The sol-gel route to advanced silica-based materials and recent applications. Chem. Rev. 2013, 113, 6592–6620. [Google Scholar] [CrossRef] [PubMed]
- Serra, A.; Ramis, X.; Fernández-Francos, X. Epoxy Sol-Gel Hybrid Thermosets. Coatings 2016, 6, 8. [Google Scholar] [CrossRef]
- Lazo, M.A.G.; Blank, M.; Leterrier, Y.; Månson, J.A.E. Superhard transparent hybrid nanocomposites for high fidelity UV-nanoimprint lithography. Polymer 2013, 54, 6177–6183. [Google Scholar] [CrossRef]
- Sangermano, M.; Gaspari, E.; Vescovo, L.; Messori, M. Enhancement of scratch-resistance properties of methacrylated UV-cured coatings. Prog. Org. Coat. 2011, 72, 287–291. [Google Scholar] [CrossRef]
- Xie, H.; Shi, W. Polymer/SiO2 hybrid nanocomposites prepared through the photoinitiator-free UV curing and sol-gel processes. Compos. Sci. Technol. 2014, 93, 90–96. [Google Scholar] [CrossRef]
- Gigot, A.; Sangermano, M.; Capozzi, L.C.; Dietliker, K. In-situ synthesis of organic-inorganic coatings via a photolatent base catalyzed Michael-addition reaction. Polymer 2015, 68, 195–201. [Google Scholar] [CrossRef]
- Chemtob, A.; de Paz-Simon, H.; Sibeaud, M.; el Fouhali, B.; Croutxé-Barghorn, C.; Jacomine, L.; Gauthier, C.; Le Houérou, V. An orthogonal, one-pot, simultaneous UV-mediated route to thiol-ene/sol-gel film. Express Polym. Lett. 2016, 10, 439–449. [Google Scholar] [CrossRef]
- Belmonte, A.; Russo, C.; Ambrogi, V.; Fernández-Francos, X.; la Flor, S.D. Epoxy-based shape-memory actuators obtained via dual-curing of off-stoichiometric “thiol-epoxy” mixtures. Polymers 2017, 9, 113. [Google Scholar] [CrossRef]
- Belmonte, A.; Guzmán, D.; Fernández-Francos, X.; De la Flor, S. Effect of the network structure and programming temperature on the shape-memory response of thiol-epoxy “click” systems. Polymers 2015, 7, 2146–2164. [Google Scholar] [CrossRef]
- Karger-Kocsis, J.; Kéki, S. Review of progress in shape memory epoxies and their composites. Polymers 2017, 10, 34. [Google Scholar] [CrossRef]
- Duerig, T.; Pelton, A.; Stöckel, D. An overview of nitinol medical applications. Mater. Sci. Eng. A 1999, 273–275, 149–160. [Google Scholar] [CrossRef]
- Efimenko, K.; Finlay, J.; Callow, M.E.; Callow, J.A.; Genzer, J. Development and testing of hierarchically wrinkled coatings for marine antifouling. ACS Appl. Mater. Interfaces 2009, 1, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.S.; Martina, D.; Creton, C.; Lindner, A.; Crosby, A.J. Enhanced adhesion of elastic materials to small-scale wrinkles. Langmuir 2012, 28, 14899–14908. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Quiroga, R.A.; Calixto, S.; Lougnot, D.J. Optical characterization and applications of a dual-cure photopolymerizable system. Appl. Opt. 2003, 42, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Acik, G.; Yildiran, S.; Kok, G.; Salman, Y.; Tasdelen, M.A. Synthesis and characterization of sugar-based methacrylates and their random copolymers by ATRP. Express Polym. Lett. 2017, 11, 799–808. [Google Scholar] [CrossRef]
- Kaya, N.U.; Onen, A.; Guvenilir, Y. Photopolymerization of acrylates by enzymatically synthesized PCL based macrophotoinitiator. Express Polym. Lett. 2017, 11, 493–503. [Google Scholar] [CrossRef]
- Llevot, A. Sustainable Synthetic Approaches for the Preparation of Plant Oil-Based Thermosets. J. Am. Oil Chem. Soc. 2017, 94, 169–186. [Google Scholar] [CrossRef]















© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konuray, O.; Fernández-Francos, X.; Ramis, X.; Serra, À. State of the Art in Dual-Curing Acrylate Systems. Polymers 2018, 10, 178. https://doi.org/10.3390/polym10020178
Konuray O, Fernández-Francos X, Ramis X, Serra À. State of the Art in Dual-Curing Acrylate Systems. Polymers. 2018; 10(2):178. https://doi.org/10.3390/polym10020178
Chicago/Turabian StyleKonuray, Osman, Xavier Fernández-Francos, Xavier Ramis, and Àngels Serra. 2018. "State of the Art in Dual-Curing Acrylate Systems" Polymers 10, no. 2: 178. https://doi.org/10.3390/polym10020178
APA StyleKonuray, O., Fernández-Francos, X., Ramis, X., & Serra, À. (2018). State of the Art in Dual-Curing Acrylate Systems. Polymers, 10(2), 178. https://doi.org/10.3390/polym10020178