Abstract
Proper inclusion of van der Waals interactions (vdW) in ab initio calculations based on the density functional theory (DFT) is crucial to describe soft, organic, layered solids such as -(BEDT-TTF). Since no consensus has been reached on the reliability of available vdW DFT functionals, most of the first principles calculations have been based on experimental crystal structure data without any structural optimization. Here, we explore optimal DFT-based schemes that account for the effects of vdW interactions on the structural and electronic band properties of three paradigmatic charge transfer salts, -(BEDT-TTF)Cu(CN), -(BEDT-TTF)Ag(CN), and -(BEDT-TTF)Cu[N(CN)]Cl, for which a unified optimization of the structure is possible. Detailed examination of the prototype test system -(BEDT-TTF)Cu[N(CN)]Cl shows that the optB88-vdW functional performs slightly better than the PBE-vdW and that the choice of pseudopotentials is critical to obtaining realistic results.
1. Introduction
In the past two decades, layered solids -(BEDT-TTF) (X = Cu(CN), Ag(CN), and Cu[N(CN)]Cl), based on the organic molecule bis(ethylenedithio)tetrathiafulvalene (abbreviated as BEDT-TTF), have attracted unprecedented scientific attention worldwide. These materials are charge-transfer salts formed by assembling of two components, the conducting BEDT-TTF layers (electron donors) separated by the non-conducting anion (X) layers (electron acceptors), as illustrated in Figure 1. In conducting layers, the BEDT-TTF molecules form face-to-face pairs (dimers) arranged on anisotropic triangular lattices. According to stoichiometry, half of an electron per BEDT-TTF molecule is transferred to the anion in each compound.
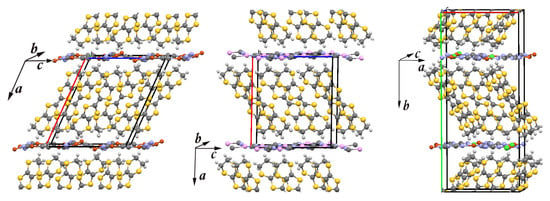
Figure 1.
The crystal structure of three layered organic charge-transfer salts -(BEDT-TTF) (X = Cu(CN), Ag(CN), and Cu[N(CN)]Cl). Carbon, sulfur, and hydrogen atoms of the bis(ethylenedithio)tetrathiafulvalene [CH)]CS (BEDT-TTF) molecule are denoted in dark gray, yellow, and light gray, respectively. In the anion (X) network, chlorine, cooper, silver, carbon, and nitrogen are denoted in green, red, pink, dark grey, and violet, respectively. The lines mark the unit cell. Left panel: The unit cell of -CuCN contains 4 BEDT-TTF molecules and 2 Cu(CN) anions. Middle panel: The unit cell of -AgCN contains 4 BEDT-TTF molecules and 2 Ag(CN) anions. Right panel: The unit cell of -Cl contains 8 BEDT-TTF molecules and 4 Cu[N(CN)]Cl anions.
The major interest in the physics and chemistry of these very complex compounds stems from the fact that they exhibit large ionicity due to the well-defined anions and organic cations [1,2], ferroelectric-like [3] and relaxor effects [4] despite the extremely small charge rearrangement [5,6,7,8], quantum spin liquid (QSL) behavior, and antiferromagnetic (AF) and Mott insulator properties [9,10,11], which in turn strongly affect their electronic, optical, and dynamical characteristics [12,13,14]. Under moderate pressures, -(BEDT-TTF) systems exhibit unconventional superconductivity at relatively high critical temperatures 12.8 K. The proximity of superconductivity to AF and QSL states and the layered structure of these organic solids indicates important similarities with high-Tc cuprates [15].
Over the past five years, we have carried out a number of comparative experimental and theoretical studies of the structural, optical, electronic, and dynamical properties of charge transfer salts -(BEDT-TTF)Cu(CN), -(BEDT-TTF)Ag(CN), and -(BEDT-TTF)Cu[N(CN)]Cl (in this paper abbreviated as -CuCN, -AgCN, and -Cl) [4,7,16,17,18] and -EtMeSb[Pd(dmit)] (abbreviated as EtMeSb) [19]. A particularly intriguing aspect of these compounds is an interplay between the chemical and van der Waals (vdW) bonding in the determination of their crystal and electronic structure and the ensuing dynamical properties. Notably, while anions and organic cations are bound also via hydrogen bonds, the non-local vdW interactions are expected to contribute to interdimer interactions within the conducting planes, as well as to the total cohesion energy of these organic solids. Despite a tacit consent on the importance of vdW interactions, the previous density functional theory (DFT) studies of these systems undertaken by the other authors have not explicitly taken into account the vdW interactions but rather used standard semilocal functionals [8,20,21]. Koretsune and Hotta [22] were the first to show to what extent the optimized values of these DFT calculations deviate from the experimental ones and demonstrated that even small structural changes of several percents invalidate the estimates of realistic values of the model parameters. Being aware of the relevance of vdW interactions, these authors performed DFT calculations by relaxing atomic positions only, while the unit cell parameters based on crystallographic data obtained in X-ray diffraction experiments were kept fixed. The obtained results of the electronic band structure were claimed to be basically similar to each other, the small differences probably being due to disparate refinements applied in the analysis of the -CuCN and -AgCN crystallographic data arising from the two possible orientations of the anionic cyanides.
Interpretations of the results of our sequential measurements of the various properties of -CuCN, -AgCN, -Cl, and EtMeSb compounds have been supported by extensive DFT-based calculations of their crystal, electronic, and vibrational structure using, for the first time, the functionals with non-local vdW interactions included [4,7,17,19]. We have elaborated impacts of the interplay of vdW interactions and cation–anion coupling on the electronic structure and charge redistributions in these systems, proposed interpretations, and drawn conclusions on the important role of vdW forces in the studied systems. Since there had not been prior DFT studies including vdW within the layered organic charge transfer solids community, we have resorted to the most standard non-local vdW functionals widely used and permanently upgraded in the treatment of systems with reduced dimensionality such as solid surfaces, surface layers, and graphene. The development, and in particular, the adoption of non-local functionals has been rather slow because the semi-empirical vdW corrections added to the standard semi-local density functionals [23,24] used in the density functional description of molecular structures and layered crystals seemed quite satisfactory since they lead to rather realistic predictions of lattice parameters [25,26]. However, as we have elaborated in [27], these approaches have fundamental limitations that are not present in vdW functionals. Significant testing of the vdW-DF functional was undertaken in [27,28,29,30,31,32], where the improvement of DFT calculations using vdW-DF [33] in comparison to GGA calculations (which were considered state of the art at the time) was proven. Since in the course of our studies [4,7,17,19], the available state-of-the-art algorithms have continuously been upgraded, the role of vdW interactions in the considered systems has not been treated on a fully unified footing. To remedy this shortcoming of the previous calculations and the possible ambiguities arising thereof, we present in this work a unified assessment of the effects of vdW forces on the structural and electronic properties of the paradigmatic charge transfer compounds -(BEDT-TTF), X = Cu(CN), and Ag(CN) and Cu[N(CN)]Cl, and discuss the obtained results in relation to the ones obtained earlier.
2. vdW Interactions and Structural Properties
In this section, we investigate the formation and structure of unit cells of -CuCN, -AgCN, and -Cl as calculated with and without the inclusion of vdW interactions among the cell constituents. Throughout the present calculations, the experimental crystal structure obtained from X-ray diffraction measurements of -CuCN, -AgCN, and -Cl at 100 K, 150 K, and 100 K, respectively, was used for the input unrelaxed atomic coordinates [7,17,34]. Two types of DFT functionals were employed in the derivation of the corresponding structures, viz. the standard semilocal PBE functional and the nonlocal vdW-DF functional. Each structure was relaxed (for computational details, see Section 5) to allow for the change of volume and shape of the unit cell together with the atomic positions. Specific structure parametrizations for the three studied systems are summarized below:
- -CuCN: For this system, the point of departure are the X-ray structural data at 100 K [17], which have been solved in symmetry and reduced to , which turned out to be the relaxed structure of lowest energy.
- -AgCN: In this compound, the electronic structure DFT calculations are based on the X-ray data taken at 150 K [7], solved in symmetry, and reduced to . Here, the higher symmetry group is found to be the relaxed structure of lowest energy.
- -Cl: In this third example, the crystal structure based on the data at 100 K [34] was solved in symmetry, reduced to , and the relaxed structure with symmetry corresponding to the experimental one was regained.
This section is divided in two subsections. In the first, we present the results obtained for three different systems, -CuCN, -AgCN, and -Cl, using the same DFT functionals and pseudopotentials (for details see Section 5). In the second, we compare the results for the -Cl system obtained from calculations that use different vdW functionals and pseudopotentials.
2.1. -CuCN, -AgCN, and -Cl
2.1.1. vdW Interactions Absent
We first studied all three systems, -CuCN, -AgCN, and -Cl, in the absence of vdW interactions. Energies and structure were determined from DFT calculations using the PBE functional [35]. As for the pseudopotentials, which replace the core orbitals, they were the same for the common atoms in the three systems, viz. C, S,H, N, Cu for the system -CuCN; C,S,H, N, Ag for the system -AgCN; and C,S,H, N, Cu, Cl for the system -Cl.
The calculations yielded the volume of the unit cell enhanced by 9.7%, 14.1%, and 15.3%, respectively, relative to the experimental ones (see Table 1). Here, we find different effects on the unit cell parameters: for -CuCN the most affected parameter was the one pointing out of plane (see Table 1 in [4]), while for the -AgCN system, all three cell parameters were similarly affected (see Table 2 in [4]). Finally, for -Cl, we find the in-plane parameter to be most affected (see Section 2.2.).

Table 1.
Unit cell volume enhancement (in %), defined as relative deviation from the experimental value, of -CuCN, -AgCN, and -Cl, obtained from ab initio calculations without van der Waals (vdW, based on PBE functional) and with vdW (based on the PBE-vdW functional). Pseudopotentials used for each system are indicated.
2.1.2. vdW Interactions Included
The inclusion of vdW interactions for all three systems, -CuCN, -AgCN, and -Cl, was implemented through the nonlocal van der Waals density functional PBE-vdW incorporating PBE for exchange [35] and correlation from [33]. The same pseudopotentials as in Section 2.1.1 were used. The obtained results are summarized in Table 1. It is evident that the inclusion of vdW interactions gives results for the unit cell size much closer to the experimental values. We note that the effects of vibrations on the computed geometry may be considered small since the thermal effect on the experimental volumes measured at higher temperatures is lower than 1% [11,21]. The parameters of the optimized and experimental structures are presented in Tables 1 and 2 of [4].
2.2. -Cl
In this subsection, we present the analysis of the -Cl system by carrying out calculations with the vdW functionals and pseudopotentials different from those used in Section 2.1.2. Again, we use for reference the energies and structure of the -Cl system in the absence of vdW interactions as determined previously by employing the PBE functional. On the other hand, we revisit the role of vdW interactions through the implementation of the optB88-vdW functional incorporating optB88 for exchange [36] and correlation from [30,33,37]. The same pseudoptentials for S, H, N, and Cl as in the previous section were used. However, this time we varied the pseudopotentials for carbon and copper atoms since we did not know a priori which chemical situation would materialize in our systems.
The obtained results are presented in Table 2. The results closest to the experimental values were obtained with C, Cu. Here, a very good agreement with the measured value of the unit cell volume was achieved, the deviation being only −0.48%. However, as we explained above, the found deviation could be slightly larger since the vibrational effects, here lower than 1%, have not been taken into account [11,21]. On the other hand, the choice of C, Cu pseudopotentials seems to be less favorable since it diminishes (choice (2)) or completely overwhelms (choice (3)) the role of vdW forces. This result indicates that the core orbitals, despite their presumably small overlap, cannot be neglected in the calculations. The parameters of the optimized structures for choice (1) and (2) of the pseudopotentials are presented in Table 3 and Table 4, while the parameters for choice (3) are given in Table 3 of [4].
Table 2.
Unit cell volume enhancement (in %), defined as relative deviation from the experimental value, of -Cl obtained from ab initio calculations without vdW (based on the PBE functional) and with vdW (based on the optB88-vdW functional), using a different set of pseudopotentials denoted as choices (1), (2), and (3).
Table 3.
Unit cell parameters of -Cl obtained from X-ray diffraction measurements at 100 K (left column), ab initio calculations based on the PBE functional (central column) and on the optB88-vdW functional (right column). In both types of calculations, the pseudopotentials denoted as choice (1) were used. Relative deviations from experimental values are given in parentheses.
Table 4.
Unit cell parameters of -Cl obtained from X-ray diffraction measurements at 100 K (left column), ab initio calculations based on the PBE functional (central column) and on the optB88-vdW functional (right column). In both types of calculations, the pseudopotentials denoted as choice (2) were used. Relative deviations from experimental values are given in parentheses.
2.3. Discussion
Inspection of the presented results confirms the findings from our previous studies [4,7,17], showing the significant role of nonlocal vdW interactions in these ionically cation–anion bonded solids. Results of the work suggest that the optimal choice of the vdW functional for studying the structure of -CuCN, -AgCN, and -Cl would be optB88-vdW. Likewise, we find the optimal choice of pseudopotentials for carbon and copper to be C and Cu. In fact, it turns out that this combination is crucial. These conclusions are drawn from the computed relaxed volumes closest to the experimental ones and the undesired, but expected, trends of large volume enhancements in the absence of vdW forces.
The role of pseudopotentials in the calculations without vdW is nicely revealed in our studies of the -Cl system using the same DFT functional for correlation and exchange in the absence of vdW, i.e., PBE, while varying the pseudopotentials for copper and carbon (Table 2, choices (1), (2), and (3)). We find the largest increase of volume (about 15%) relative to the experimental one with C and Cu pseudopotentials (Table 2, choice (1)). Interestingly enough, the least satisfactory choice of pseudopotential for copper is Cu because it deceptively substitutes the role of vdW forces (Table 2, choice (3)). This corrects our previous statement on the irrelevance of vdW in -Cl (see Table 3 in [4]). Note that its unrealistic influence is partly reduced by the use of C instead of the C pseudopotential for carbon (Table 2, choice (2)).
In the following, we address possible reasons for the different performance of the used pseudopotentials [38]. The first important result is that Cu performs better than the Cu pseudopotential. While the Cu pseudopotential is constructed with 11 valence electrons (the remaining ones are closed in core orbitals and as such cannot participate in chemical bonds), the Cu means that the p-shell has also been included, making altogether 17 valence electrons. Such a pseudopotential is required when the role of deeper p-shells is expected in chemical bonding. However, the result that Cu performs better than he Cu pseudopotential does not necessarily indicate the important role of p-electrons [39]. The second result that the C pseudopotential performs better than the C one may indicate that dimers with short bonds are present in the -Cl compound. Indeed, the BEDT-TTF molecule contains three C=C double bonds, two outer ring C=C, and one central C=C bond (see Figure 1). Thus, we conclude that the choice of C instead of the C pseudopotential may appear to be more relevant for -CuCN and -AgCN as well.
We reiterate that the change of correlation in going from the PBE to the PBE-vdW functional yields volumes enhanced by about 2.5% relative to the measured values for all three systems studied, viz. -CuCN, -AgCN, and -Cl (Table 1). This is somewhat different from the value of −0.48% obtained with optB88-vdW with the same choice of pseudopotentials Cu and C in all three cases, which yields the unit cell size for -Cl closest to the experiment (see Table 1 and Table 2, choice (1) for -Cl). The result that optB88-vdW performs better than PBE-vdW indicates that the former functional, which is less repulsive at short distances, is better adapted for the studied systems than the latter. Our conclusion is based on the study of graphene on Ni(111) [36], which showed that strong repulsion present in PBE-vdW can be alleviated by another choice of DFT-vdW functional such as optB88-vdW. Namely, this work showed that while vdW-DF and vdW-DF2 functionals still result in nonbonding behavior in the vicinity of the surface, the opt-vdW functionals recently developed by Klimes and Michaelides [30] lead to improved binding at short distances.
What remains to be understood is why the cell parameters change unevenly in different systems when vdW forces are not included. Since a significant role of the vdW binding contribution is expected in the effective interdimer interactions, the changes obtained for -Cl seem to be the most reasonable ones as they are observed along the direction that separates organic dimers. On the other hand, the changes obtained for -CuCN are more difficult to understand because they take place along the direction that separates ionic and organic layers. The latter result might be due to the ethylene-end groups of BEDT-TTF molecules, which are not yet fully ordered at 100 K, since for -CuCN at low temperatures, the most affected parameters were found to be the in-plane parameters, as we would expect [22].
3. vdW Interactions and Electronic Band Structure
Identification of the optimal state-of-the-art functionals for implementation of vdW interactions in the studies of structural properties of -CuCN, -AgCN, and -Cl charge transfer salts presented in the previous section also enables a unified determination of the analogously optimal DFT-generated electronic band structure of these compounds. To this end, we allowed for full relaxation of the unit cell (with vdW) before performing band structure calculations. This step is crucial for the electronic band structure calculations since the inclusion of vdW interaction influences the system mostly through the total energy and ground state geometry [27]. The calculations for -CuCN, -AgCN, and -Cl were performed using the same PBE-vdW functional as used in the calculations of unit cell parameters (Section 2), while for -Cl, they were supplemented by the ones using the optB88-vdW functional (Table 2, choice (1)). The obtained results are illustrated in Figure 2, Figure 3, Figure 4 and Figure 5.
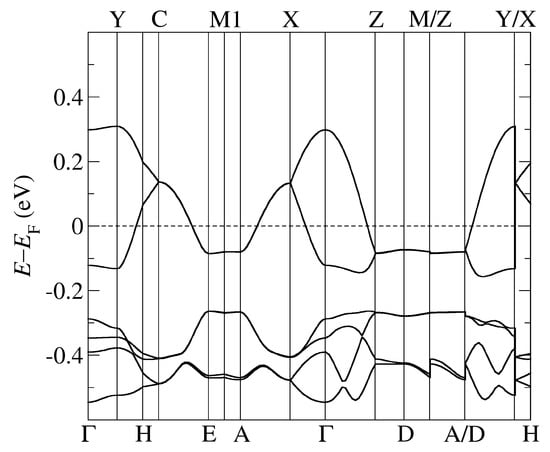
Figure 2.
The band structure of -CuCN obtained by using PBE-vdW and the pseudopotentials C, Cu. = (0,0,0); X = (0,0,1/2); Y = (1/2,0,0), Z = (0,1/2,0) in the units of reciprocal lattice vectors.
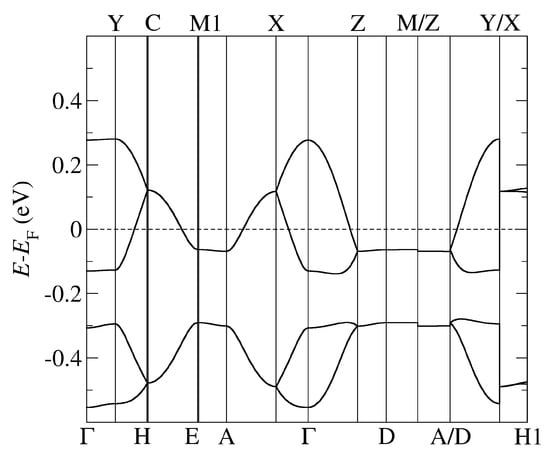
Figure 3.
The band structure of -AgCN obtained by using PBE-vdW and the pseudopotentials C, Ag. = (0,0,0); X = (0,0,1/2); Y = (1/2,0,0), Z = (0,1/2,0) in the units of reciprocal lattice vectors.
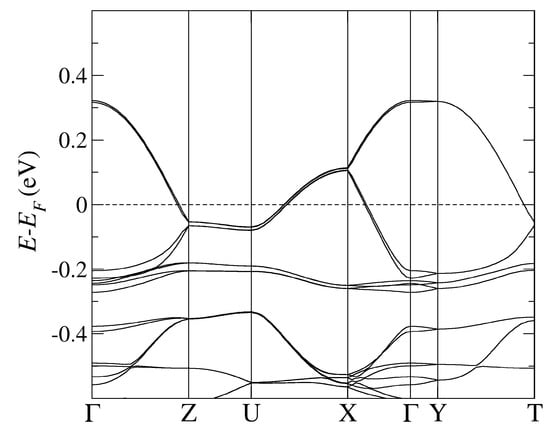
Figure 4.
The band structure of -Cl obtained by using PBE-vdW and the pseudopotentials C, Cu. = (0,0,0); X = (1/2,0,0); Y = (0,1/2,0), Z = (0,0,1/2) in the units of reciprocal lattice vectors.
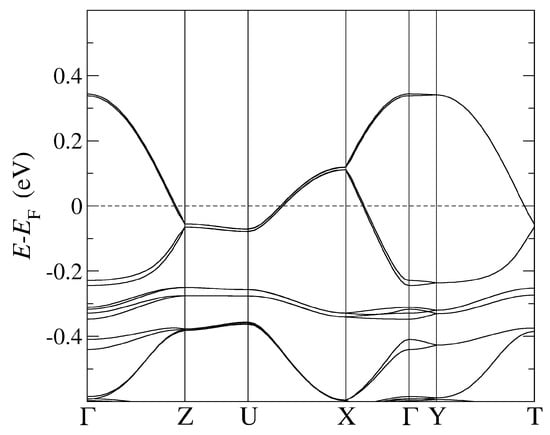
Figure 5.
The band structure of -Cl obtained by using optB88-vdW and the pseudopotentials C, Cu. = (0,0,0); X = (1/2,0,0); Y = (0,1/2,0), Z = (0,0,1/2) in the units of reciprocal lattice vectors.
In Figure 2 and Figure 3, we followed the standard MLC path commonly employed with the monoclinic space group [40]. The high symmetry points are identified as follows: Y corresponds to the -axis, X to the -axis, and Z to the -axis, while A corresponds to the diagonal in the (, ) plane. On the other hand, in Figure 4 and Figure 5, we followed the standard ORC path commonly employed with the monoclinic space group [40]. The high symmetry points are identified as follows: X corresponds to the -axis, Z to the -axis, and Y to the -axis, while U corresponds to the diagonal in the (, ) plane.
We first note that the general properties of the obtained band structures, in particular their shape, are similar to the ones obtained earlier in the calculations using standard DFT functionals without vdW forces and based on the X-ray data refined in different space groups and with fixed experimental unit cell parameters. The earlier DFT calculations were performed for the -CuCN and -Cl systems [8,20,21,22], while only the extended Hüeckel calculations for -AgCN have been reported [11]. The band structure of these systems consists of an anti-bonding combination of the highest occupied molecular orbitals (HOMO) of the BEDT-TTF dimers crossing the Fermi level (FL) (two in -CuCN and -AgCN and four in -Cl) and the bonding combination below the FL (two in -CuCN and -AgCN and four in -Cl). The two bands in -CuCN and -AgCN are fully degenerate along the C–E–M1–A–X and Z–D–M/Z–A/D lines. Note that A–X and Z–D lines correspond to M–Z and Y–M lines in the path used in the calculations of -CuCN [8,20]. More importantly, the degeneracy is found as in other calculations despite the different space group symmetries related to two possible orientations of the anionic cyanides: (this work), P[8], and [20].
Table 5 summarizes our antibonding bandwidth results together with the results obtained by other authors using either the standard DFT functionals without vdW forces or the extended Hüeckel method.
Table 5.
Direct bandwidth of -CuCN, -AgCN, and -Cl, defined as the width of antibonding bands straddling the Fermi level at the point obtained from ab initio calculations that include vdW interactions (this work) or without them ([8,11,20]). Space groups are also denoted.
Comparing the results from our fully self-consistent approach using the vdW DFT functional with the results obtained by standard DFT functional [8,11,20,21,22] in which the absence of vdW is neglected and their omission ad hoc compensated by fixing the unit cell parameters, we find slight deviations in dispersion for all three systems -CuCN, -AgCN, and -Cl: the band shape around the Fermi level is preserved, but the width and details of lower-lying bands are different. In particular, we note that the shape of the bands below the FL and originating from the anions is slightly different, and these bands are located either closer to (-CuCN) or more below the FL (-AgCN) than in the band structures previously obtained in the DFT calculations without inclusion vdW forces. We also note that for -AgCN, we find smaller bandwidths of both the antibonding and bonding bands than in the work based on the extended Hückel calculations [11].
Lastly, the most relevant and direct comparison between the two types of calculations (with and without vdW) can be made in the case of -Cl (see Table 6) because of the absence of orientational ambiguities of the anionic cyanides otherwise present in -CuCN and -AgCN. Importantly, for -Cl we find a larger bandwidth of antibonding bands crossing the FL than in the DFT calculations without vdW forces, the deviation being more pronounced for the optimal choice of the optB88-vdW functional than for the PBE-vdW one. On the other hand, bonding bands below the FL obtained in calculations using either PBE-vdW or optB88-vdW exhibit rather similar width when compared to the band structure results from [20]. However, in the latter calculations without vdW, the bonding bands as well as anionic bands are located closer to the FL than found in our DFT calculations with vdW. Specifically, our calculations show that the bonding bands and anionic bands are located further below the FL when the optimal vdW functional (optB88-vdW) instead of the PBE-vdW functional is used.
Table 6.
Direct antibonding and bonding bandwidths of -Cl defined as the width at the point obtained from ab initio calculations that include vdW interactions (this work), or without them ([20]).
4. Charge Transfer
Electron () and hole () occupations per unit cell (u.c.) were calculated by integrating the density of states of charge transfer bands, viz. the two upper HOMO bands for -CuCN and -AgCN and four bands for -Cl, respectively (see calculated band structures in Figure 2, Figure 3 and Figure 4). From this, we obtain the values presented in Table 7.
Table 7.
Hole () and electron () occupations per unit cell of -CuCN, -AgCN, and -Cl, obtained from ab initio calculations with vdW interactions included via the PBE-vdW functional.
In -AgCN and -Cl, the full charge transfer from BEDT-TTF molecular to anionic subsystem takes place, in accord with stechiometric expectations. By contrast, in -CuCN, the charge transfer is incomplete due to the stronger effect of Cu d-orbitals than in -AgCN. For the -Cl system, we present only the hole occupation per u.c. because the BEDT-TTF band states below the FL are strongly hybridized with anionic Cu states (see Figure 4), which precludes clear identification of the charge transfer. If, by contrast, the optB88-vdW functional is used instead of the PBE-vdW, the resulting BEDT-TTF and Cu-anionic-derived band states do not overlap around the point anymore (cf. Figure 5), and an electron occupation of 4 per u.c. is retrieved here as well.
5. Materials and Methods
First-principles calculations of the electronic structure were carried out in the framework of density functional theory as implemented in the VASP code (VASP version 5.4.1.) [41,42] using the projector augmented-wave method [41,42,43,44,45,46]. Several pseudopotentials were used, as described in [38]. To fulfill the convergence requirements for the plane wave codes, the expansion in the plane waves was consistently performed with the highest cutoff energy of 700 eV. The k-grid was fully converged, which was checked through the energy convergence. The Brillouin zone was sampled by a Monkhorst–Pack choice of k-points [47]. We have used the semilocal functional PBE [35] and the two kinds of self-consistently implemented nonlocal van der Waals density functionals. The first vdW-DF functional was PBE-vdW, including PBE for exchange [35] and correlation from [33]. The second vdW-DF functional was optB88-vdW, including optB88 for exchange [36] and correlation from [30,33,37].
For the input atomic coordinates, the experimental structure previously obtained by X-ray diffraction measurements was used [7,17,34]. The structure was optimized allowing for a change of volume and shape of the unit cell together with atomic positions within, until the forces on atoms dropped below 1 meV/Å.
6. Conclusions
In this communication, we have analyzed the performance of several DFT-based schemes for treating the effects of vdW interactions on the structural and electronic band properties of layered organic charge transfer salts -CuCN, -AgCN, and -Cl. We showed that in addition to ionic bonding between molecular cations and ionic anions, weak vdW interdimer interactions resulting from dynamical correlations between BEDT-TTF molecular pairs play an important role in the determination of their structural, and thereby their electronic, properties.
We have identified the optimal choice of the vdW-DF functional and pseudopotentials for achieving the computed relaxed volumes closest to the experimental ones. By contrast, the DFT calculations that do not take into account the vdW interactions produce large volume enhancements at the level of 10–15%. We also found that the shape of bands straddling the Fermi level is preserved in all cases, but their bandwidth as well as the structural details of low-lying bands differ when compared to the calculations in which the vdW interactions are neglected and their omission ad hoc compensated by fixing the unit cell parameters.
The question remains whether the optimizations using the available vdW-DF functionals could also satisfactorily account for the different aspects of physics and chemistry of these complex systems. Strong local electron correlations and hydrogen bonding represent important phenomena responsible for the stabilization of diverse electronic phases of the above studied systems [48,49]. Detailed theoretical analysis of these aspects of the electronic structure poses a challenge for future work.
Author Contributions
DFT calculations were performed by P.L. Structural data analysis was performed by O.M. The interpretation of the data and the text of the manuscript were drafted by B.G. and S.T., who also conceived the work. All authors contributed to the discussion and the final manuscript.
Funding
Center of Excellence for Advanced Materials and Sensing Devices (ERDF Grant No. KK.01.1.1.01.0001).
Acknowledgments
P.L. was supported by the H2020 CSA Twinning Project No. 692194, RBI-T-WINNING and by the Center of Excellence for Advanced Materials and Sensing Devices (ERDF Grant No. KK.01.1.1.01.0001). We thank I. Lončarić and T. Ivek for helpful comments. S.T. acknowledges very useful discussions with E. Canadell, P. Foury, V. Ilakovac, and J-P. Pouget.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Williams, J.M.; Kini, A.M.; Wang, H.H.; Carlson, K.D.; Geiser, U.; Montgomery, L.K.; Pyrka, G.J.; Watkins, D.M.; Kommers, J.M.; Boryschuk, S.J.; et al. From Semiconductor-Semiconductor Transition (42 K) to the Highest-Tc Organic Superconductor, κ-(BEDT-TTF)2Cu[N(CN)2]Cl (Tc = 12.5 K). Inorg. Chem. 1990, 29, 3272–3274. [Google Scholar] [CrossRef]
- Geiser, U.; Wang, H.H.; Carlson, K.D.; Williams, J.M.; Charlier, H.A.; Heindl, J.E.; Yaconi, G.A.; Love, B.J.; Lathrop, M.W.; Schirber, J.E.; et al. Superconductivity at 2.8 K and 1.5 kbar in κ-(BEDT-TTF)2Cu2(CN)3: First Organic Superconductor Containing a Polymeric Copper Cyanide Anion. Inorg. Chem. 1991, 30, 2586–2588. [Google Scholar] [CrossRef]
- Lunkenheimer, P.; Müller, J.; Krohns, S.; Schrettle, F.; Loidl, A.; Hartmann, B.; Rommel, R.; de Souza, M.; Hotta, C.; Schlueter, J.A.; et al. Multiferroicity in an organic charge-transfer salt that is suggestive of electric-dipole-driven magnetism. Nat. Mater. 2012, 11, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Pinterić, M.; Rivas Góngora, D.; Rapljenović, Z.; Ivek, T.; Čulo, M.; Korin-Hamzić, B.; Milat, O.; Gumhalter, B.; Lazić, P.; Sanz Alonso, M.; et al. Electrodynamics in organic dimer insulators close to Mott Critical point. Crystals 2018, 8, 190. [Google Scholar] [CrossRef]
- Sedlmeier, K.; Elsässer, S.; Neubauer, D.; Beyer, R.; Wu, D.; Ivek, T.; Tomić, S.; Schlueter, J.A.; Dressel, M. Absence of charge order in the dimerized κ-phase BEDT-TTF salts. Phys. Rev. B 2012, 86, 245103. [Google Scholar] [CrossRef]
- Tomić, S.; Pinterić, M.; Ivek, T.; Sedlmeier, K.; Beyer, R.; Wu, D.; Schlueter, J.A.; Schweitzer, D.; Dressel, M. Magnetic ordering and charge dynamics in κ-(BEDT-TTF)2Cu[N(CN)2]Cl. J. Phys. Condens. Matter 2013, 25, 436004. [Google Scholar] [CrossRef] [PubMed]
- Pinterić, M.; Lazić, P.; Pustogow, A.; Ivek, T.; Kuveždić, M.; Milat, O.; Gumhalter, B.; Basletić, M.; Čulo, M.; Korin-Hamzić, B.; et al. Anions effects on the electronic structure and electrodynamic properties of the Mott insulator κ-(BEDT-TTF)2Ag2(CN)3. Phys. Rev. B 2016, 94, 161105(R). [Google Scholar] [CrossRef]
- Foury-Leylekian, P.; Ilakovac, V.; Balédent, V.; Fertey, P.; Arakcheeva, A.; Milat, O.; Petermann, D.; Guillier, G.; Miyagawa, K.; Kanoda, K.; et al. (BEDT-TTF)2Cu2(CN)3 spin liquid: Beyond the average structure. Crystals 2018, 8, 158. [Google Scholar] [CrossRef]
- Kanoda, K.; Kato, R. Mott Physics in Organic Conductors with Triangular Lattices. Annu. Rev. Condens. Matter Phys. 2011, 2, 167. [Google Scholar] [CrossRef]
- Zhou, Y.; Kanoda, K.; Ng, T.-K. Quantum spin liquid states. Rev. Mod. Phys. 2017, 89, 025003. [Google Scholar] [CrossRef]
- Hiramatsu, T.; Yoshida, Y.; Saito, G.; Otsuka, A.; Yamochi, H.; Maesato, M.; Shimizu, Y.; Ito, H.; Nakamura, Y.; Kishida, H.; et al. Design and preparation of a new quantum spin liquid candidate, κ-(ET)2Ag2(CN)3, having a nearby superconductivity. Bull. Chem. Soc. Jpn. 2017, 90, 1073–1082. [Google Scholar] [CrossRef]
- Tomić, S.; Dressel, M. Ferroelectricity in molecular solids: A review of electrodynamic properties. Rep. Prog. Phys. 2015, 78, 096501. [Google Scholar] [CrossRef] [PubMed]
- Dressel, M.; Pustogow, A. Electrodynamics of quantum spin liquids. J. Phys. Condens. Matter 2018, 30, 203001. [Google Scholar] [CrossRef] [PubMed]
- Pustogow, A.; Bories, M.; Löhle, A.; Rösslhuber, R.; Zhukova, E.; Gorshunov, B.; Tomić, S.; Schlueter, J.A.; Hübner, R.; Hiramatsu, T.; et al. Quantum spin liquids unveil the genuine Mott state. Nat. Mater. 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Clay, R.T.; Mazumdar, S. From charge- and spin-ordering to superconductivity in the organic charge-transfer solids. Phys. Rep. 2019, 788, 1–89. [Google Scholar] [CrossRef]
- Pinterić, M.; Čulo, M.; Milat, O.; Basletić, M.; Korin-Hamzić, B.; Tafra, E.; Hamzić, A.; Ivek, T.; Peterseim, T.; Miyagawa, K.; et al. Anisotropic charge dynamics in the spin-Liquid candidate κ-(BEDT-TTF)2Cu2(CN)3. Phys. Rev. B 2014, 90, 195139. [Google Scholar] [CrossRef]
- Dressel, M.; Lazić, P.; Pustogow, A.; Zhukova, E.; Gorshunov, B.; Schlueter, J.A.; Milat, O.; Gumhalter, B.; Tomić, S. Lattice vibrations of the charge-transfer salt κ-(BEDT-TTF)2Cu2(CN)3: Comprehensive explanation of the electrodynamic response in a spin-liquid compound. Phys. Rev. B 2016, 93, 081201(R). [Google Scholar] [CrossRef]
- Čulo, M.; Tafra, E.; Mihaljević, B.; Basletić, M.; Kuveždić, M.; Ivek, T.; Hamzić, A.; Tomić, S.; Hiramatsu, T.; Yoshida, Y.; et al. Hall effect study of the κ-(ET)2X family: Evidence for Mott-Anderson localization. Phys. Rev. B 2019, 99, 045114. [Google Scholar] [CrossRef]
- Lazić, P.; Pinterić, M.; Rivas Góngora, D.; Pustogow, A.; Treptow, K.; Ivek, T.; Milat, O.; Gumhalter, B.; Došlić, N.; Dressel, M.; et al. Importance of van der Waals interactions and cation-anion coupling in an organic quantum spin liquid. Phys. Rev. B 2018, 97, 245134. [Google Scholar] [CrossRef]
- Kandpal, H.C.; Opahle, I.; Zhang, Y.-Z.; Jeschke, H.O.; Valentí, R. Revision of Model Parameters for κ-Type Charge Transfer Salts: An Ab Initio Study. Phys. Rev. Lett. 2009, 103, 067004. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, H.O.; de Souza, M.; Valentí, R.; Manna, R.S.; Lang, M.; Schlueter, J.A. Temperature dependence of structural and electronic properties of the spin-liquid candidate κ-(BEDT-TTF)2Cu2(CN)3. Phys. Rev. B 2012, 85, 035125. [Google Scholar] [CrossRef]
- Koretsune, T.; Hotta, C. Evaluating model parameters of the κ and β′-type Mott insulating organic solids. Phys. Rev. B 2014, 89, 045102. [Google Scholar] [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Bučko, T.; Hafner, J.; Lebègue, S.; Angyan, J.G. Improved Description of the Structure of Molecular and Layered Crystals: Ab Initio DFT Calculations with van der Waals Corrections. J. Phys. Chem. B 2010, 114, 11814–11824. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yu, G.; Sun, Y.; Huang, X.; Guan, J.; Zhang, H.; Li, H.; Chen, W. Molecular charge transfer by adsorbing TCNQ/TTF molecules via p–p interaction: A simple and effective strategy to modulate the electronic and magnetic behaviors of zigzag SiC nanoribbons. Phys. Chem. Chem. Phys. 2015, 17, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Lazić, P.; Atodiresei, N.; Caciuc, V.; Brako, R.; Gumhalter, B.; Blügel, S. Rationale for switching to nonlocal functionals in density functional theory. J. Phys. Condens. Matter 2012, 24, 424215. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klimeš, J.; Bowler, D.R.; Michaelides, A. Chemical accuracy for the van der Waals density functional. J. Phys. Condens. Matter 2010, 22, 022201. [Google Scholar] [CrossRef]
- Lazić, P.; Alaei, M.; Atodiresei, N.; Caciuc, V.; Brako, R.; Blügel, S. Density functional theory with nonlocal correlation: A key to the solution of the CO adsorption puzzle. Phys. Rev. B 2010, 81, 045401. [Google Scholar] [CrossRef]
- Klimeš, J.; Bowler, D.R.; Michaelides, A. Van der Waals density functionals applied to solids. Phys. Rev. B 2011, 83, 195131. [Google Scholar] [CrossRef]
- Berland, K.; Cooper, V.R.; Lee, K.; Schröder, E.; Thonhauser, T.; Hyldgaard, P.; Lundqvist, B.I. Van der Waals forces in density functional theory: a review of the vdW-DF method. Rep. Prog. Phys. 2015, 78, 066501. [Google Scholar] [CrossRef] [PubMed]
- Berland, K.; Chakraborty, D.; Thonhauser, T. Van der Waals density functional with corrected C6 coefficients. Phys. Rev. B 2019, 99, 195418. [Google Scholar] [CrossRef]
- Dion, M.; Rydber, H.; Schröder, E.; Langreth, D.C.; Lundqvist, B.I. Van der Waals Density Functional for General Geometries. Phys. Rev. Lett. 2004, 92, 246401. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, T.; Yoshida, Y.; Saito, G.; Otsuka, A.; Yamochi, H.; Maesato, M.; Shimizu, Y.; Ito, H.; Kishida, H. Quantum spin liquid: Design of a quantum spin liquid next to a superconducting state based on a dimer-type ET Mott insulator. J. Mater. Chem. C 2015, 3, 1378–1388. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Mittendorfer, F.; Garhofer, A.; Redinger, J.; Klimeš, J.; Harl, J.; Kresse, G. GrapheneonNi(111): Strong interaction and weak adsorption. Phys. Rev. B 2011, 84, 201401. [Google Scholar] [CrossRef]
- Román-Pérez, G.; Soler, J.M. Efficient Implementation of a van der Waals Density Functional: Application to Double-Wall Carbon Nanotubes. Phys. Rev. Lett. 2009, 103, 096102. [Google Scholar] [CrossRef]
- Subsection: Recommended PAW Potentials for DFT Calculations Using Vasp.5.2. Available online: https://cms.mpi.univie.ac.at/vasp/vasp/PAW_potentials.html (accessed on 1 December 2018).
- Lejaeghere, K.; Bihlmayer, G.; Björkman, T.; Blaha, P.; Blügel, S.; Blum, V.; Caliste, D.; Castelli, I.E.; Clark, S.J.; Dal Corso, A.; et al. Reproducibility in density functionaltheory calculations of solids. Science 2016, 351, aad3000. [Google Scholar] [CrossRef]
- Setyawan, W.; Curtarolo, S. High-throughput electronic band structure calculations: Challenges and tools. Phys. Rev. B 1976, 13, 299–312. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. J. Phys. Rev. B 1993, 48, 13115. [Google Scholar]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. J. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758558. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillonin-zone integrations. Comput. Mater. Sci. 2010, 49, 5188. [Google Scholar]
- Guterding, D.; Valenti, R.; Jeschke, H.O. Influence of molecular conformations on the electronic structure of organic charge transfer salts. Phys. Rev. B 2015, 92, 081109(R). [Google Scholar] [CrossRef]
- Pouget, J.-P.; Alemany, P.; Canadell, E. Donor-anion interactions in quarter-filled low dimensional organic conductors. Mater. Horiz. 2018, 5, 590–640. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).