Acid Modification of the Unsupported NiMo Catalysts by Y-Zeolite Nanoclusters


Abstract
:1. Introduction
2. Experimental
2.1. Synthesis of Unsupported NiMo Catalysts
2.2. Synthesis of Y-Zeolite Nanoclusters
2.3. Synthesis of Unsupported NiMo Catalysts Modified by Y-Zeolite Nanoclusters
2.4. Characterization of Catalysts
2.5. Catalytic Activity Evaluation
3. Results and Discussion
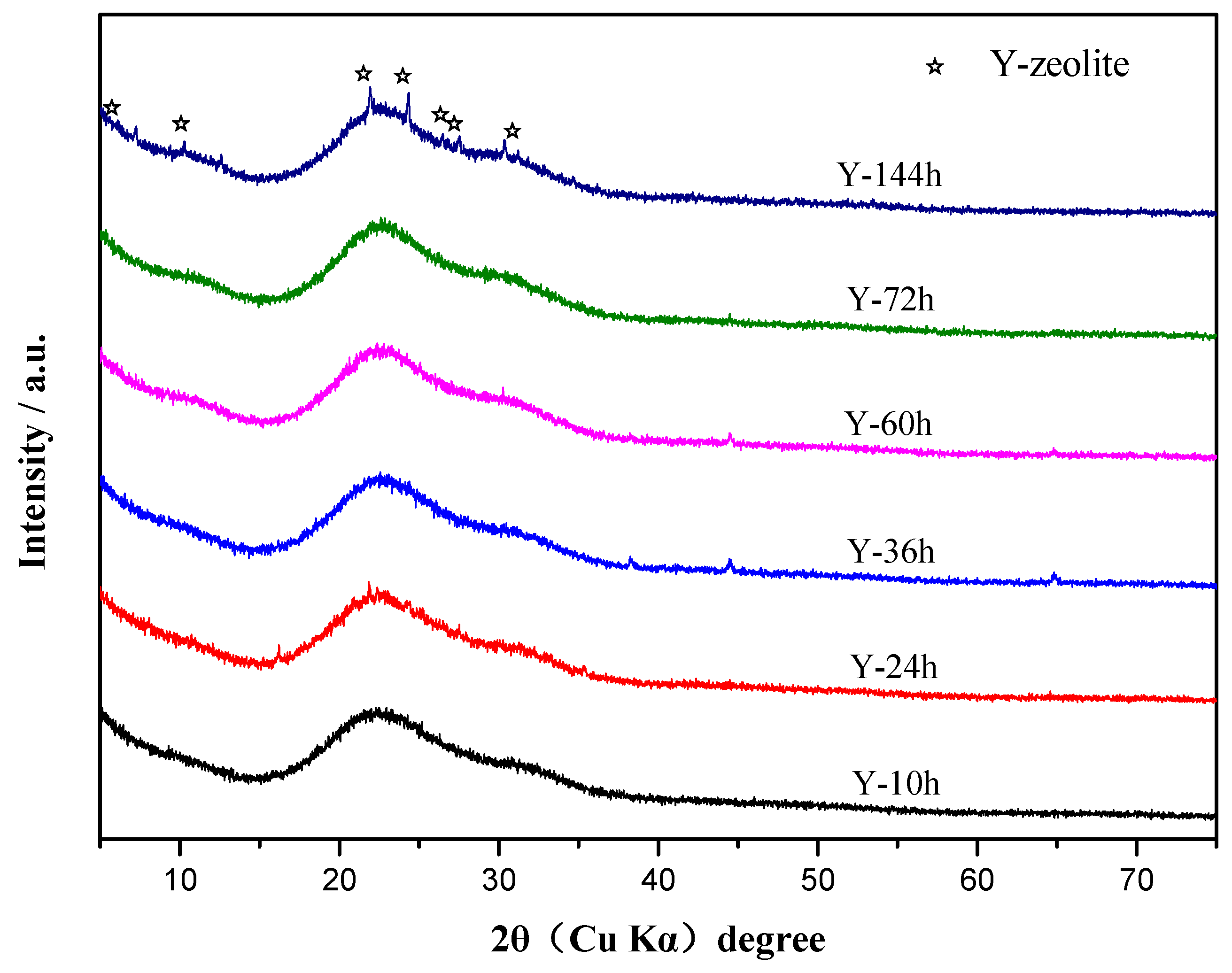
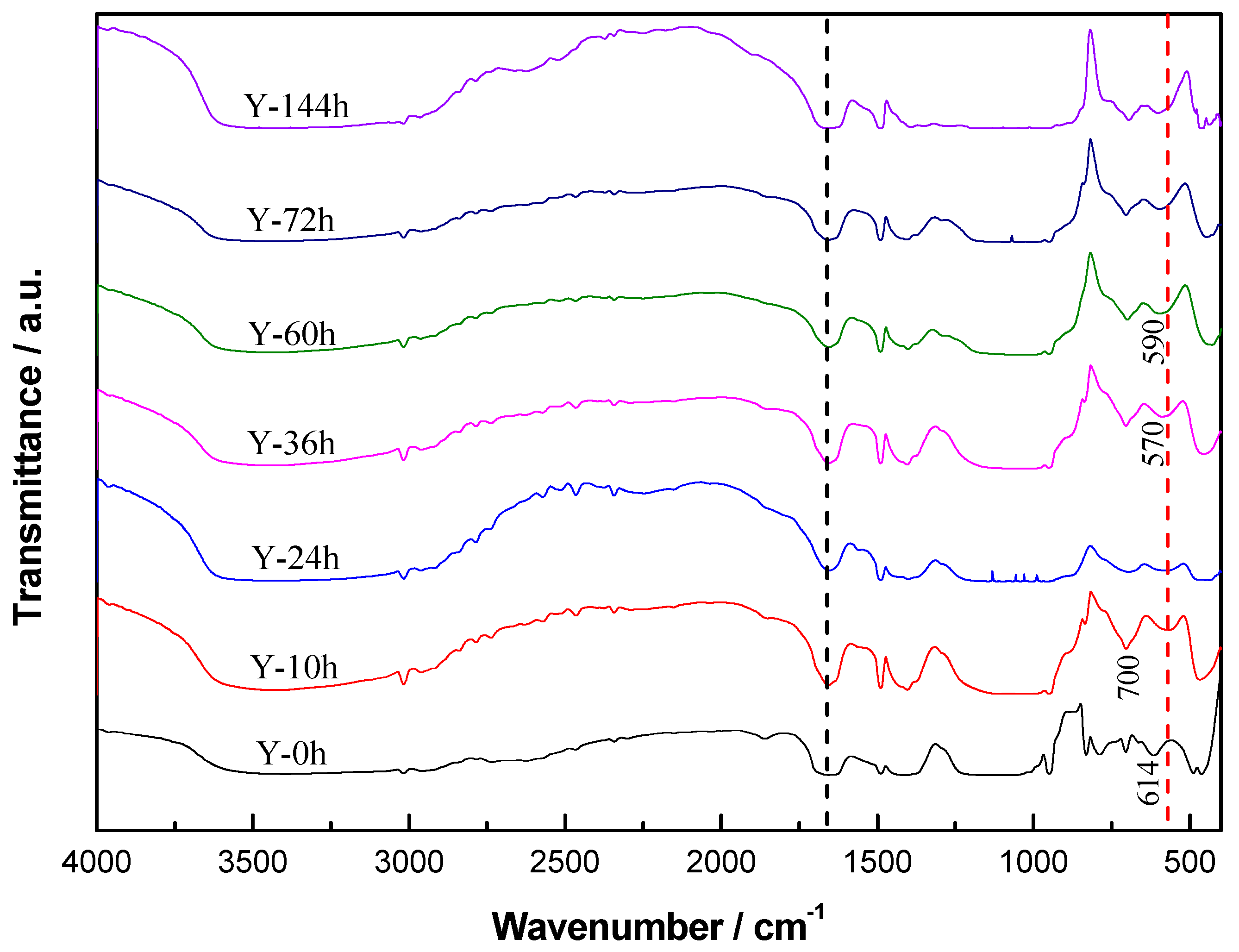
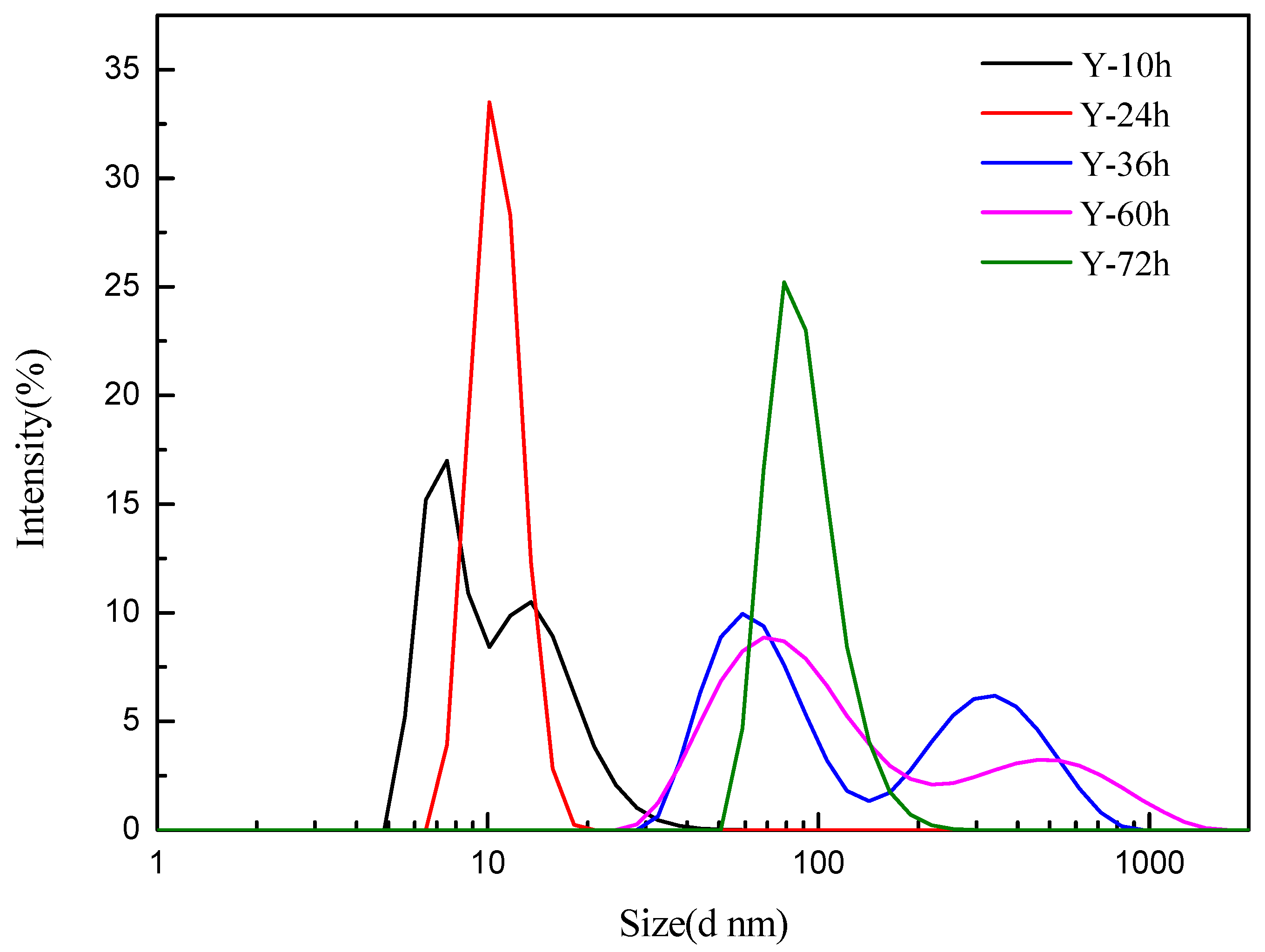
3.1. Characterization of Y-Zeolite Nanoclusters
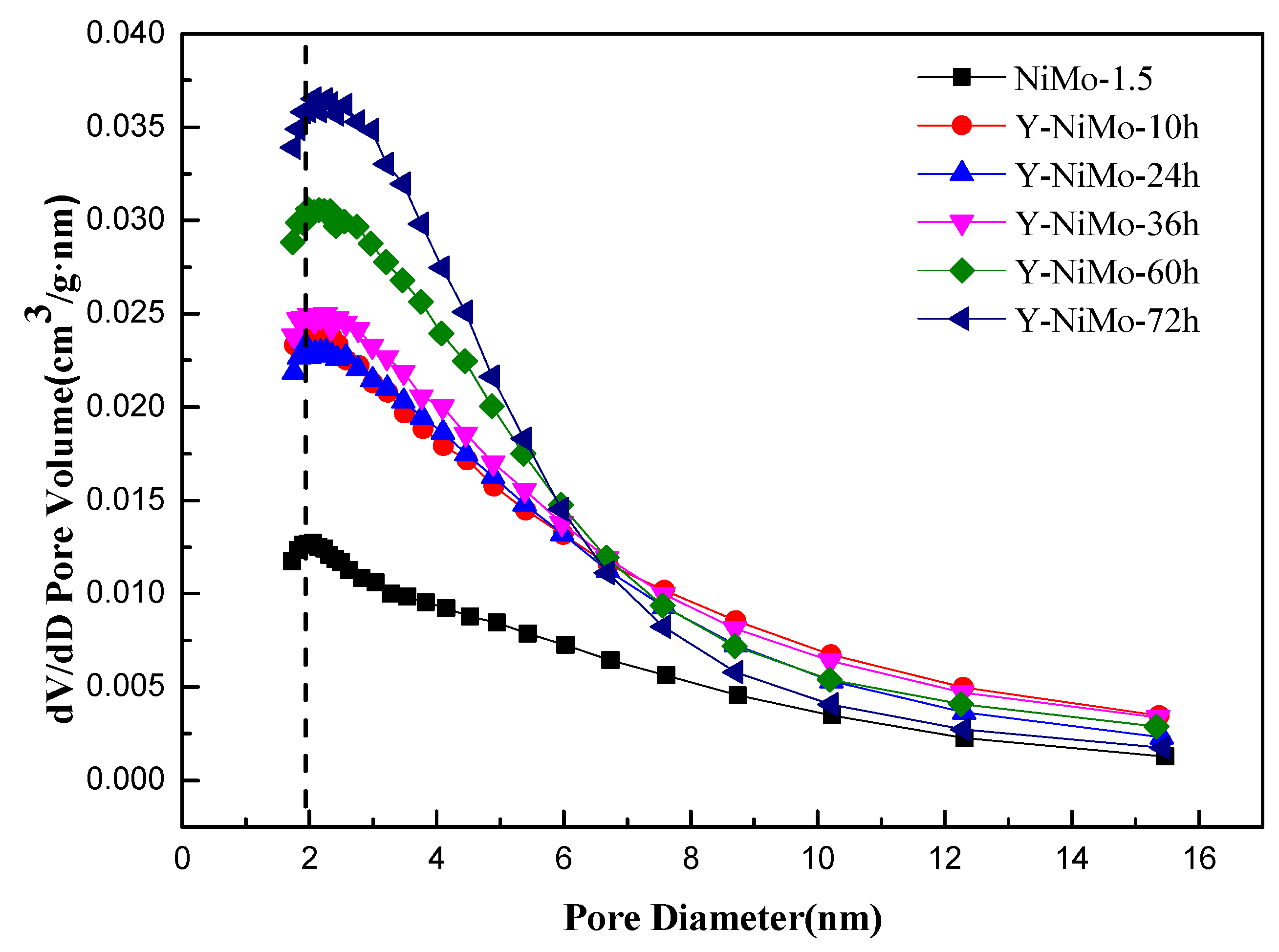
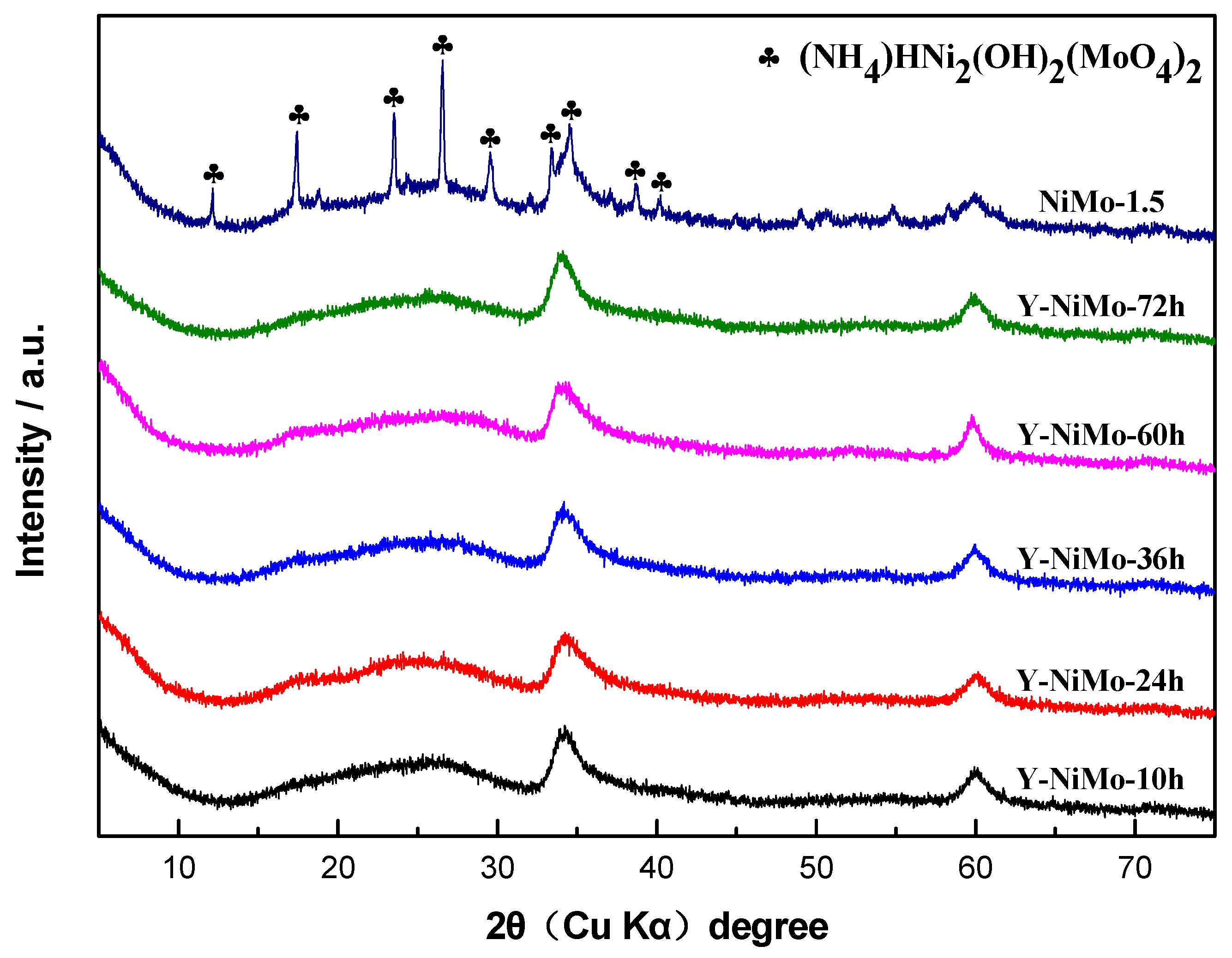
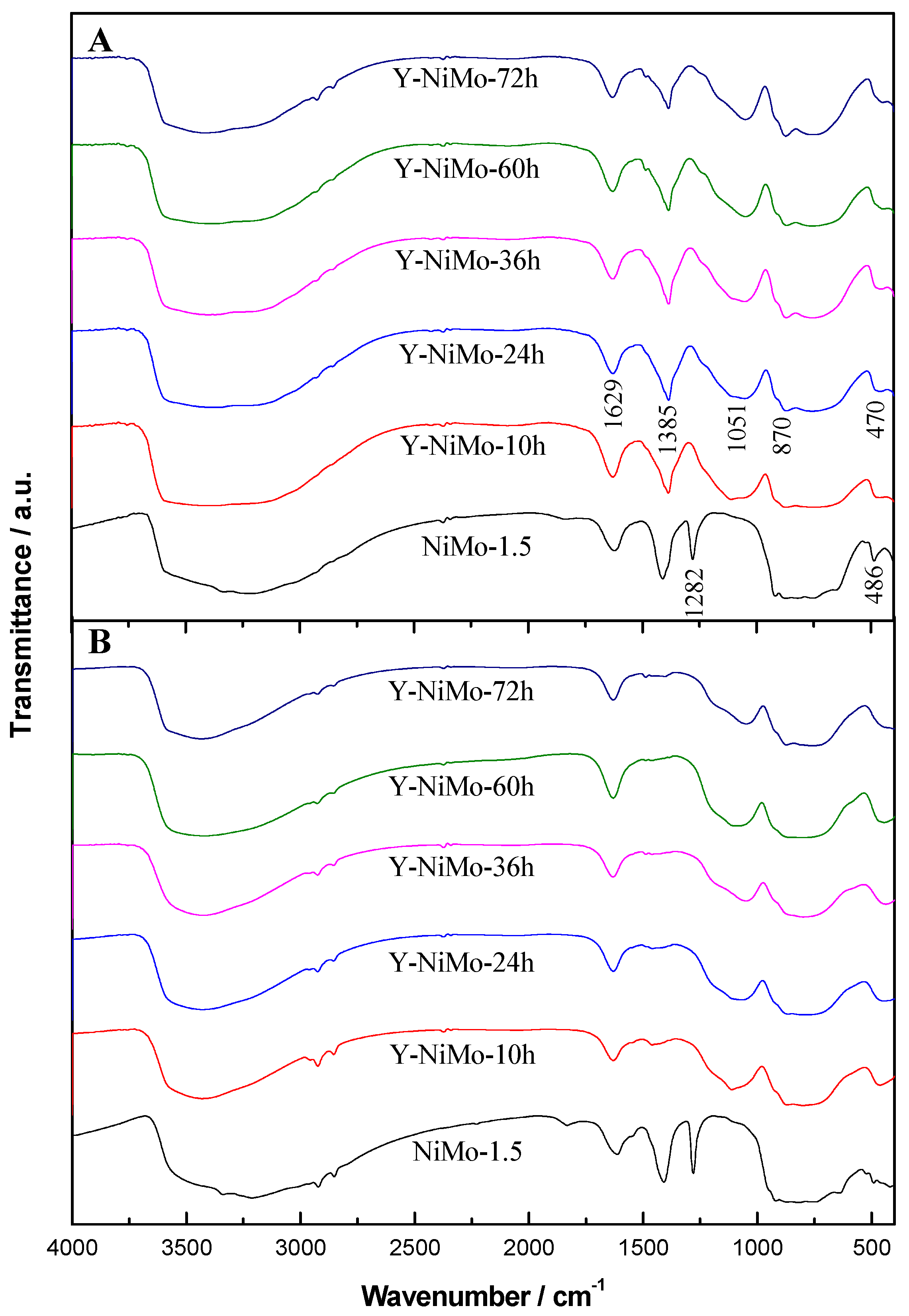
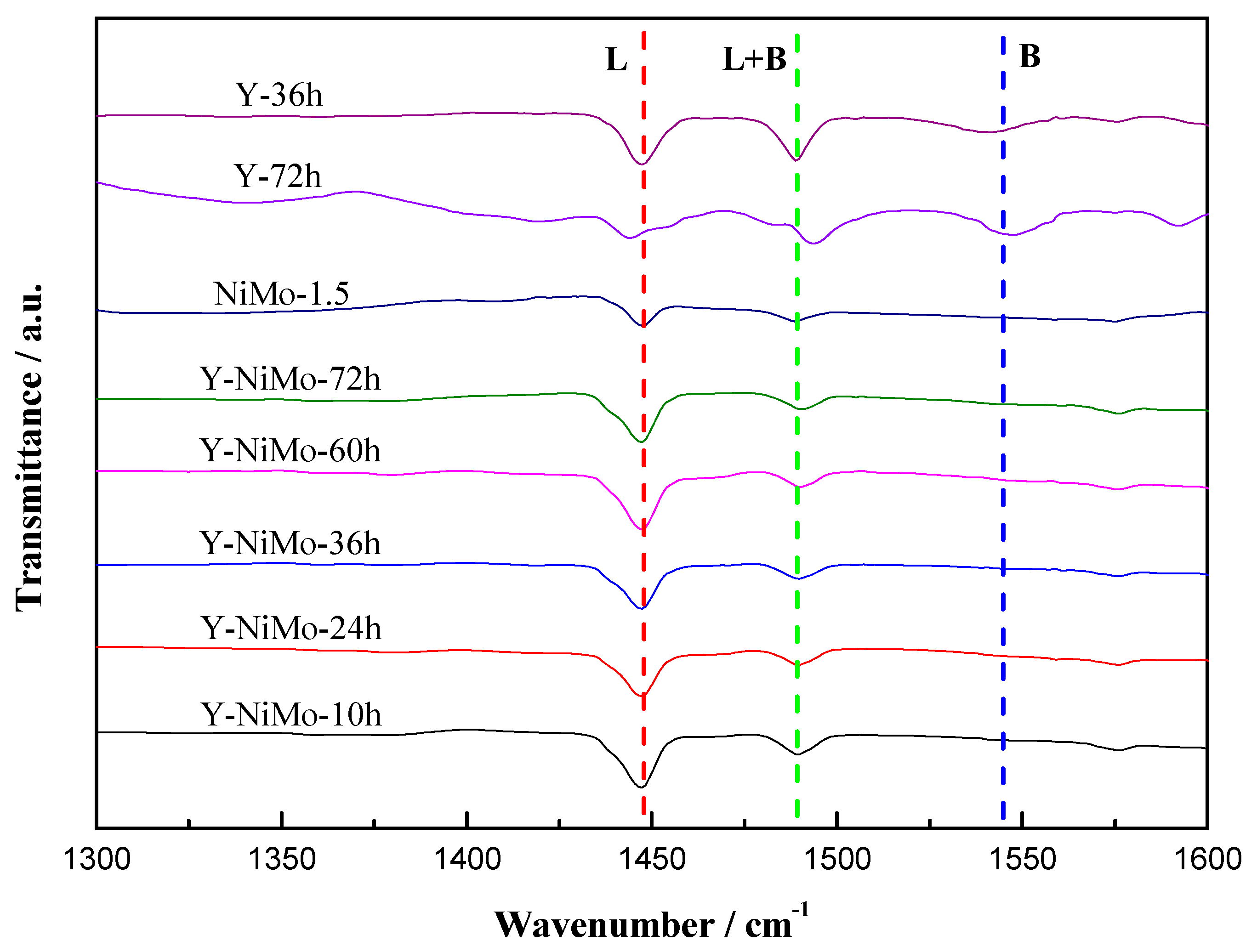
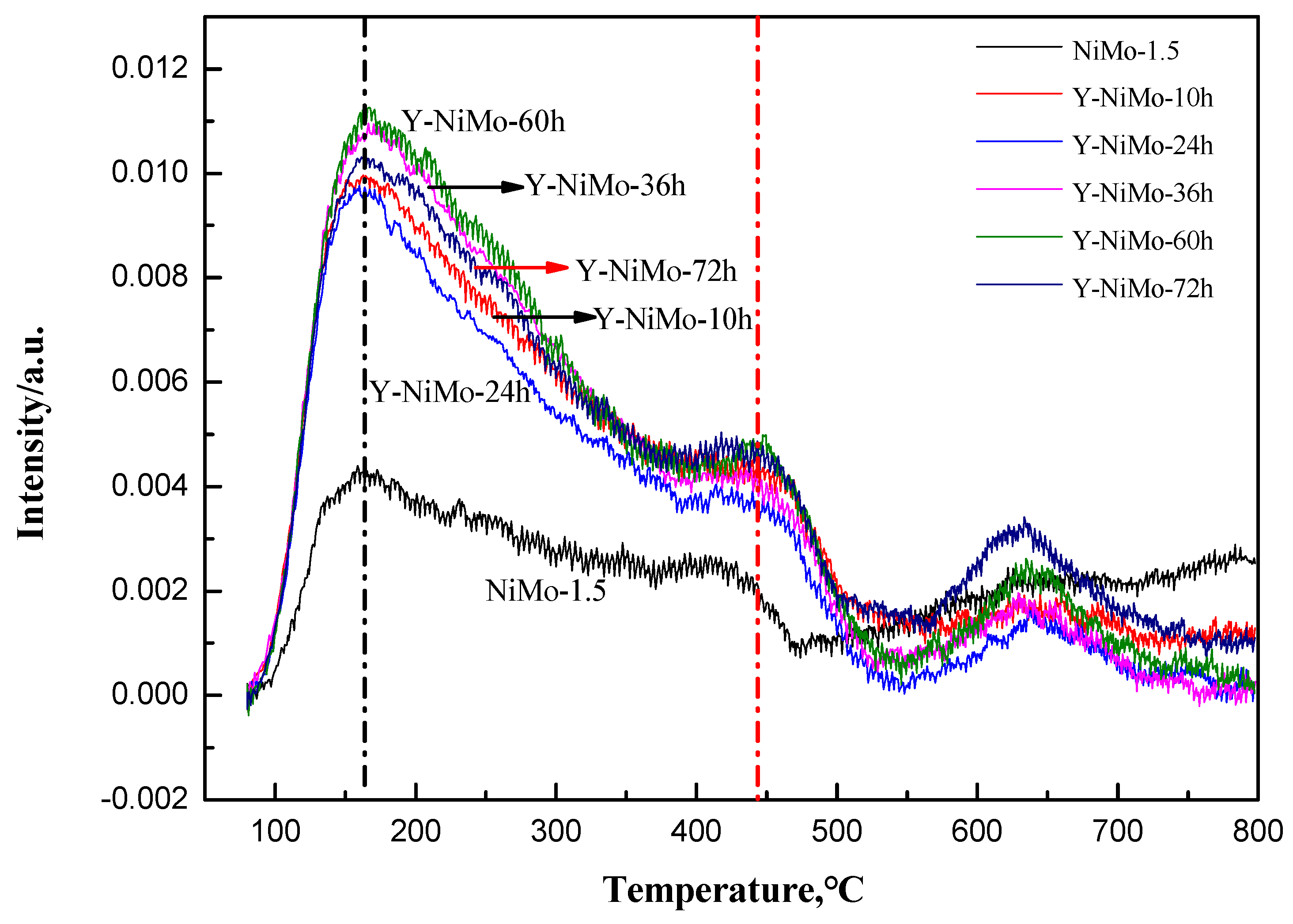
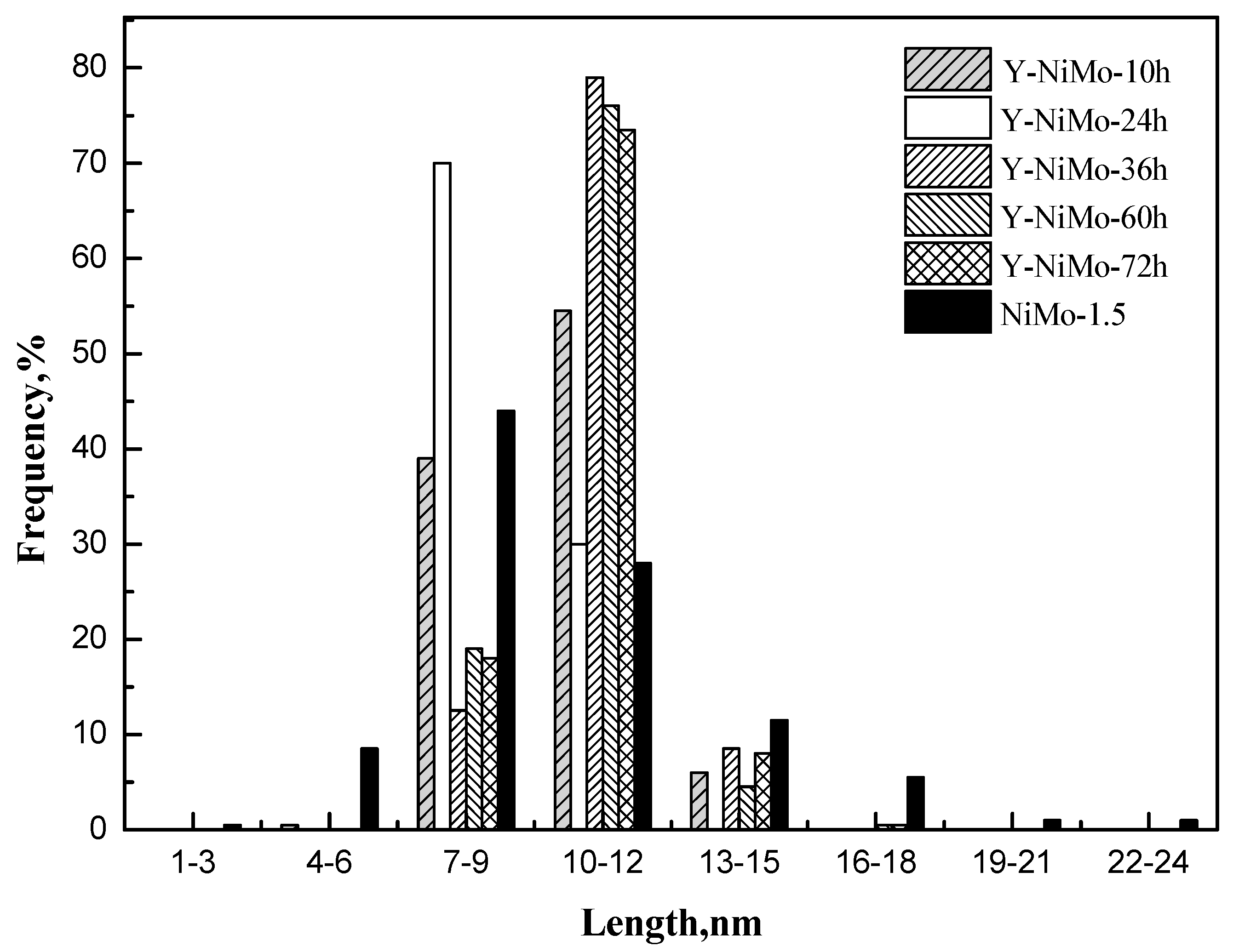
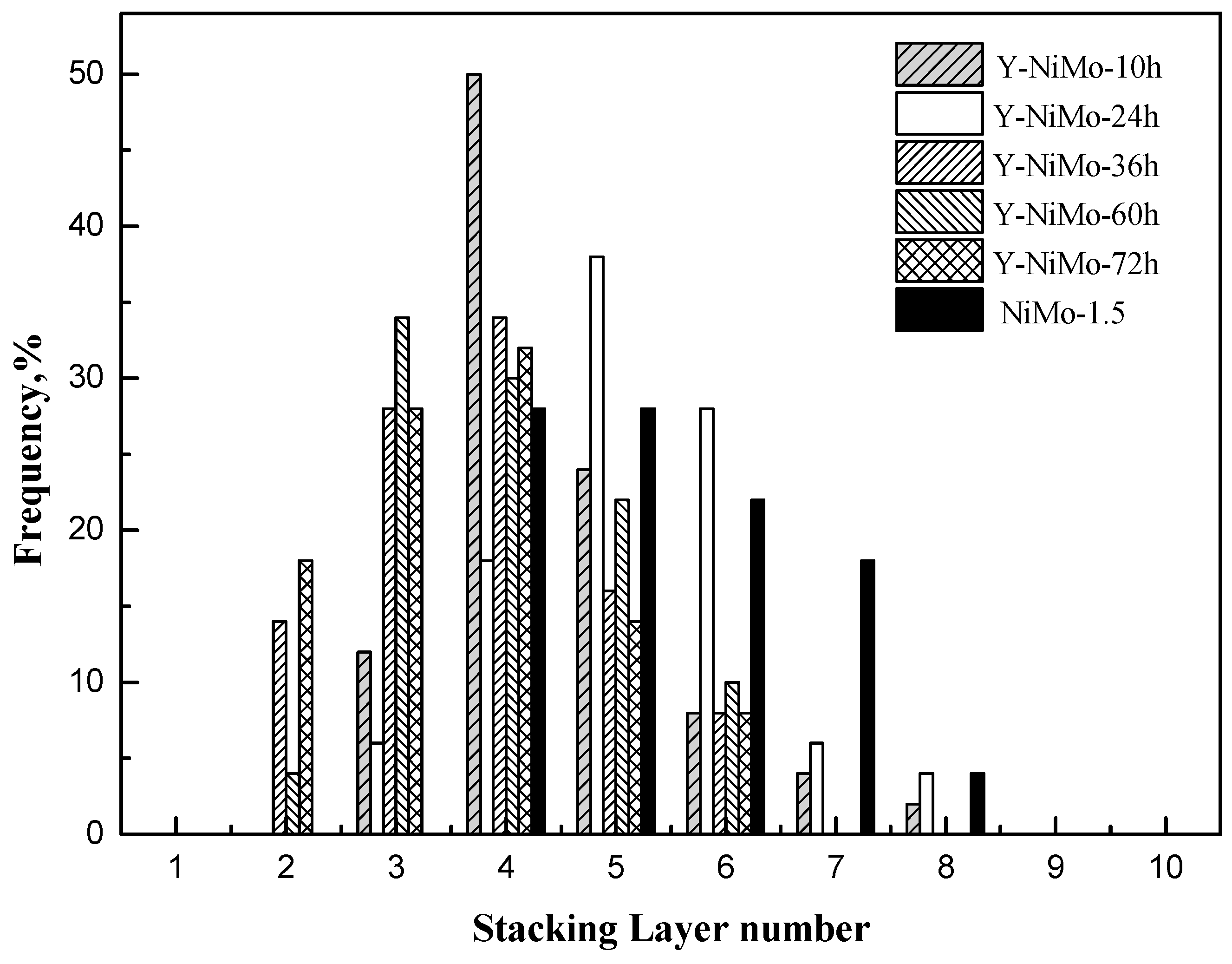
3.2. Characterization of the Modified Unsupported NiMo Catalysts
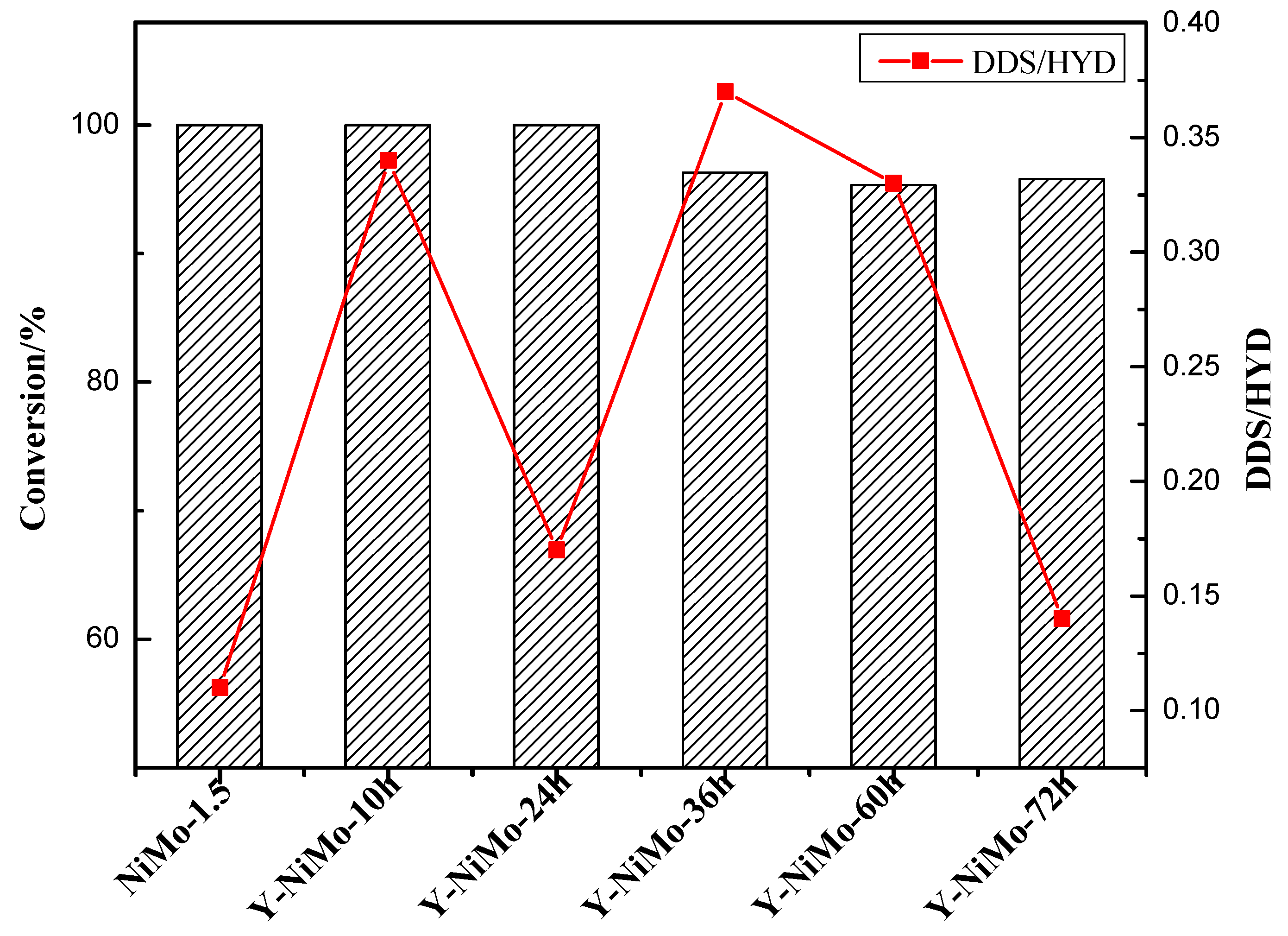
3.3. Catalytic Performance
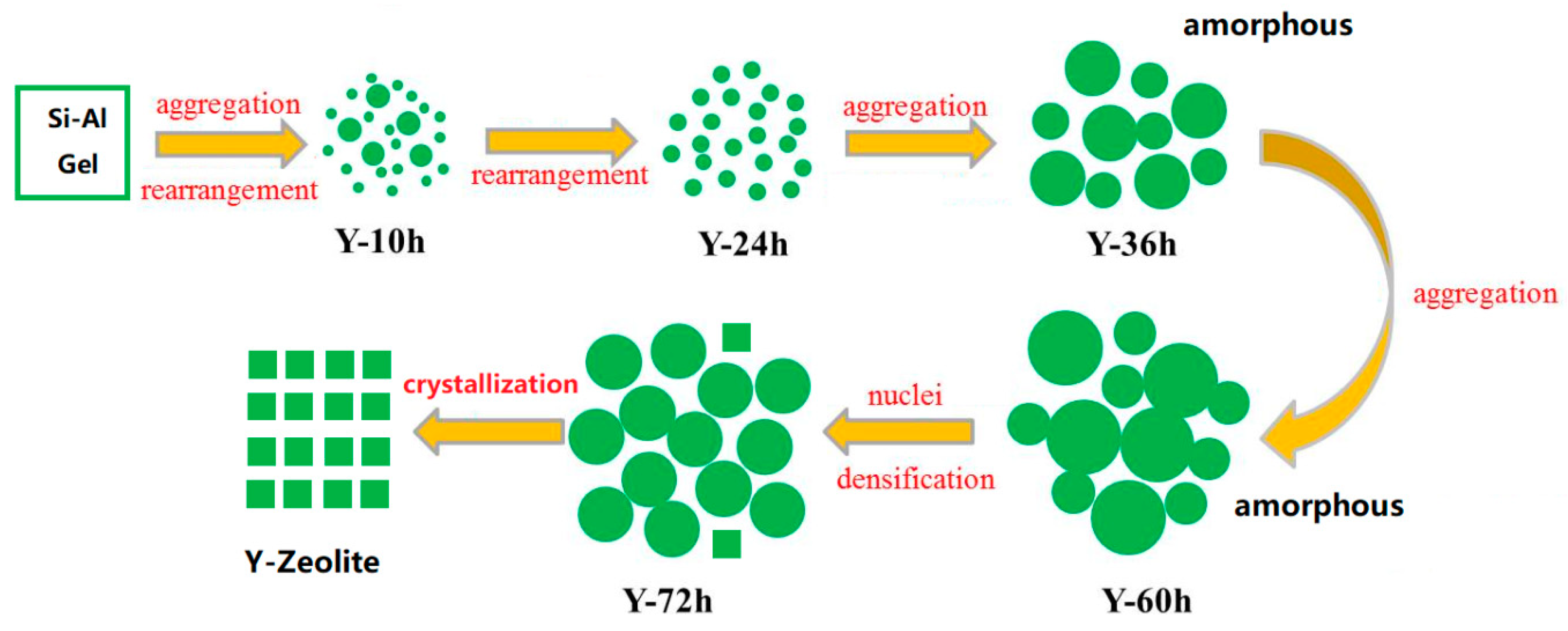
3.4. The Action Mechanism of Y-Zeolite Nanoclusters
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Olivas, A.; Galván, D.H.; Alonso, G.; Fuentes, S. Trimetallic nimow unsupported catalysts for HDS. Appl. Catal. A Gen. 2009, 352, 10–16. [Google Scholar] [CrossRef]
- Bocarando, J.; Huirache-Acuña, R.; Bensch, W.; Huang, Z.D.; Petranovskii, V.; Fuentes, S.; Alonso-Núñez, G. Unsupported Ni-Mo-W sulphide HDS catalysts with the varying nickel concentration. Appl. Catal. A: Gen. 2009, 363, 45–51. [Google Scholar] [CrossRef]
- Yoosuk, B.; Kim, J.H.; Song, C.; Ngamcharussrivichai, C.; Prasassarakich, P. Highly active MoS2, CoMoS2 and NiMoS2 unsupported catalysts prepared by hydrothermal synthesis for hydrodesulfurization of 4,6-dimethyldibenzothiophene. Catal. Today 2008, 130, 14–23. [Google Scholar] [CrossRef]
- Zhang, L.; Afanasiev, P.; Li, D.; Long, X.; Vrinat, M. Solution synthesis of unsupported Ni–W–S hydrotreating catalysts. Catal. Commun. 2007, 8, 2232–2237. [Google Scholar] [CrossRef]
- Korányi, T.I.; Manninger, I.; Paál, Z.; Marks, O.; Günter, J.R. Activation of unsupported CoMo catalysts in thiophene hydrodesulfurization. J. Catal. 1989, 116, 422–439. [Google Scholar] [CrossRef]
- González-Cortés, S.L. Comparing the hydrodesulfurization reaction of thiophene on γ-Al2O3 supported CoMo, NiMo and NiW sulfide catalysts. React. Kinet. Catal. Lett. 2009, 97, 131–139. [Google Scholar] [CrossRef]
- Yongchul, P.; Eunsuok, O.A.; Rhee, H.K. Characterization and catalytic activity of WNiMo/Al2O3 catalyst for hydrodenitrogenation of pyridine. Ind. Eng. Chem. Res. 1997, 36, 5083–5089. [Google Scholar]
- Egorova, M.; Prins, R. Hydrodesulfurization of dibenzothiophene and 4,6-dimethyldibenzothiophene over sulfided NiMo/γ-Al2O3, CoMo/γ-Al2O3, and Mo/γ-Al2O3 catalysts. J. Catal. 2004, 225, 417–427. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Q.; Zhang, J.; Yin, C.; Zhao, Y.; Yin, S.; Liu, C.; Sun, W. PVP-assisted synthesis of unsupported NiMo catalysts with enhanced hydrodesulfurization activity. Fuel Process. Technol. 2017, 160, 93–101. [Google Scholar] [CrossRef]
- Liu, H.; Yin, C.; Li, X.; Chai, Y.; Li, Y.; Liu, C. Effect of NiMo phases on the hydrodesulfurization activities of dibenzothiophene. Catal. Today 2017, 282, 222–229. [Google Scholar] [CrossRef]
- Liu, H.; Liu, C.; Yin, C.; Liu, B.; Li, X.; Li, Y.; Chai, Y.; Liu, Y. Low temperature catalytic hydrogenation naphthalene to decalin over highly-loaded NiMo, NiW and NiMoW catalysts. Catal. Today 2016, 276, 46–54. [Google Scholar] [CrossRef]
- Amaya, S.L.; Alonso-Núñez, G.; Cruz-Reyes, J.; Fuentes, S.; Echavarría, A. Influence of the sulfidation temperature in a NiMoW catalyst derived from layered structure (NH4)Ni2OH(H2O)(MoO4)2. Fuel 2015, 139, 575–583. [Google Scholar] [CrossRef]
- Espino, J.; Alvarez, L.; Ornelas, C.; Rico, J.L.; Fuentes, S.; Berhault, G.; Alonso, G. Comparative study of WS2 and Co(Ni)/WS2 HDS catalysts prepared by ex situ / in situ activation of ammonium thiotungstate. Catal. Lett. 2003, 90, 71–80. [Google Scholar] [CrossRef]
- Gochi, Y.; Ornelas, C.; Paraguay, F.; Fuentes, S.; Alvarez, L.; Rico, J.L.; Alonso-Núñez, G. Effect of sulfidation on Mo-W-Ni trimetallic catalysts in the HDS of DBT. Catal. Today 2005, 107–108, 531–536. [Google Scholar] [CrossRef]
- Wang, T.F.; Ye, Z.; Hui, G.E.; Tang, M.X.; Zhou, L.G.; Lv, Z.; Li, X. Hydrodesulfurization of thiophene over Mo/AC catalyst presulfided by ammonium thiosulfate. J. Fuel Chem. Technol. 2015, 43, 202–207. [Google Scholar] [CrossRef]
- Yin, C.; Wang, Y.; Xue, S.; Liu, H.; He, L.; Liu, C. Influence of sulfidation conditions on morphology and hydrotreating performance of unsupported Ni–Mo–W catalysts. Fuel 2016, 175, 13–19. [Google Scholar] [CrossRef]
- Yin, C.; Dong, C.; Kong, Y.; Li, K.; Zhang, H.; Liu, D.; Liu, C. Effects of aging treatment on the hydrotreating performance of the unsupported catalyst. Ind. Eng. Chem. Res. 2019, 58, 2683–2688. [Google Scholar] [CrossRef]
- Dong, C.; Yin, C.; Wu, T.; Wu, Z.; Liu, D.; Liu, C. Effect of β-zeolite nanoclusters on the acidity and hydrodesulfurization activity of an unsupported NiMo catalyst. Catal. Commun. 2019, 119, 164–169. [Google Scholar] [CrossRef]
- Abbot, J. Role of Brønsted and Lewis acid sites during cracking reactions of alkanes. Appl. Catal. 1989, 47, 33–44. [Google Scholar] [CrossRef]
- Schallmoser, S.; Ikuno, T.; Wagenhofer, M.F.; Kolvenbach, R.; Haller, G.L.; Sanchez-Sanchez, M.; Lercher, J.A. Impact of the local environment of Brønsted acid sites in ZSM-5 on the catalytic activity in n-pentane cracking. J. Catal. 2014, 316, 93–102. [Google Scholar] [CrossRef]
- D’Ippolito, S.A.; Especel, C.; Vivier, L.; Epron, F.; Pieck, C.L. Influence of the Brønsted acidity, SiO2/Al2O3 ratio and rh–pd content on the ring opening: Part I. Selective ring opening of decalin. Appl. Catal. A Gen. 2014, 469, 532–540. [Google Scholar] [CrossRef]
- Kubička, D.; Kumar, N.; Mäki-Arvela, P.; Tiitta, M.; Niemi, V.; Salmi, T.; Murzin, D.Y. Ring opening of decalin over zeolites:I. Activity and selectivity of proton-form zeolites. J. Catal. 2004, 222, 65–79. [Google Scholar] [CrossRef]
- Ferraz, S.G.A.; Zotin, F.M.Z.; Araujo, L.R.R.; Zotin, J.L. Influence of support acidity of NiMoS catalysts in the activity for hydrogenation and hydrocracking of tetralin. Appl. Catal. A Gen. 2010, 384, 51–57. [Google Scholar] [CrossRef]
- Kirschhock, C.E.A.; Ravishankar, R.; Verspeurt, F.; Grobet, P.J.; And, P.A.J.; Martens, J.A. Identification of precursor species in the formation of MFI zeolite in the TPAOH-TEOS-H2O system. J. Phys. Chem. B 1999, 103, 4965–4971. [Google Scholar] [CrossRef]
- Kirschhock, C.E.A.; Ravishankar, R.; Verspeurt, F.; Grobet, P.J.; And, P.A.J.; Martens, J.A. Reply to the comment on “identification of precursor species in the formation of MFI zeolite in the TPAOH-TEOS-H2O system”. J. Phys. Chem. B 2002, 106, 3329–3332. [Google Scholar] [CrossRef]
- Prokešová, P.; Mintova, S.; Čejka, J.; Bein, T. Preparation of nanosized micro/mesoporous composites. Mater. Sci. Eng. C 2003, 23, 1001–1005. [Google Scholar] [CrossRef]
- Frunz, L.; Prins, R.; Pirngruber, G.D. ZSM-5 precursors assembled to a mesoporous structure and its subsequent transformation into a zeolitic phase-from low to high catalytic activity. Microporous Mesoporous Mater. 2006, 88, 152–162. [Google Scholar] [CrossRef]
- Tsapatsis, M.; Lovallo, M.; Okubo, T.; Davis, M.E.; Sadakata, M. Characterization of zeolite L nanoclusters. Chem. Mater. 1995, 7, 1734–1741. [Google Scholar] [CrossRef]
- Li, Y.; Chi, K.; Zhang, H.; Du, P.; Hu, D.; Xiao, C.; Li, H.; Zhao, Z.; Duan, A.; Xu, C. The influence of hydrothermal crystallization temperature on a novel FDU-12 mesoporous composite assembled by ZSM-5 nanoclusters and its hydrodesulfurization performance for DBT and FCC diesel. Fuel Process. Technol. 2018, 180, 56–66. [Google Scholar] [CrossRef]
- Liu, H.; Yin, C.; Liu, B.; Li, X.; Li, Y.; Chai, Y.; Liu, C. Effect of calcination temperature of unsupported NiMo catalysts on the hydrodesulfurization of dibenzothiophene. Energy Fuels 2014, 28, 2429–2436. [Google Scholar] [CrossRef]
- Larlus, O.; Mintova, S.; Bein, T. Environmental syntheses of nanosized zeolites with high yield and monomodal particle size distribution. Microporous Mesoporous Mater. 2006, 96, 405–412. [Google Scholar] [CrossRef]
- Zuo, D.; Vrinat, M.; Nie, H.; Maugé, F.; Shi, Y.; Lacroix, M.; Li, D. The formation of the active phases in sulfided NiW/Al2O3 catalysts and their evolution during post-reduction treatment. Catal. Today 2004, 93, 751–760. [Google Scholar] [CrossRef]
- Yin, H.-L.; Liu, X.-L.; Zhou, T.-N.; Lin, A.-G. Effect of preparation method of nanosized zeolite HY-Al2O3 composite as NiMo catalyst support on diesel HDS. J. Fuel Chem. Technol. 2018, 46, 950–956. [Google Scholar] [CrossRef]
- Hensen, E.J.M.; Kooyman, P.J.; van der Meer, Y.; van der Kraan, A.M.; de Beer, V.H.J.; van Veen, J.A.R.; van Santen, R.A. The relation between morphology and hydrotreating activity for supported MoS2 particles. J. Catal. 2001, 199, 224–235. [Google Scholar] [CrossRef]
- Méndez, F.J.; Franco-López, O.E.; Bokhimi, X.; Solís-Casados, D.A.; Escobar-Alarcón, L.; Klimova, T.E. Dibenzothiophene hydrodesulfurization with NiMo and CoMo catalysts supported on niobium-modified MCM-41. Appl. Catal. B: Environ. 2017, 219, 479–491. [Google Scholar] [CrossRef]
- Ding, L.; Ying, Z.; Zhang, Z.; Chen, J. Hydrotreating of light cycled oil using WNi/Al2O3 catalysts containing zeolite beta and/or chemically treated zeolite Y. J. Catal. 2006, 241, 435–445. [Google Scholar] [CrossRef]
- Prokešová, P.; Mintova, S.; Čejka, J.; Bein, T. Preparation of nanosized micro/mesoporous composites via simultaneous synthesis of beta/MCM-48 phases. Microporous Mesoporous Mater. 2003, 64, 165–174. [Google Scholar] [CrossRef]
- Zhao, J.; Yin, Y.; Li, Y.; Chen, W.; Liu, B. Synthesis and characterization of mesoporous zeolite Y by using block copolymers as templates. Chem. Eng. J. 2016, 284, 405–411. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, K.; Dong, D.; Li, D.; Hill, M.R.; Hill, A.J.; Wang, H. Synthesis of hierarchical porous zeolite NaY particles with controllable particle sizes. Microporous Mesoporous Mater. 2010, 127, 167–175. [Google Scholar] [CrossRef]
- Ikuno, T.; Chaikittisilp, W.; Liu, Z.; Iida, T.; Yanaba, Y.; Yoshikawa, T.; Kohara, S.; Wakihara, T.; Okubo, T. Structure-directing behaviors of tetraethylammonium cations toward zeolite beta revealed by the evolution of aluminosilicate species formed during the crystallization process. JACS J. Am. Chem. Soc. 2015, 137, 14533–14544. [Google Scholar] [CrossRef]
- Levin, D.; Ying, J.Y. Oxidative dehydrogenation of propane by non-stoichiometric nickel molybdates. Stud. Surf. Sci. Catal. 1997, 110, 367–373. [Google Scholar]
- Levin, D.; Soled, S.L.; Ying, J.Y. Crystal structure of an ammonium nickel molybdate prepared by chemical precipitation. Inorg. Chem. 1996, 35, 4191–4197. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Hwang, C.S.; Lee, J.F. Characterization of CeO–Al2O3–SiO2 glasses by infrared and X-Ray absorption near edge structure spectroscopies. J. Mater. Res. 1996, 11, 2641–2650. [Google Scholar] [CrossRef]
- Pelmenschikov, A.G.; Wolput, J.H.M.C.V.; Jaenchen, J.; Santen, R.A.V. (a,b,c) triplet of infrared OH bands of zeolitic H-complexes. J. Phys. Chem. 1995, 99, 3612–3617. [Google Scholar] [CrossRef]
- Flanigen, E.M. Infrared structural studies of zeolite frameworks. Adv. Chem. 1971, 101, 201–229. [Google Scholar]
- Wang, Y.; Ma, J.; Liang, D.; Zhou, M.; Li, F.; Li, R. Lewis and Brønsted acids in super-acid catalyst SO42−/ZrO2–SiO2. J. Mater. Sci. 2009, 44, 6736–6740. [Google Scholar] [CrossRef]
- Weissman, J.G. Niobia-alumina supported hydroprocessing catalysts: Relationship between activity and support surface acidity. Catal.Today 1996, 28, 159–166. [Google Scholar] [CrossRef]
- Zhao, R.Y.; Zeng, L.Y.; Liang, J.; Liu, C.G. Interaction between Ni promoter and Al2O3 support and its effect on the performance of NiMo/γ-Al2O3 catalyst in hydrodesulphurization. J. Fuel Chem. Technol. 2016, 44, 564–569. [Google Scholar] [CrossRef]
- Liu, X.; Li, X.; Yan, Z. Facile route to prepare bimodal mesoporous γ-Al2O3 as support for highly active CoMo-based hydrodesulfurization catalyst. Appl. Catal. B Environ. 2012, 121–122, 50–56. [Google Scholar] [CrossRef]
- Vradman, L.; Landau, M.V. Structure–function relations in supported Ni–W sulfide hydrogenation catalysts. Catal. Lett. 2001, 77, 47–54. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | SBET a/m2·g−1 | Vp b/cm3·g−1 | Dp c/nm |
|---|---|---|---|
| NiMo-1.5 | 67 | 0.09 | 3.99 |
| Y-zeolite nanoclusters | 577 | 0.13 | 2.75 |
| Y-NiMo-10h | 131 | 0.20 | 4.66 |
| Y-NiMo-24h | 126 | 0.17 | 4.10 |
| Y-NiMo-36h | 137 | 0.20 | 4.45 |
| Y-NiMo-60h | 158 | 0.21 | 4.04 |
| Y-NiMo-72h | 176 | 0.20 | 3.65 |
| Samples | L (1447 cm−1) | B (1540 cm−1) | L + B(1490 cm−1) |
|---|---|---|---|
| Y-36h | 0.357 | 0.113 | 0.295 |
| Y-72h | 0.157 | 0.227 | 0.260 |
| NiMo-1.5 | 0.137 | 0 | 0.083 |
| Y-NiMo-10h | 0.390 | 0 | 0.132 |
| Y-NiMo-24h | 0.302 | 0 | 0.101 |
| Y-NiMo-36h | 0.305 | 0 | 0.105 |
| Y-NiMo-60h | 0.382 | 0 | 0.104 |
| Y-NiMo-72h | 0.342 | 0 | 0.117 |
| Samples | 100–300 °C | 300–400 °C | 400–550 °C |
|---|---|---|---|
| NiMo-1.5 | 61.034 | 23.269 | 15.697 |
| Y-NiMo-10h | 144.208 | 46.715 | 43.576 |
| Y-NiMo-24h | 134.164 | 41.182 | 31.856 |
| Y-NiMo-36h | 156.233 | 46.907 | 37.488 |
| Y-NiMo-60h | 160.110 | 47.368 | 42.105 |
| Y-NiMo-72h | 149.215 | 47.091 | 45.245 |
| Samples | DBT a | BP b | CHB c | BCH d | DHP e |
|---|---|---|---|---|---|
| NiMo-1.5 | 0 | 4.1 | 12.9 | 23.2 | 59.8 |
| Y-NiMo-10h | 0 | 18.0 | 38.0 | 15.2 | 28.9 |
| Y-NiMo-24h | 0 | 7.4 | 26.1 | 18.7 | 47.8 |
| Y-NiMo-36h | 3.7 | 17.3 | 33.0 | 14.0 | 31.9 |
| Y-NiMo-60h | 4.8 | 13.6 | 25.1 | 16.0 | 40.5 |
| Y-NiMo-72h | 4.2 | 6.6 | 28.9 | 18.8 | 41.5 |
| Samples | LA/nm | NA | fMo |
|---|---|---|---|
| NiMo-1.5 | 10.2 | 5.4 | 0.11 |
| Y-NiMo-10h | 9.9 | 4.5 | 0.12 |
| Y-NiMo-24h | 9.1 | 5.2 | 0.13 |
| Y-NiMo-36h | 10.8 | 3.8 | 0.11 |
| Y-NiMo-60h | 10.5 | 4.0 | 0.12 |
| Y-NiMo-72h | 10.7 | 3.7 | 0.11 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, C.; Yin, C.; Wu, T.; Wu, Z.; Liu, D.; Liu, C. Acid Modification of the Unsupported NiMo Catalysts by Y-Zeolite Nanoclusters. Crystals 2019, 9, 344. https://doi.org/10.3390/cryst9070344
Dong C, Yin C, Wu T, Wu Z, Liu D, Liu C. Acid Modification of the Unsupported NiMo Catalysts by Y-Zeolite Nanoclusters. Crystals. 2019; 9(7):344. https://doi.org/10.3390/cryst9070344
Chicago/Turabian StyleDong, Chengwu, Changlong Yin, Tongtong Wu, Zhuyan Wu, Dong Liu, and Chenguang Liu. 2019. "Acid Modification of the Unsupported NiMo Catalysts by Y-Zeolite Nanoclusters" Crystals 9, no. 7: 344. https://doi.org/10.3390/cryst9070344
APA StyleDong, C., Yin, C., Wu, T., Wu, Z., Liu, D., & Liu, C. (2019). Acid Modification of the Unsupported NiMo Catalysts by Y-Zeolite Nanoclusters. Crystals, 9(7), 344. https://doi.org/10.3390/cryst9070344