Comparative Study of Different Crystallization Methods in the Case of Cilostazol Crystal Habit Optimization


Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Crystallization Methods
2.2.1. Conventional Crystallization Methods
2.2.2. Impinging Jet Crystallization
2.3. Characterization of the Cilostazol Particles
2.3.1. Determination of Crystal Morphology
2.3.2. Particle Size Distribution Analysis
2.3.3. Identification of Polymorphism
2.4. Determination of Wettability by Contact Angle Measurement
2.5. Investigation of Dissolution Rate
2.6. Statistical Analysis
3. Results and Discussions
3.1. Crystal Morphology
3.2. Polymorphism
3.3. Wettability
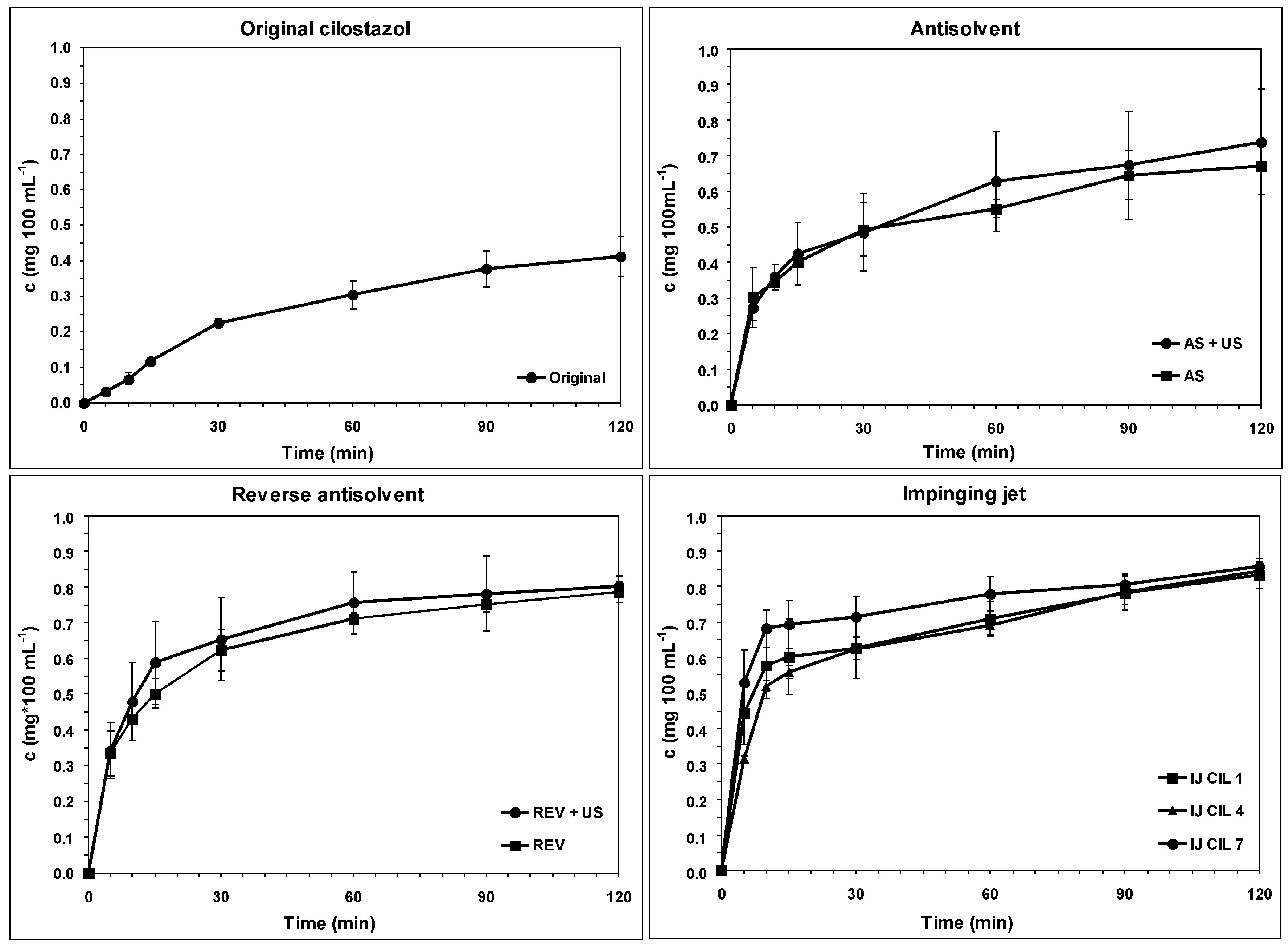
3.4. Dissolution Rate
3.5. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miletic, T.; Kyriakos, K.; Graovac, A.; Ibric, S. Spray-dried voriconazole–cyclodextrin complexes: Solubility, dissolution rate and chemical stability. Carbohydr. Polym. 2013, 98, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Tang, N.; Lian, R.; Qi, J.; Wu, W. Understanding the relationship between wettability and dissolution of solid dispersion. Int. J. Pharm. 2014, 465, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Rasenack, N.; Müller, B.W. Micron-size drug particles: Common and novel micronization techniques. Pharm. Dev. Technol. 2004, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Patole, T.; Deshpande, A. Co-crystallization—A technique for solubility enhancement. Int. J. Pharm. Sci. Res. 2014, 5, 3566–3576. [Google Scholar]
- Gao, Z.; Rohani, S.; Gong, J.; Wang, J. Recent developments in the crystallization process: Toward the pharmaceutical industry. Engineering 2017, 3, 343–353. [Google Scholar] [CrossRef]
- Pitt, K.; Peña, R.; Tew, J.D.; Pal, K.; Smith, R.; Nagy, Z.K.; Litster, J.D. Particle design via spherical agglomeration: A critical review of controlling parameters, rate processes and modelling. Powder Technol. 2018, 326, 327–343. [Google Scholar] [CrossRef]
- Lovette, M.A.; Doherty, M.F. Needle-shaped crystals: Causality and solvent selection guidance based on periodic bond chains. Cryst. Growth Des. 2013, 13, 3341–3352. [Google Scholar] [CrossRef]
- Simone, E.; Othman, R.; Vladisavljevic, G.T.; Nagy, K.Z. Preventing crystal agglomeration of pharmaceutical crystals using temperature cycling and a novel membrane crystallization procedure for seed crystal generation. Pharmaceutics 2018, 10, 17. [Google Scholar] [CrossRef]
- Han, X.; Ghoroi, C.; To, D.; Chen, Y.; Davé, R. Simultaneous micronization and surface modification for improvement of flow and dissolution of drug particles. Int. J. Pharm. 2011, 415, 185–195. [Google Scholar] [CrossRef]
- Vandana, K.; Raju, Y.P.; Chowdary, V.H.; Sushma, M.; Kumar, N.V. An overview on in situ micronization technique—An emerging novel concept in advanced drug delivery. Saudi Pharm. J. 2014, 22, 283–289. [Google Scholar] [CrossRef]
- An, J.-H.; Kiyonga, A.N.; Lee, E.H.; Jung, K. Simple and efficient spherical crystallization of clopidogrel bisulfate form-I via anti-solvent crystallization method. Crystals 2019, 9, 53. [Google Scholar] [CrossRef]
- Am Ende, D.J.; Brenek, S.J. Strategies to control particle size during crystallization processes. Am. Pharm. Rev. 2004, 7, 98–104. [Google Scholar]
- Aigner, Z.; Szegedi, Á.; Szabadi, V.; Ambrus, R.; Sovány, T.; Szabó-Révész, P. Comparative study of crystallization processes in case of glycine crystallization. Acta Pharm. Hung. 2012, 82, 61–68. [Google Scholar] [PubMed]
- Matsumoto, M.; Wada, Y.; Onoe, K. Change in glycine polymorphs induced by minute-bubble injection during antisolvent crystallization. Adv. Powder Technol. 2015, 26, 415–421. [Google Scholar] [CrossRef]
- Rimez, B.; Debuysschère, R.; Conté, J.; Lecomte-Norrant, E.; Gourdon, C.; Cognet, P.; Scheid, B. Continuous-flow tubular crystallization to discriminate between two competing crystal polymorphs. 1. Cooling crystallization. Cryst. Growth Des. 2018, 18, 6431–6439. [Google Scholar] [CrossRef]
- Chester, E.; Markwalter, C.E.; Prud’homme, R.K. Design of a small-scale multi-inlet vortex mixer for scalable nanoparticle production and application to the encapsulation of biologics by inverse flash nanoprecipitation. J. Pharm. Sci. 2018, 107, 2465–2471. [Google Scholar]
- Alvarez, A.J.; Myerson, A.S. Continuous plug flow crystallization of pharmaceutical compounds. Cryst. Growth Des. 2010, 10, 2219–2228. [Google Scholar] [CrossRef]
- Su, C.-S.; Liao, C.-Y.; Jheng, W.-D. Particle size control and crystal habit modification of phenacetin using ultrasonic crystallization. Chem. Eng. Technol. 2015, 38, 181–186. [Google Scholar] [CrossRef]
- Kim, H.N.; Suslick, K.S. The effects of ultrasound on crystals: Sonocrystallization and sonofragmentation. Crystals 2018, 8, 280. [Google Scholar] [CrossRef]
- Dhumal, R.S.; Biradar, S.V.; Paradkar, A.R.; York, P. Particle engineering using sonocrystallization: Salbutamol sulphate for pulmonary delivery. Int. J. Pharm. 2009, 368, 129–137. [Google Scholar] [CrossRef]
- Gielen, B.; Claes, T.; Janssens, J.; Jordens, J.; Thomassen, L.C.J.; Gerven, T.V.; Braeken, L. Particle size control during ultrasonic cooling crystallization of paracetamol. Chem. Eng. Technol. 2017, 40, 1300–1308. [Google Scholar] [CrossRef]
- Yang, Z.-Y.; Yen, S.-K.; Hu, W.-S.; Huang, Y.-Z.; Yang, T.-M.; Su, C.-S. Sonocrystallization—Case studies of salicylamide particle size reduction and isoniazid derivative synthesis and crystallization. Crystals 2018, 8, 249. [Google Scholar] [CrossRef]
- Cheng, J.; Yang, C.; Jiang, M.; Li, Q.; Mao, Z.-S. Simulation of antisolvent crystallization in impinging jets with coupled multiphase flow-micromixing-PBE. Chem. Eng. Sci. 2017, 171, 500–512. [Google Scholar] [CrossRef]
- Tari, T.; Fekete, Z.; Szabó-Révész, P.; Aigner, Z. Reduction of glycine particle size by impinging jet crystallization. Int. J. Pharm. 2015, 478, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Calvignac, B.; Boutin, O. The impinging jets technology: A contacting device using a SAS process type. Powder Technol. 2009, 191, 200–205. [Google Scholar] [CrossRef]
- Tamura, H.; Kadota, K.; Shirakawa, Y.; Tozuka, Y.; Shimosaka, A.; Hidaka, J. Morphology control of amino acid particles in interfacial crystallization using inkjet nozzle. Adv. Powder Technol. 2014, 25, 847–852. [Google Scholar] [CrossRef]
- Tari, T.; Ambrus, R.; Szakonyi, G.; Madarász, D.; Frohberg, P.; Csóka, I.; Szabó-Révész, P.; Ulrich, J.; Aigner, Z. Optimizing the crystal habit of glycine by using additive for impinging jet crystallization. Chem. Eng. Technol. 2017, 40, 1323–1331. [Google Scholar] [CrossRef]
- Jiang, M.; Li, Y.-E.D.; Tung, H.-H.; Braatz, R.D. Effect of jet velocity on crystal size distribution from antisolvent and cooling crystallizations in a dual impinging jet mixer. Chem. Eng. Process. 2015, 97, 242–247. [Google Scholar] [CrossRef]
- Dubbini, A.; Censi, R.; Martena, V.; Hoti, E.; Ricciutelli, M.; Malaj, L.; Martino, P.D. Influence of pH and method of crystallization on the solid physical form of indomethacin. Int. J. Pharm. 2014, 473, 536–544. [Google Scholar] [CrossRef]
- Blagden, N.; de Matas, M.; Gavan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef]
- Kakran, M.; Sahoo, N.G.; Li, L.; Judeh, Z. Particle size reduction of poorly water soluble artemisinin via antisolvent precipitation with a syringe pump. Powder Technol. 2013, 237, 468–476. [Google Scholar] [CrossRef]
- Xu, J.; Luo, K.Q. Enhancing the solubility and bioavailability of isoflavone by particle size reduction using a supercritical carbondioxide-based precipitation. Chem. Eng. Res. Des. 2014, 92, 2542–2549. [Google Scholar] [CrossRef]
- Szabó-Révész, P.; Göcző, H.; Pintye-Hódi, K.; Kása, P.; Erős, I.; Hasznos-Nezdei, M.; Farkas, B. Development of spherical crystal agglomerates of an aspartic acid salt for direct tablet making. Powder Technol. 2011, 114, 118–124. [Google Scholar] [CrossRef]
- Lin, R.; Liu, W.; Woo, M.W.; Chen, X.D.; Selomulya, C. On the formation of “coral-like” spherical α-glycine crystalline particles. Powder Technol. 2015, 279, 310–316. [Google Scholar] [CrossRef]
- Tanaka, M.; Yamanaka, S.; Shirakawa, Y.; Shimosaka, A.; Hidaka, J. Preparation of porous particles by liquid–liquid interfacial crystallization. Adv. Powder Technol. 2011, 22, 125–130. [Google Scholar] [CrossRef]
- Hesselbach, J.; Barth, N.; Lippe, K.; Schilde, C.; Kwade, A. Process chain and characterisation of nanoparticle enhanced composite coatings. Adv. Powder Technol. 2015, 26, 1624–1632. [Google Scholar] [CrossRef]
- Hayato, U.; Toshio, T.; Yukio, K.; Hiroyoshi, H. Purification of cyclic adenosine monophosphate phosphodiesterase from human platelets using new-inhibitor sepharose chromatography. Biochem. Pharmacol. 1984, 33, 3339–3344. [Google Scholar] [CrossRef]
- Mahmoud, D.B.; Shukr, H.; Bendas, E.R. In vitro and in vivo evaluation of self-nanoemulsifying drug delivery systems of cilostazol for oral and parenteral administration. Int. J. Pharm. 2014, 476, 60–69. [Google Scholar] [CrossRef]
- Ha, E.-S.; Ha, D.-H.; Kuk, D.-H.; Sim, W.-Y.; Baek, I.-H.; Kim, J.-S.; Park, H.J.; Kim, M.-S. Solubility of cilostazol in the presence of polyethylene glycol 4000, polyethylene glycol 6000, polyvinylpyrrolidone K30, and poly(1-vinylpyrrolidone-co-vinyl acetate) at different temperatures. J. Chem. Thermodyn. 2017, 113, 6–10. [Google Scholar] [CrossRef]
- Stowell, G.W.; Behme, R.J.; Denton, S.M.; Pfeiffer, I.; Sancilio, F.D.; Whittall, L.B.; Whittle, R.R. Thermally-prepared polymorphic forms of cilostazol. J. Pharm. Sci. 2002, 91, 2481–2488. [Google Scholar] [CrossRef]
- Mustapha, O.; Kim, K.S.; Shafique, S.; Kim, D.S.; Jin, S.G.; Seo, Y.G.; Youn, Y.S.; Oh, K.T.; Lee, B.J.; Park, Y.J.; et al. Development of novel cilostazol–loaded solid SNEDDS using a SPG membrane emulsification technique: Physicochemical characterization and in vivo evaluation. Colloids Surf. B 2017, 150, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Mustapha, O.; Kim, K.S.; Shafique, S.; Kim, D.S.; Jin, S.G.; Seo, Y.G.; Youn, Y.S.; Oh, K.T.; Yong, C.S.; Kim, J.O.; et al. Comparison of three different types of cilostazol-loaded solid dispersion: Physicochemical characterization and pharmacokinetics in rats. Colloids Surf. B 2017, 154, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Sun, C.; Jiang, T.; Zheng, L.; Wang, T.; Wang, S. Investigation of nanosized crystalline form to improve the oral bioavailability of poorly water soluble cilostazol. J. Pharm. Pharm. Sci. 2011, 14, 196–214. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gouthami, K.S.; Kumar, D.; Thipparaboina, R.; Chavan, R.B.; Shastri, N.R. Can crystal engineering be as beneficial as micronisation and overcome its pitfalls?: A case study with cilostazol. Int. J. Pharm. 2015, 491, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Lee, S.; Park, J.-S.; Woo, J.-S.; Hwang, S.-J. Micronization of cilostazol using supercritical antisolvent (SAS) process: Effect of process parameters. Powder Technol. 2007, 177, 64–70. [Google Scholar] [CrossRef]
- Jinno, J.-I.; Kamada, N.; Miyake, M.; Yamada, K.; Mukai, T.; Odomi, M.; Toguchi, H.; Liversidge, G.G.; Higaki, K.; Kimura, T. Effect of particle size reduction on dissolution and oral absorption of a poorly water-soluble drug, cilostazol, in beagle dogs. J. Control. Release 2006, 111, 56–64. [Google Scholar] [CrossRef]
- Dahlberg, C.; Millqvist-Fureby, A.; Schuleit, M. Surface composition and contact angle relationships for differently prepared solid dispersions. Eur. J. Pharm. Biopharm. 2008, 70, 478–485. [Google Scholar] [CrossRef]
- Tian, F.; Sandler, N.; Aaltonen, J.; Lang, C.; Saville, D.J.; Gordon, K.C.; Strachan, C.J.; Rantanen, J.; Rades, T. Influence of polymorphic form, morphology, and excipient interactions on the dissolution of carbamazepine compacts. J. Pharm. Sci. 2007, 96, 584–594. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | US Amplitude | US Cycle Time | Percentage Yield | Roundness | Particle Size | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| (%) | (s) | Mean (m%) | SD | Mean | SD | d(0.5) (µm) | SD | D[4,3] (µm) | SD | |
| AS | 0 | 0.0 | 88.70 | 5.35 | 4.39 | 0.37 | 14.411 | 1.761 | 19.218 | 1.268 |
| AS+US | 70 | 0.3 | 90.39 | 3.22 | 2.95 | 0.19 | 11.246 | 1.025 | 15.738 | 1.149 |
| REV | 0 | 0.0 | 55.97 | 6.70 | 2.36 | 0.23 | 9.906 | 0.309 | 12.802 | 0.521 |
| REV+US | 70 | 0.3 | 92.93 | 1.03 | 2.27 | 0.27 | 8.028 | 0.567 | 12.006 | 3.115 |
| Original | - | - | - | - | 2.08 | 0.86 | 23.563 | 4.158 | 60.639 | 10.294 |
| Sample Code | ΔT | Post Mixing Time | Percentage Yield | Roundness | Particle Size | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| (°C) | (min) | Mean (m%) | SD | Mean | SD | d(0.5) (µm) | SD | D[4,3] (µm) | SD | |
| IJ CIL 1 | 0 | 0 | 79.62 | 1.21 | 1.56 | 0.11 | 3.887 | 0.032 | 5.281 | 0.046 |
| IJ CIL 2 | 0 | 5 | 81.94 | 0.95 | 1.57 | 0.08 | 4.259 | 0.095 | 5.653 | 0.084 |
| IJ CIL 3 | 0 | 10 | 87.01 | 3.58 | 1.58 | 0.10 | 4.801 | 0.099 | 6.027 | 0.077 |
| IJ CIL 4 | 10 | 0 | 79.41 | 2.70 | 1.63 | 0.03 | 3.810 | 0.027 | 5.171 | 0.029 |
| IJ CIL 5 | 10 | 5 | 85.53 | 0.82 | 1.67 | 0.05 | 4.134 | 0.025 | 5.423 | 0.057 |
| IJ CIL 6 | 10 | 10 | 86.80 | 0.29 | 1.71 | 0.02 | 4.557 | 0.106 | 6.578 | 0.091 |
| IJ CIL 7 | 20 | 0 | 75.40 | 2.30 | 1.53 | 0.02 | 3.626 | 0.054 | 5.158 | 0.028 |
| IJ CIL 8 | 20 | 5 | 84.05 | 0.23 | 1.66 | 0.05 | 3.714 | 0.019 | 5.200 | 0.011 |
| IJ CIL 9 | 20 | 10 | 83.63 | 0.17 | 1.63 | 0.07 | 3.759 | 0.018 | 4.824 | 0.122 |
| Original | AS | AS + US | REV | REV + US | IJ CIL 1 | IJ CIL 4 | IJ CIL 7 | |
|---|---|---|---|---|---|---|---|---|
| Mean (°) | 62.4 | 61.7 | 59.9 | 58.5 | 54.6 | 52.1 | 53.3 | 46.9 |
| SD | 1.3 | 1.1 | 0.6 | 1.1 | 0.9 | 0.4 | 0.2 | 0.2 |
| Dependent Variable | Polynomial Function | r2 |
|---|---|---|
| d(0.5) | y = 4.06 + 0.30x1 − 0.31x2 − 0.02x12 + 0.08x22 − 0.20x1x2 | 0.870 |
| D[4,3] | y = 5.48 + 0.30x1 − 0.30x2 − 0.04x12 + 0.18x22 − 0.27x1x2 | 0.596 |
| Roundness | y = 1.2 − 0.02x1 − 0.002x2 + 0.01x12 + 0.03x22 − 0.01x1x2 | 0.401 |
| Dissolution rate | y = 7.38 − 0.09x1 + 0.16x2 + 0.09x12 − 0.22x22 − 0.04x1x2 | 0.797 |
| Percentage yield | y = 82.60 + 3.84x1 − 0.92x2 + 0.93x12 − 0.99x22 + 0.21x1x2 | 0.899 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tari, T.; Szabó-Révész, P.; Aigner, Z. Comparative Study of Different Crystallization Methods in the Case of Cilostazol Crystal Habit Optimization. Crystals 2019, 9, 295. https://doi.org/10.3390/cryst9060295
Tari T, Szabó-Révész P, Aigner Z. Comparative Study of Different Crystallization Methods in the Case of Cilostazol Crystal Habit Optimization. Crystals. 2019; 9(6):295. https://doi.org/10.3390/cryst9060295
Chicago/Turabian StyleTari, Tímea, Piroska Szabó-Révész, and Zoltán Aigner. 2019. "Comparative Study of Different Crystallization Methods in the Case of Cilostazol Crystal Habit Optimization" Crystals 9, no. 6: 295. https://doi.org/10.3390/cryst9060295
APA StyleTari, T., Szabó-Révész, P., & Aigner, Z. (2019). Comparative Study of Different Crystallization Methods in the Case of Cilostazol Crystal Habit Optimization. Crystals, 9(6), 295. https://doi.org/10.3390/cryst9060295