Substituent Effects in the Crystal Packing of Derivatives of 4′-Phenyl-2,2′:6′,2″-Terpyridine

 , ,
, ,  and
and 
Abstract
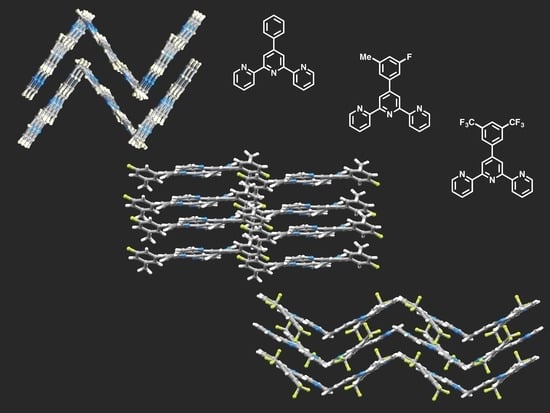
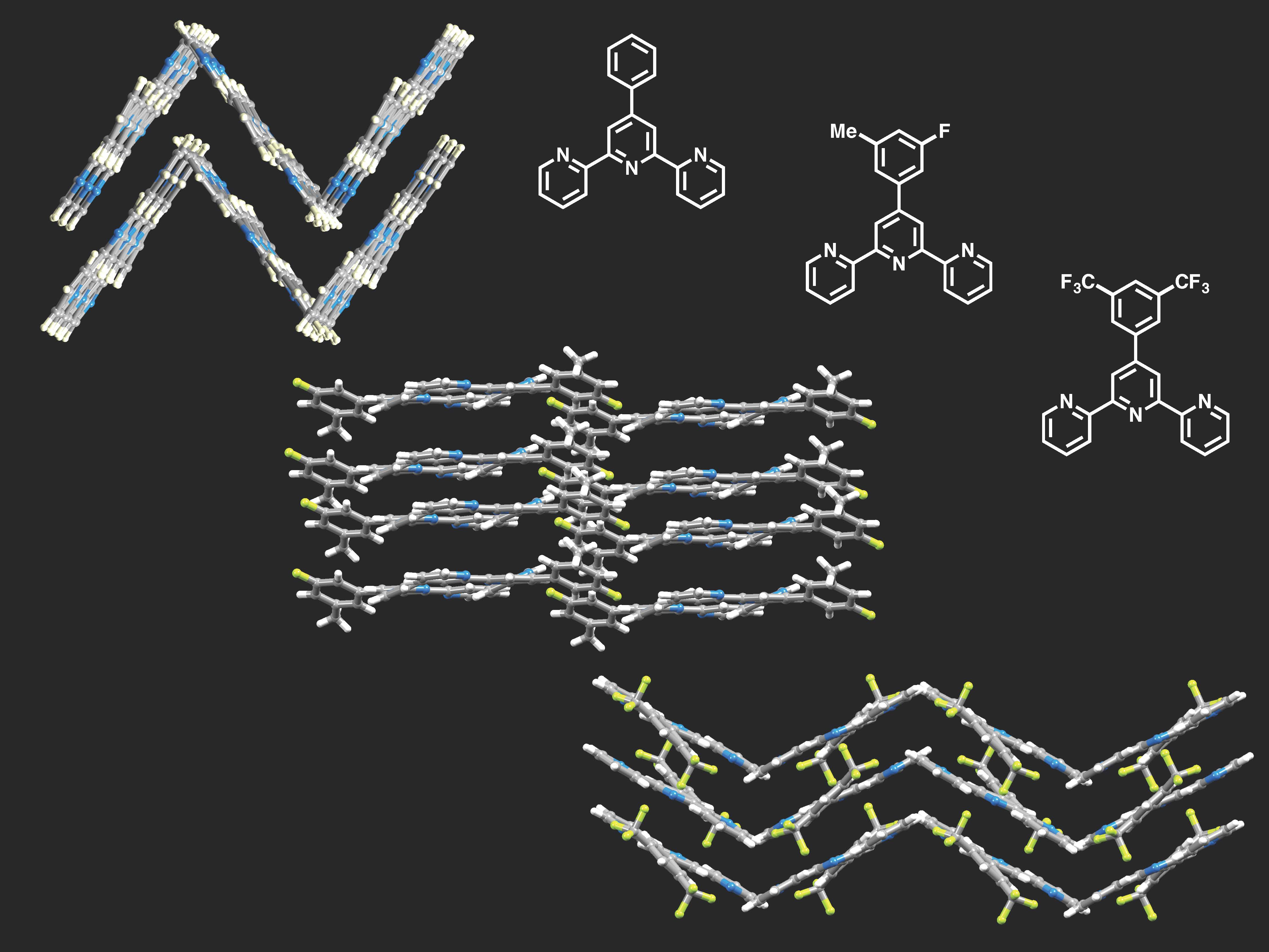{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
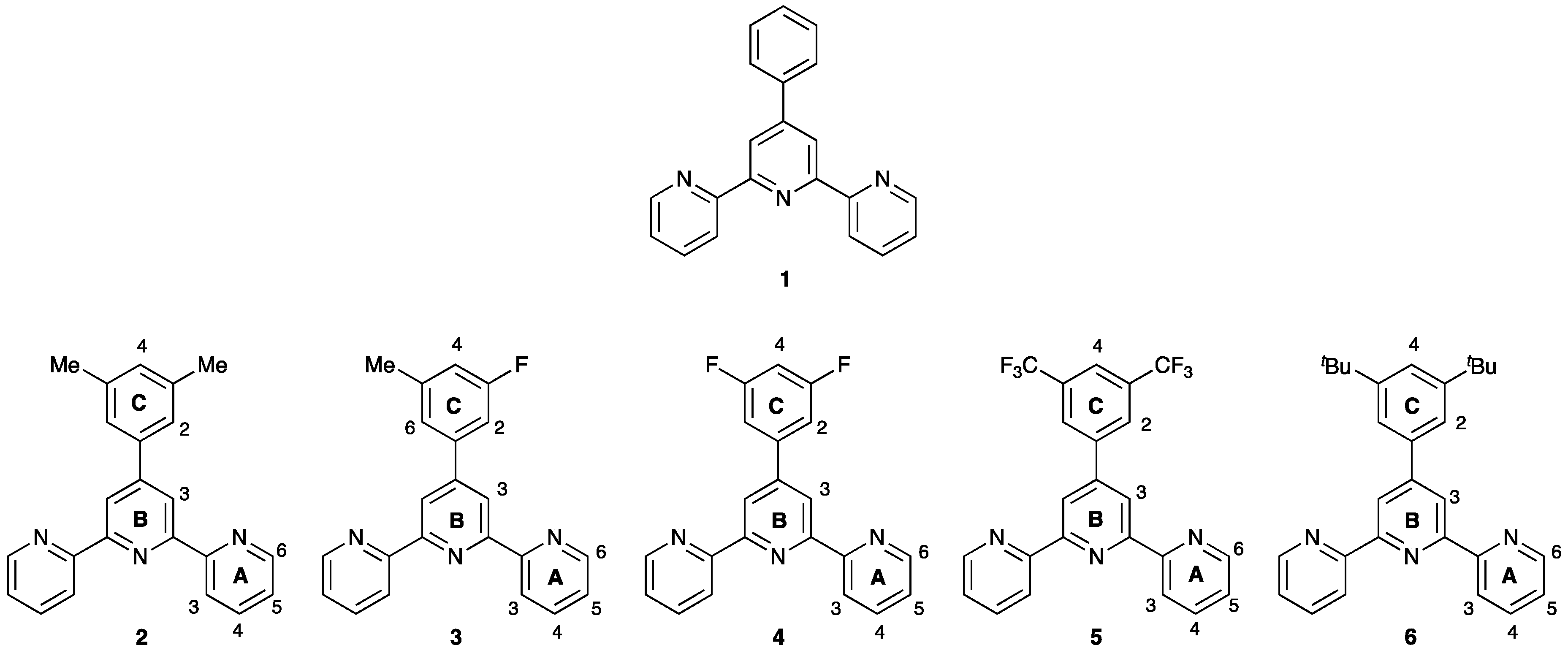
1. Introduction
2. Materials and Methods
2.1. General
2.2. Compound 2
2.3. Compound 3
2.4. Compound 4
2.5. Compound 5
2.6. Crystallography
3. Results and Discussion
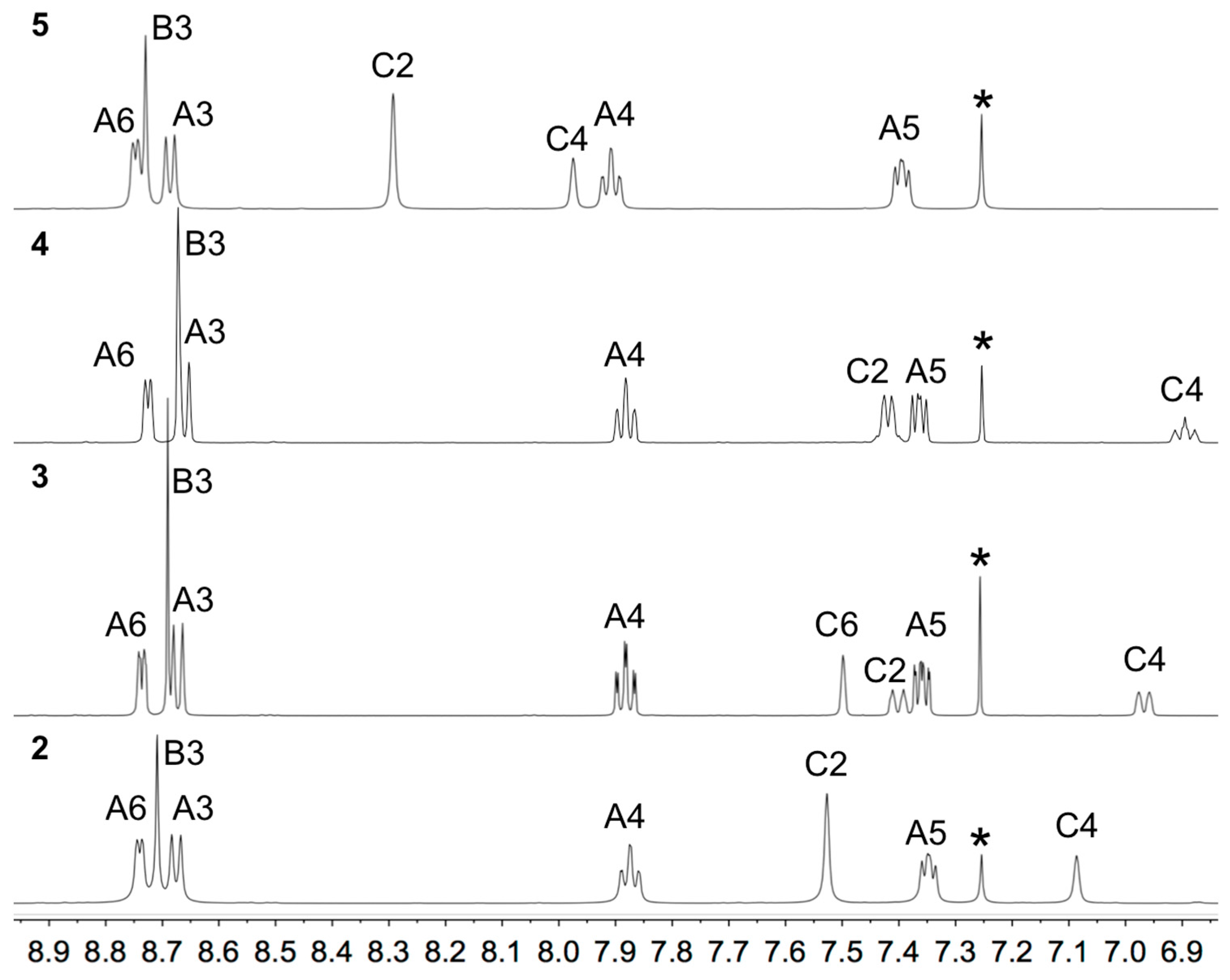
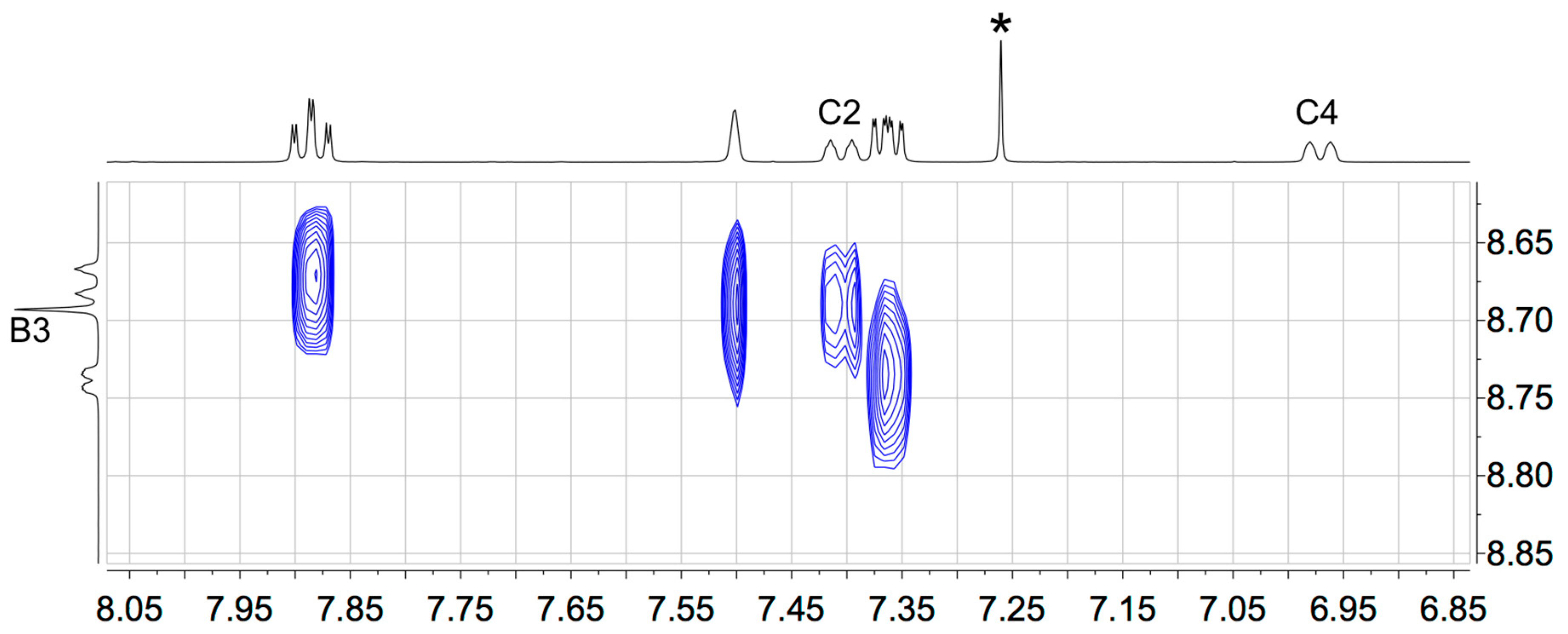
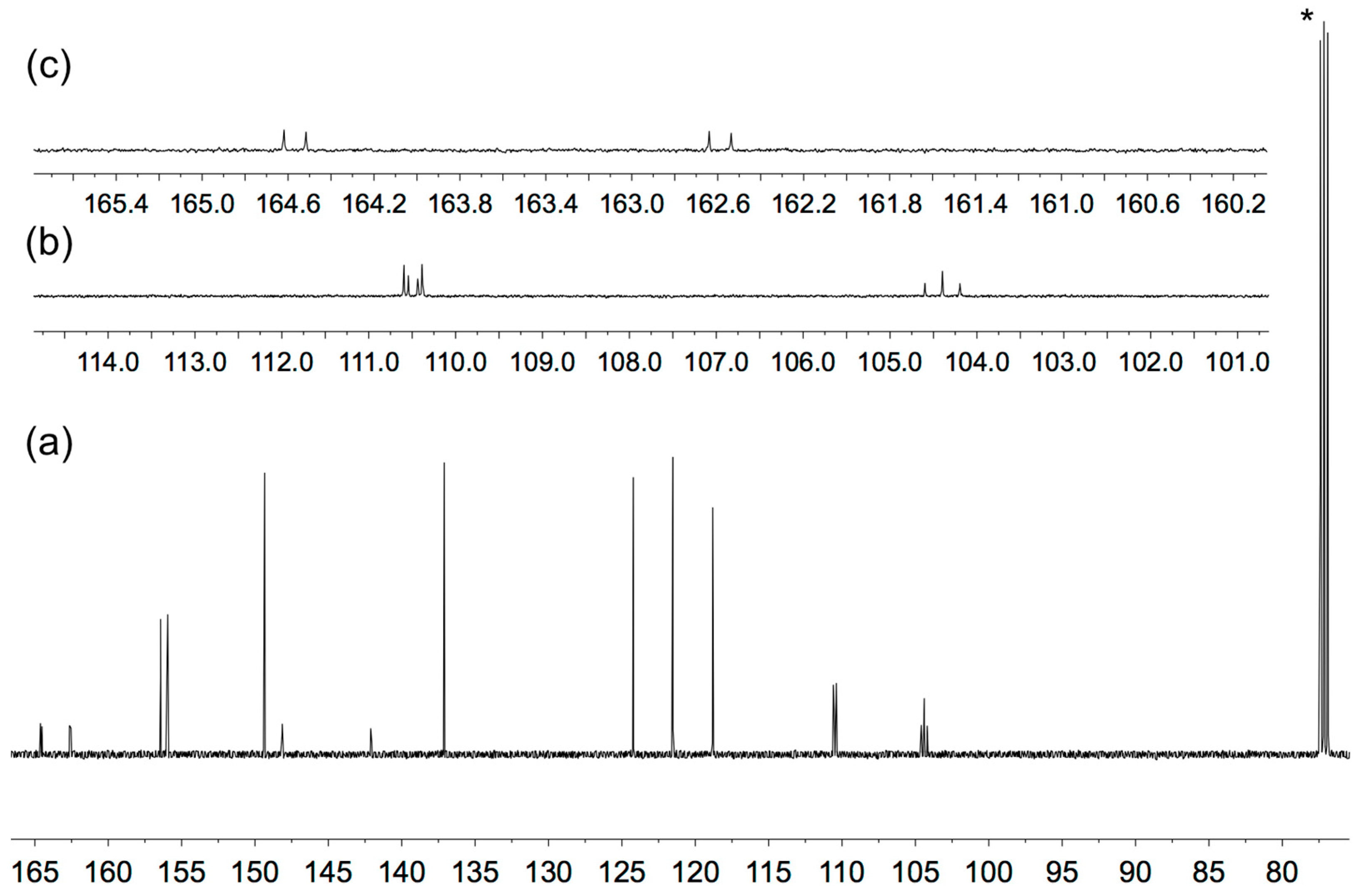
3.1. Synthesis and Solution Characterization of Complexes
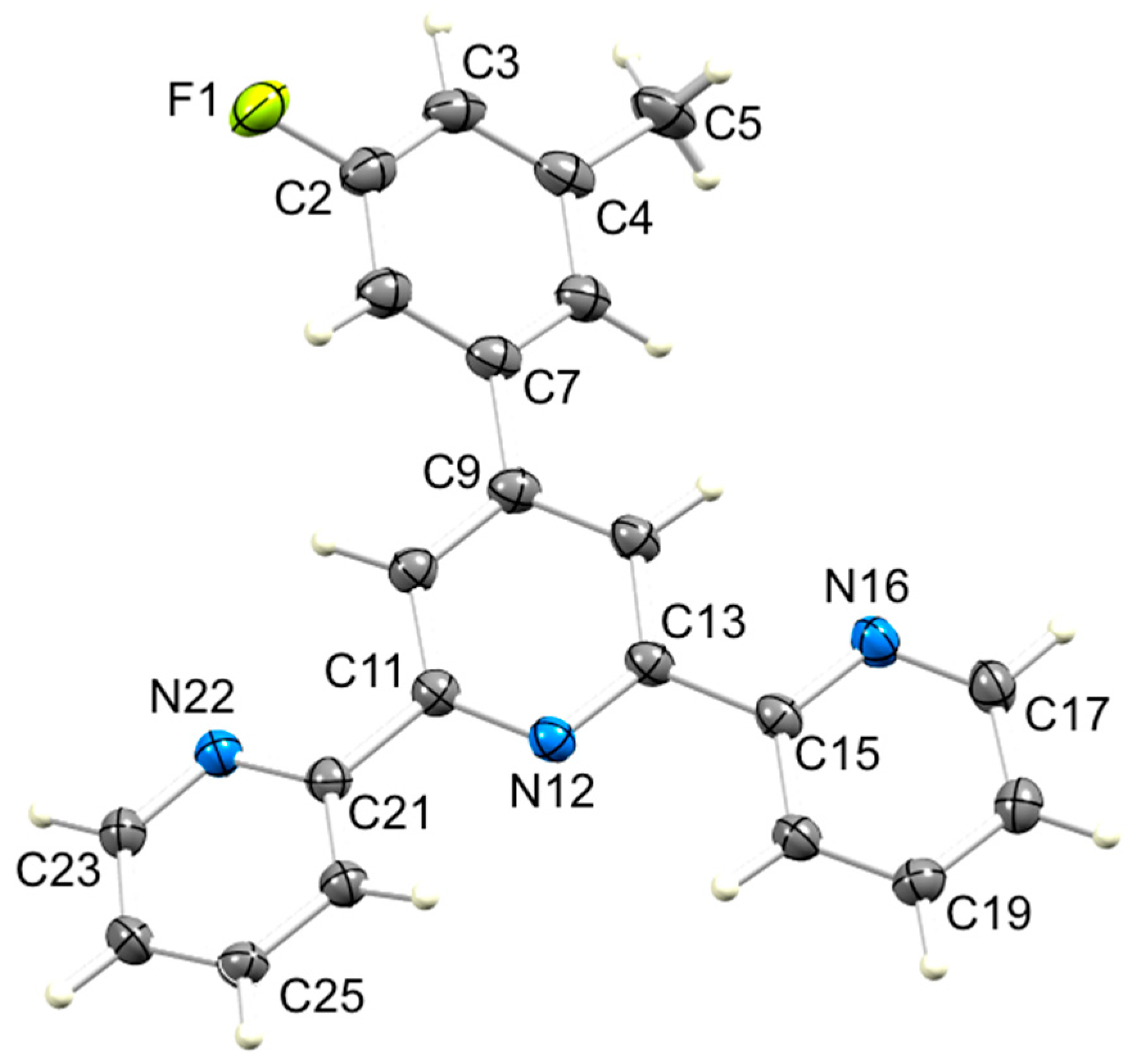
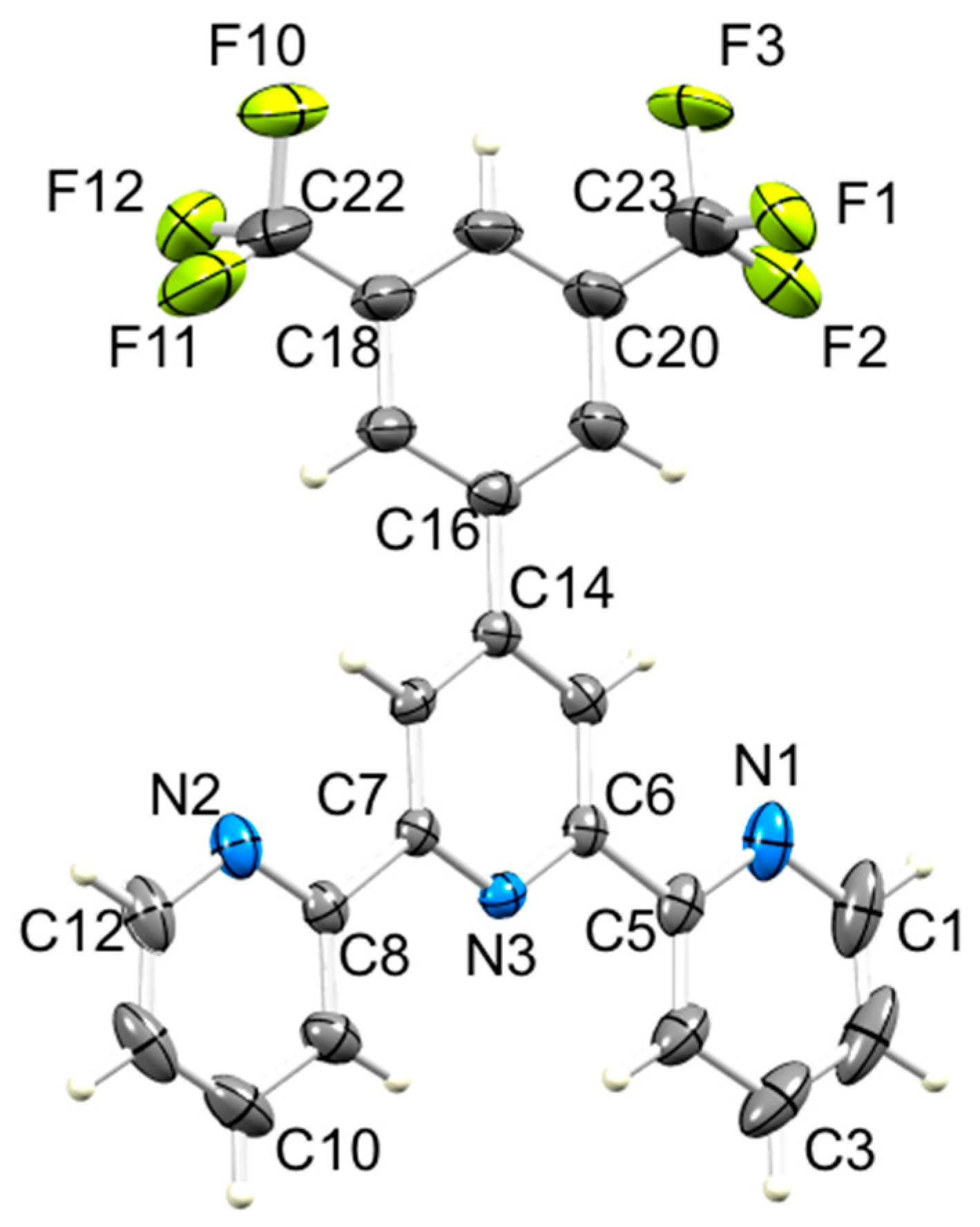
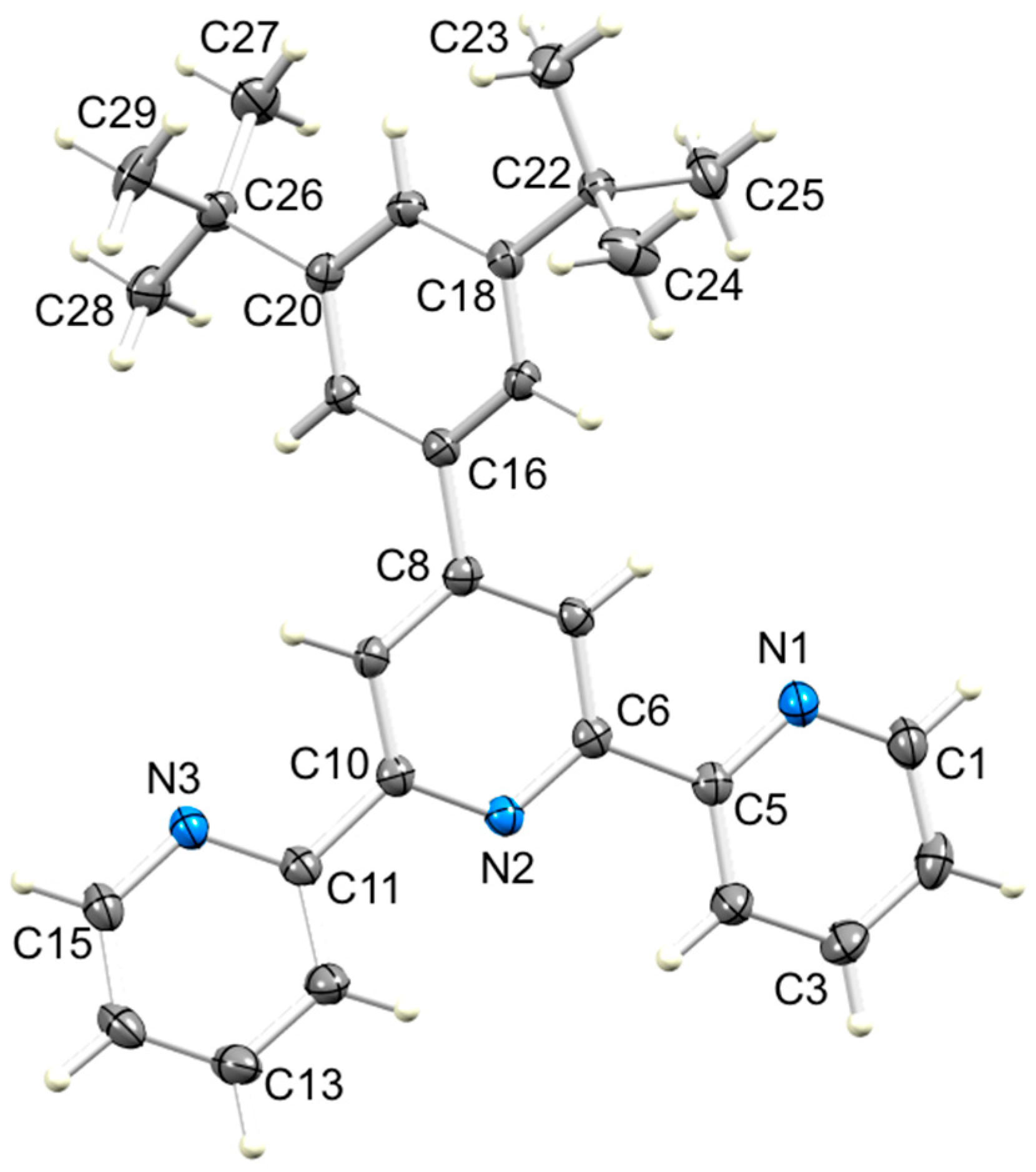
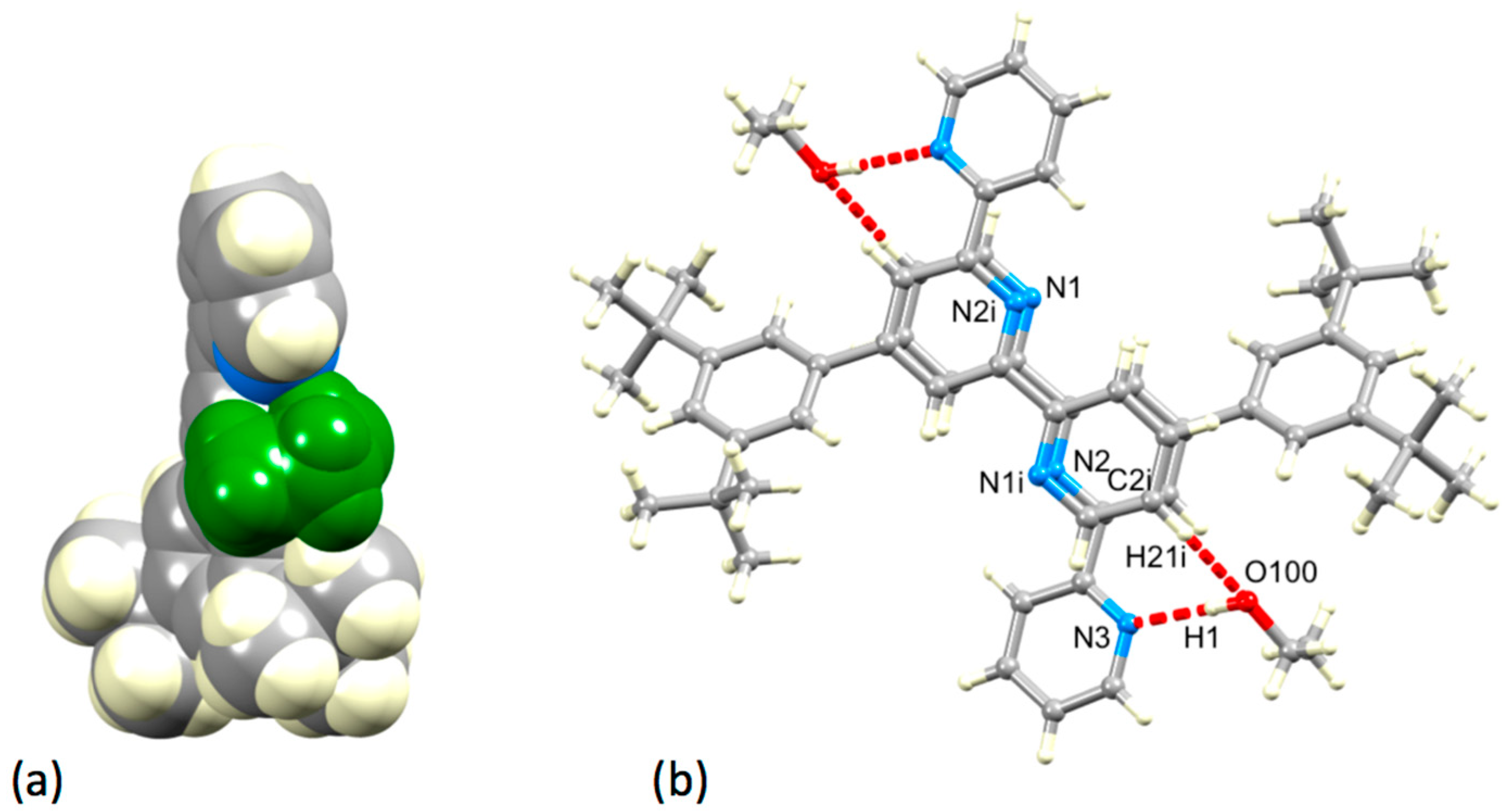
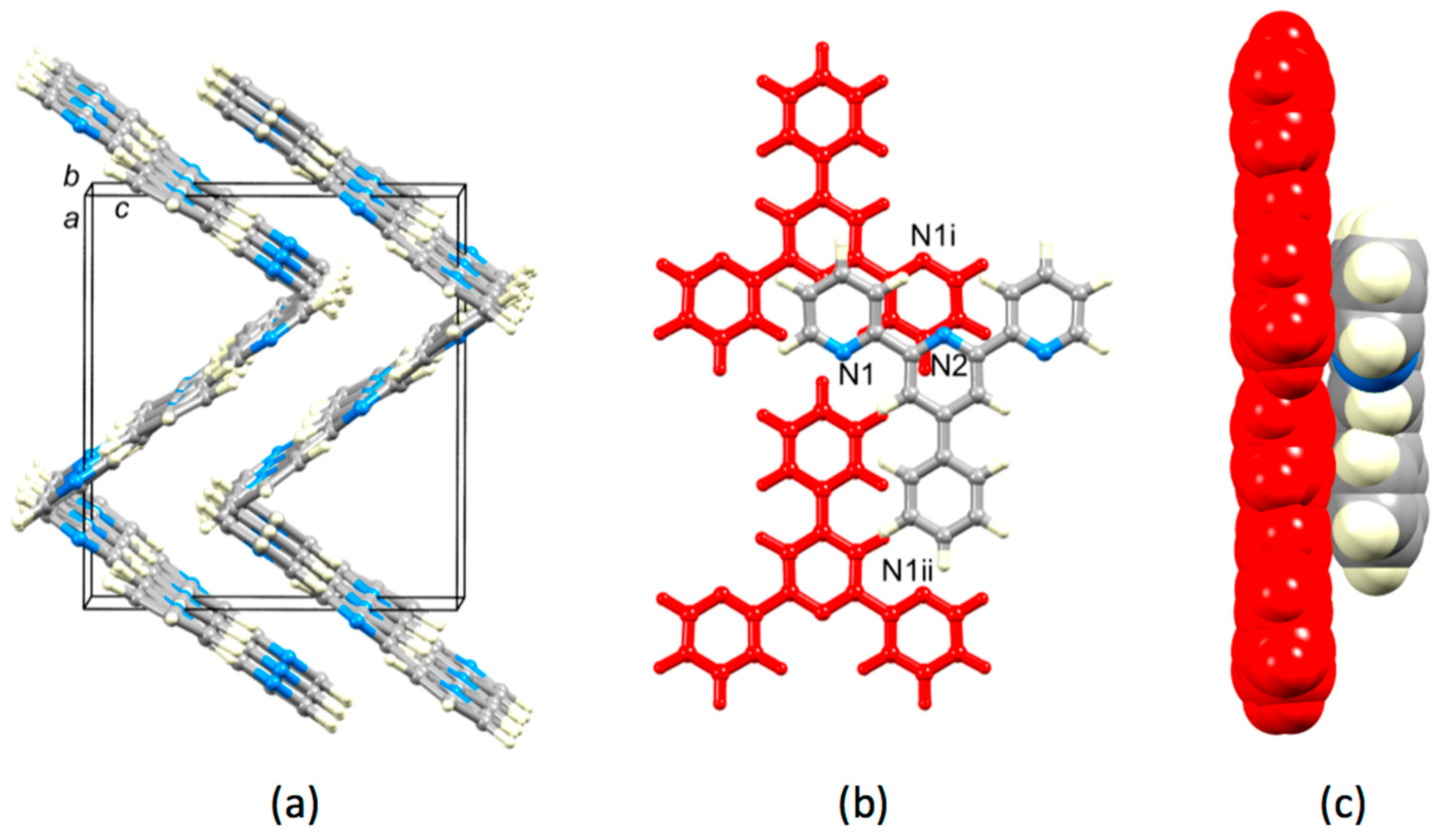
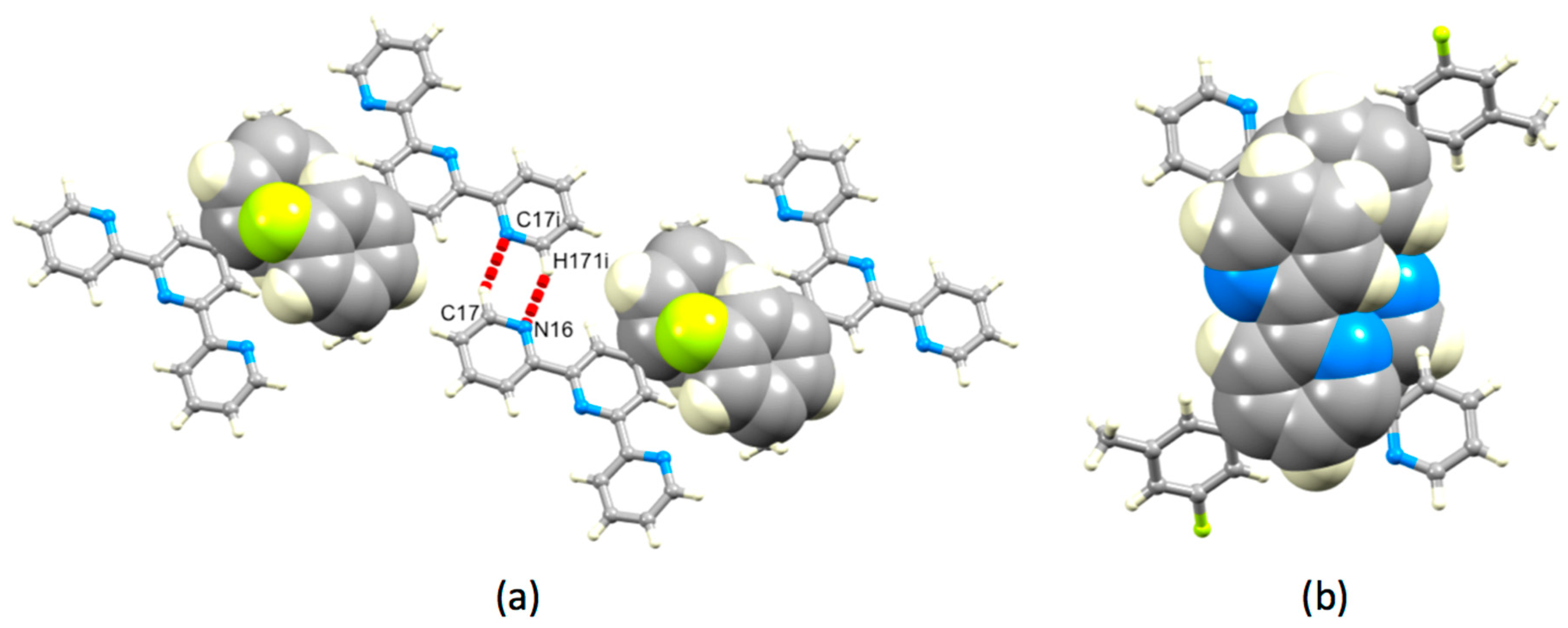
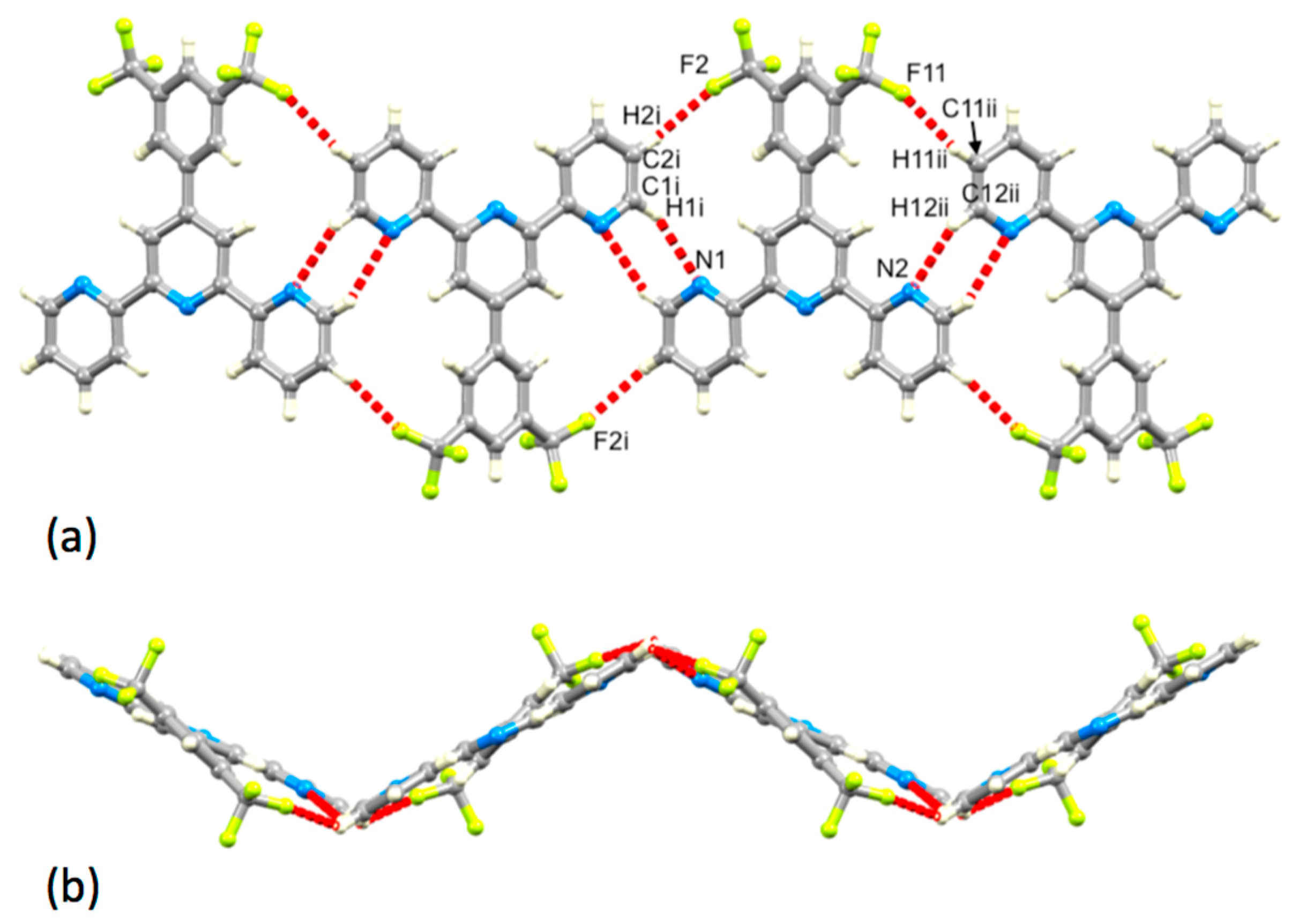
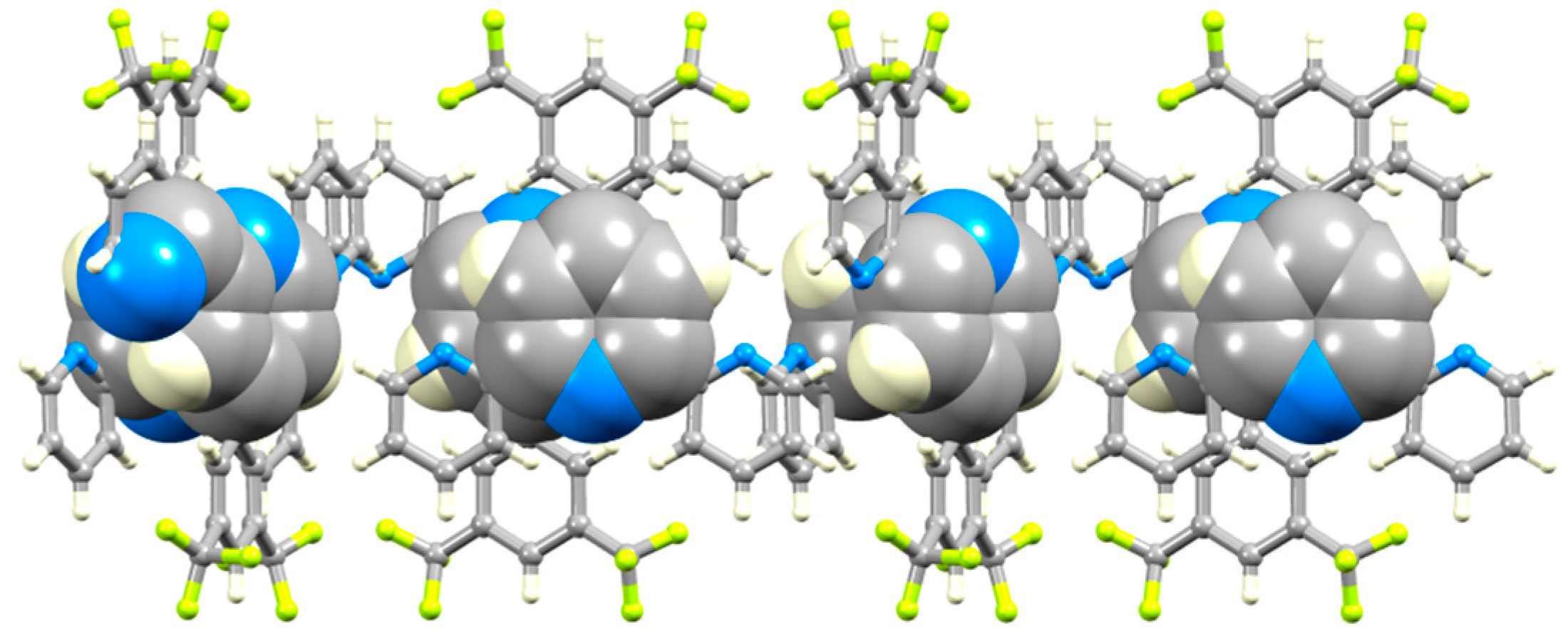
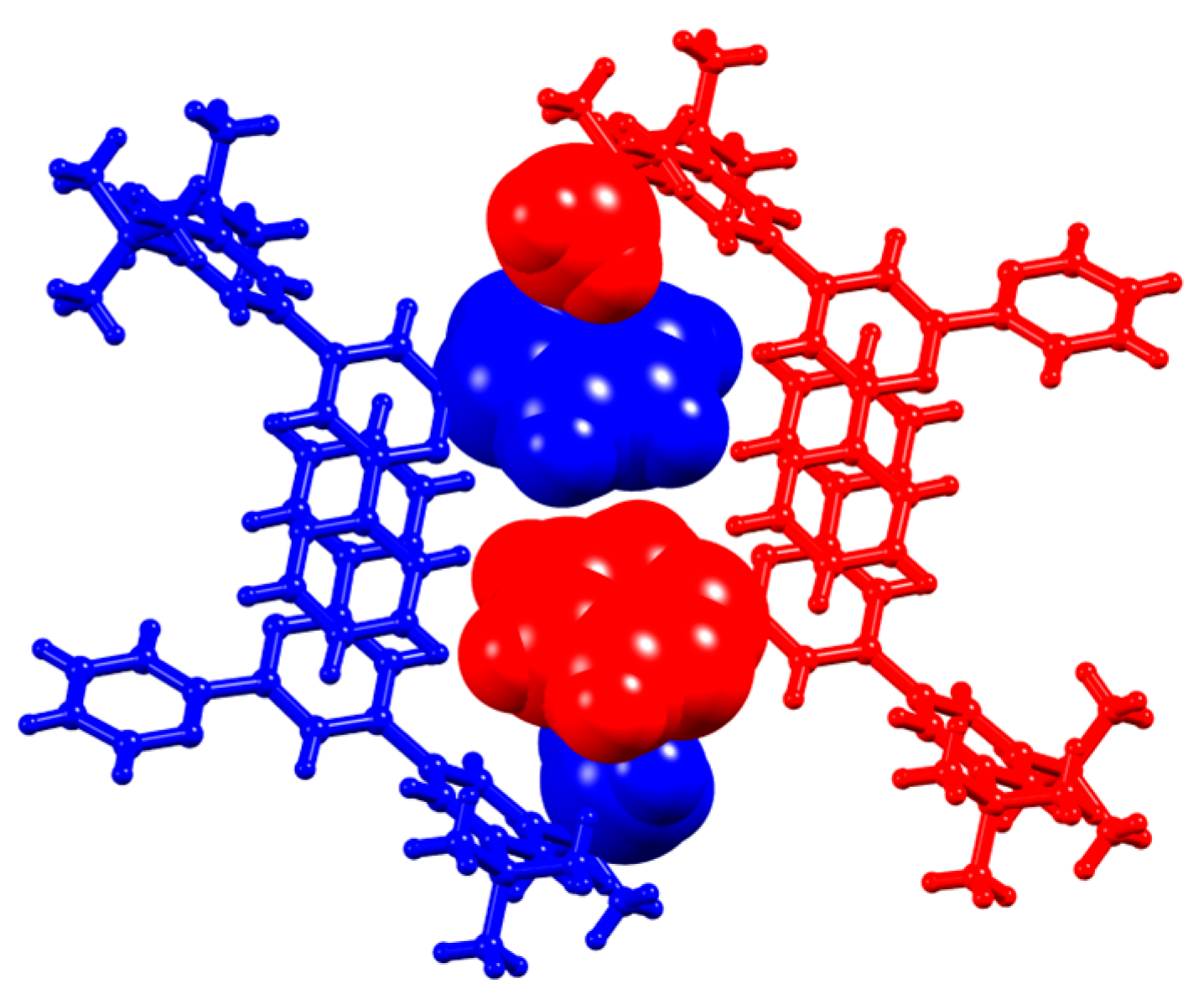
3.2. Single Crystal Structures of 3, 5 and 6
3.3. Comparison of Packing Interactions in 1, 3, 5 and 6
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Desiraju, G.R. Crystal Engineering: From Molecule to Crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: Oxford, UK, 1999; ISBN 9780198509707. [Google Scholar]
- Grabowski, S.J. [FHF]−—The Strongest Hydrogen Bond under the Influence of External Interactions. Crystals 2016, 6, 3. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Thakuria, R.; Sarma, B.; Nangia, A. Hydrogen bonding in molecular crystals. In Comprehensive Supramolecular Chemistry II; Atwood, J.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; Chapter 7.03; pp. 25–48. ISBN 978-0-12-803199-5. [Google Scholar]
- Nishio, M. CH/π hydrogen bonds in crystals. CrystEngComm 2004, 6, 130–158. [Google Scholar] [CrossRef]
- Suezawa, H.; Yoshida, T.; Umezawa, Y.; Tsuboyama, S.; Nishio, M. CH/π Interactions Implicated in the Crystal Structure of Transition Metal Compounds—A Database Study. Eur. J. Inorg. Chem. 2002, 3148–3155. [Google Scholar] [CrossRef]
- Taylor, R. It Isn’t, It Is: The C–H…X (X = O, N, F, Cl) Interaction Really is Significant in Crystal Packing. Cryst. Growth Des. 2016, 16, 4165–4168. [Google Scholar] [CrossRef]
- Gavezzotti, A.; Lo Presti, L. Building Blocks of Crystal Engineering: A Large Database Study of the Intermolecular Approach between C–H Donor groups and O, N, Cl, or F Acceptors in Organic Crystals. Cryst. Growth Des. 2016, 16, 2952–2962. [Google Scholar] [CrossRef]
- Lo Presti, L. On the significance of weak hydrogen bonds in crystal packing: A large databank comparison of polymorphic structures. CrystEngComm 2018, 20, 5976–5989. [Google Scholar] [CrossRef]
- Gatsiou, C.A.; Adjiman, C.S.; Pantelides, C.C. Repulsion–dispersion parameters for the modelling of organic molecular crystals containing N, O, S and Cl. Faraday Discuss. 2018, 211, 297–323. [Google Scholar] [CrossRef] [PubMed]
- Nyman, J.; Reutzel-Edens, S.M. Crystal structure prediction is changing from basic science to applied technology. Faraday Discuss. 2018, 211, 459–476. [Google Scholar] [CrossRef]
- Price, S.L. Is zeroth order crystal structure prediction (CSP_0) coming to maturity? What should we aim for in an ideal crystal structure prediction code? Faraday Discuss. 2018, 211, 9–30. [Google Scholar] [CrossRef] [PubMed]
- Price, S.L. Why don’t we find more polymorphs? Acta Cryst. 2013, B69, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Price, S.L. From crystal structure prediction to polymorph prediction: Interpreting the crystal energy landscape. Phys. Chem. Chem. Phys. 2008, 10, 1996–2009. [Google Scholar] [CrossRef] [PubMed]
- Constable, E.C. 2,2′:6′,2″-Terpyridines: From chemical obscurity to common supramolecular motifs. Chem. Soc. Rev. 2007, 36, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Constable, E.C. The coordination chemistry of 2,2′:6′,2″-terpyridine and higher oligopyridines. Adv. Inorg. Chem. Radiochem. 1986, 30, 69–121. [Google Scholar] [CrossRef]
- Housecroft, C.E. Divergent 4,2′:6′,4″- and 3,2′:6′,3″-terpyridines as linkers in 2- and 3-dimensional architectures. CrystEngComm 2015, 17, 7461–7468. [Google Scholar] [CrossRef]
- Constable, E.C.; Housecroft, C.E. Tetratopic bis(4,2′:6′,4″-terpyridine) and bis(3,2′:6′,3″-terpyridine) ligands as 4-connecting nodes in 2D-coordination networks and 3D-frameworks. J. Inorg. Organomet. Polym. Mater. 2018, 28, 414–427. [Google Scholar] [CrossRef]
- Hunter, C.A.; Lawson, K.R.; Perkins, J.; Urch, C.J. Aromatic Interactions. J. Chem. Soc. Perkin Trans. 2 2001, 651–669. [Google Scholar] [CrossRef]
- Janiak, C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. Dalton Trans. 2000, 3885–3896. [Google Scholar] [CrossRef]
- McMurtrie, J.; Dance, I. Crystal packing in metal complexes of 4′-phenylterpyridine and related ligands: Occurrence of the 2D and 1D terpy embrace arrays. CrystEngComm 2009, 11, 1141–1149. [Google Scholar] [CrossRef]
- McMurtrie, J.; Dance, I. Alternative two-dimensional embrace nets formed by metal complexes of 4′-phenylterpyridine crystallised with hydrophilic anions. CrystEngComm 2010, 12, 3207–3217. [Google Scholar] [CrossRef]
- Constable, E.C.; Lewis, J.; Liptrot, M.C.; Raithby, P.R. The coordination chemistry of 4′-phenyl-2,2′:6′,2″-terpyridine; the synthesis, crystal and molecular structures of 4′-phenyl-2,2′:6′,2″-terpyridine and bis(4′-phenyl-2,2′:6′,2″-terpyridine)nickel(II) chloride decahydrate. Inorg. Chim. Acta 1990, 178, 47–54. [Google Scholar] [CrossRef]
- Tu, S.; Jia, R.; Zhang, J.; Zhang, Y.; Yao, C.; Ji, S. Kröhnke reaction in aqueous media: One-pot clean synthesis of 4′-aryl-2,2′:6′,2″-terpyridines. Tetrahedron 2007, 63, 381–388. [Google Scholar] [CrossRef]
- Nayak, S.K.; Sathishkumar, R.; Row, T.N.G. Directing role of functional groups in selective generation of C–H⋯π interactions: In situ cryo-crystallographic studies on benzyl derivatives. CrystEngComm 2010, 12, 3112–3118. [Google Scholar] [CrossRef]
- Boden, N.; Davis, P.P.; Stam, C.H.; Wesselink, G.A. Solid hexafluorobenzene I. The crystal structure at 120 K. Mol. Phys. 1973, 25, 81–86. [Google Scholar] [CrossRef]
- Prasanna, M.D.; Row, T.N.G. C–halogen···π interactions and their influence on molecular conformation and crystal packing: A database study. Cryst. Eng. 2000, 3, 135–154. [Google Scholar] [CrossRef]
- Overell, J.S.W.; Pawley, G.S. An X-ray single-crystal study of the molecular system C6F6.C6D6. Acta Cryst. 1982, B38, 1966–1972. [Google Scholar] [CrossRef]
- Williams, J.H.; Cockcroft, J.K.; Fitch, A.N. Structure of the Lowest Temperature Phase of the Solid Benzene–Hexafluorobenzene Adduct. Angew. Chem. Int. Ed. 1992, 31, 1655–1657. [Google Scholar] [CrossRef]
- Reichenbächer, K.; Süssa, H.I.; Hulliger, J. Fluorine in crystal engineering—“The little atom that could”. Chem. Soc. Rev. 2005, 34, 22–30. [Google Scholar] [CrossRef]
- Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Organic fluorine compounds: A great opportunity for enhanced materials properties. Chem. Soc. Rev. 2011, 40, 3496–3508. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hanan, G.S. A facile route to sterically hindered and non-hindered 4′-aryl-2,2′:6′,2″-terpyridines. Synlett 2005, 1251–1254. [Google Scholar] [CrossRef]
- Collin, J.-P.; Dixon, I.M.; Sauvage, J.-P.; Williams, J.A.G.; Barigelletti, F.; Flamigni, L. Synthesis and photophysical properties of iridium(III) bisterpyridine and its homologues: A family of complexes with a long-lived excited state. J. Am. Chem. Soc. 1999, 121, 5009–5016. [Google Scholar] [CrossRef]
- APEX2, Version 2 User Manual, M86-E01078; Bruker Analytical X-ray Systems Madison Inc: Madison, WI, USA, 2006.
- Palatinus, L.; Chapuis, G. SUPERFLIP—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Cryst. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Betteridge, P.W.; Carruthers, J.R.; Cooper, R.I.; Prout, K.; Watkin, D.J. CRYSTALS Version 12: Software for Guided Crystal Structure Analysis. J. Appl. Cryst. 2003, 36, 1487. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Emsley, J. The Elements, 3rd ed.; Clarendon Press: Oxford, UK, 1998; ISBN 0-198-55818-X. [Google Scholar]















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, Y.M.; Prescimone, A.; Karpacheva, M.; Constable, E.C.; Housecroft, C.E. Substituent Effects in the Crystal Packing of Derivatives of 4′-Phenyl-2,2′:6′,2″-Terpyridine. Crystals 2019, 9, 110. https://doi.org/10.3390/cryst9020110
Klein YM, Prescimone A, Karpacheva M, Constable EC, Housecroft CE. Substituent Effects in the Crystal Packing of Derivatives of 4′-Phenyl-2,2′:6′,2″-Terpyridine. Crystals. 2019; 9(2):110. https://doi.org/10.3390/cryst9020110
Chicago/Turabian StyleKlein, Y. Maximilian, Alessandro Prescimone, Mariia Karpacheva, Edwin C. Constable, and Catherine E. Housecroft. 2019. "Substituent Effects in the Crystal Packing of Derivatives of 4′-Phenyl-2,2′:6′,2″-Terpyridine" Crystals 9, no. 2: 110. https://doi.org/10.3390/cryst9020110
APA StyleKlein, Y. M., Prescimone, A., Karpacheva, M., Constable, E. C., & Housecroft, C. E. (2019). Substituent Effects in the Crystal Packing of Derivatives of 4′-Phenyl-2,2′:6′,2″-Terpyridine. Crystals, 9(2), 110. https://doi.org/10.3390/cryst9020110