The Crystal Structure of the Plasmodium falciparum PdxK Provides an Experimental Model for Pro-Drug Activation


Abstract
1. Introduction
2. Materials and Methods
2.1. Purification and Crystallization
2.2. Data Collection and Structure Determination
2.3. Molecular Docking
3. Results
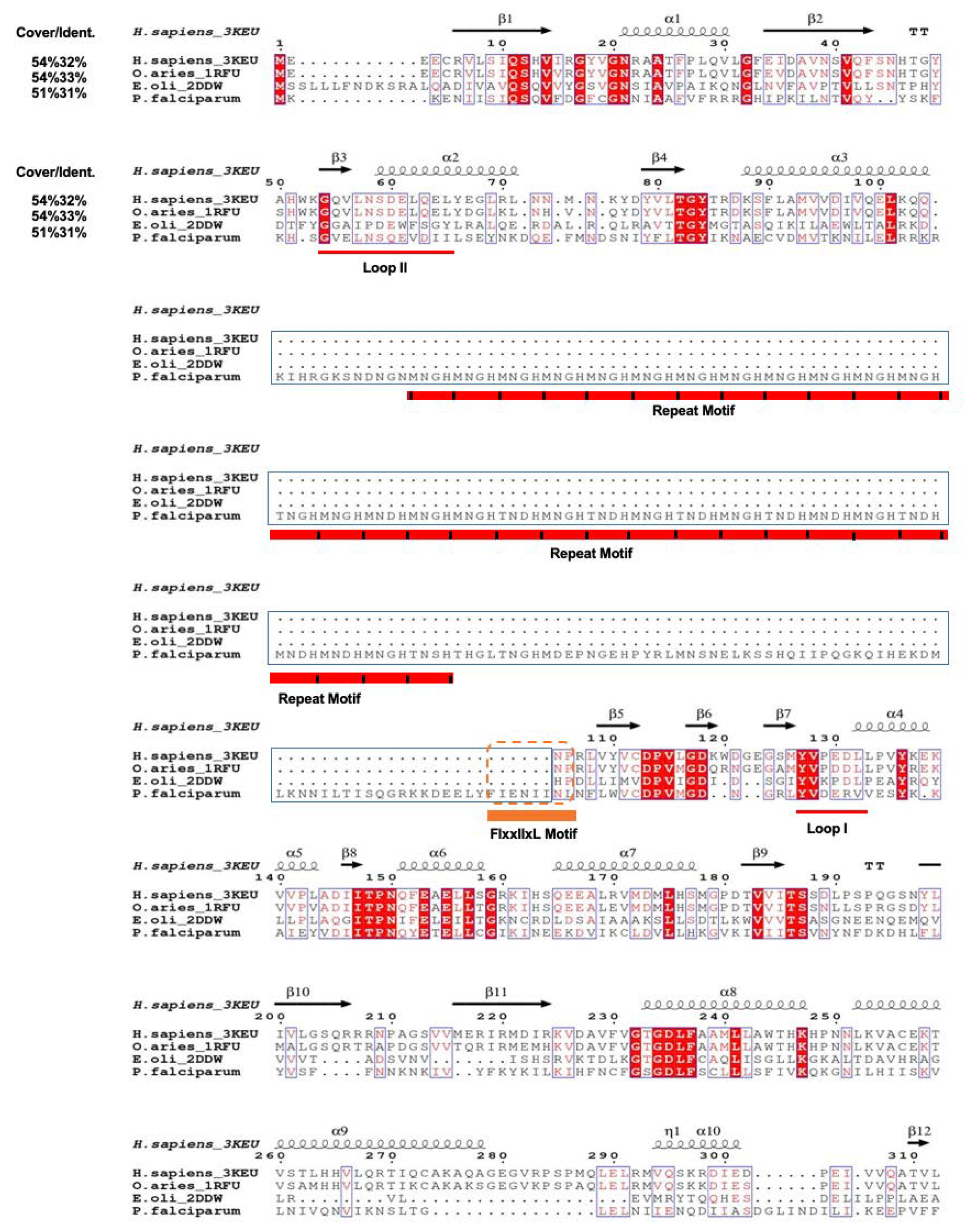
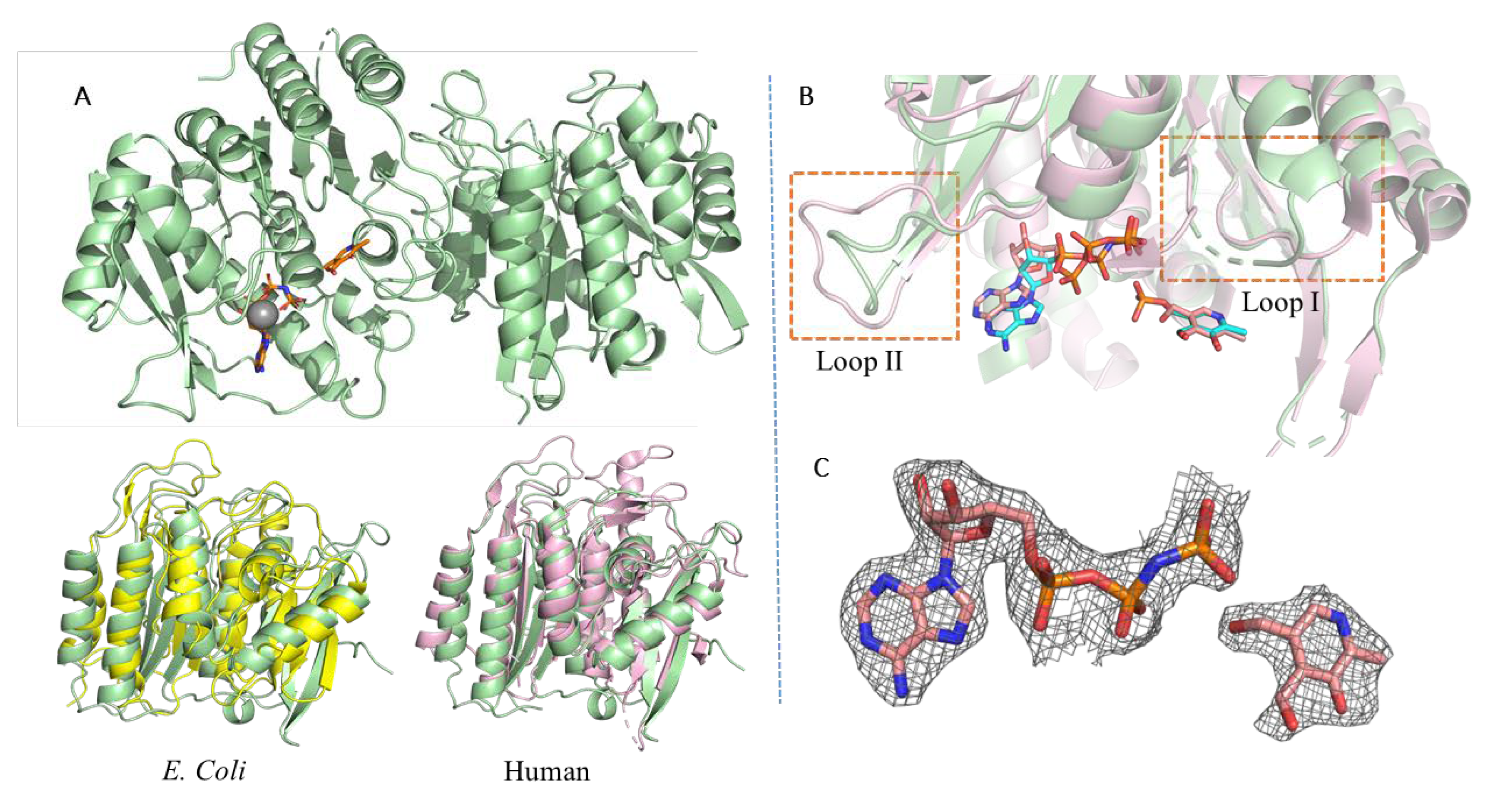
3.1. PfPdxK Adopts a Highly Similar Structure to that Seen in Different Species
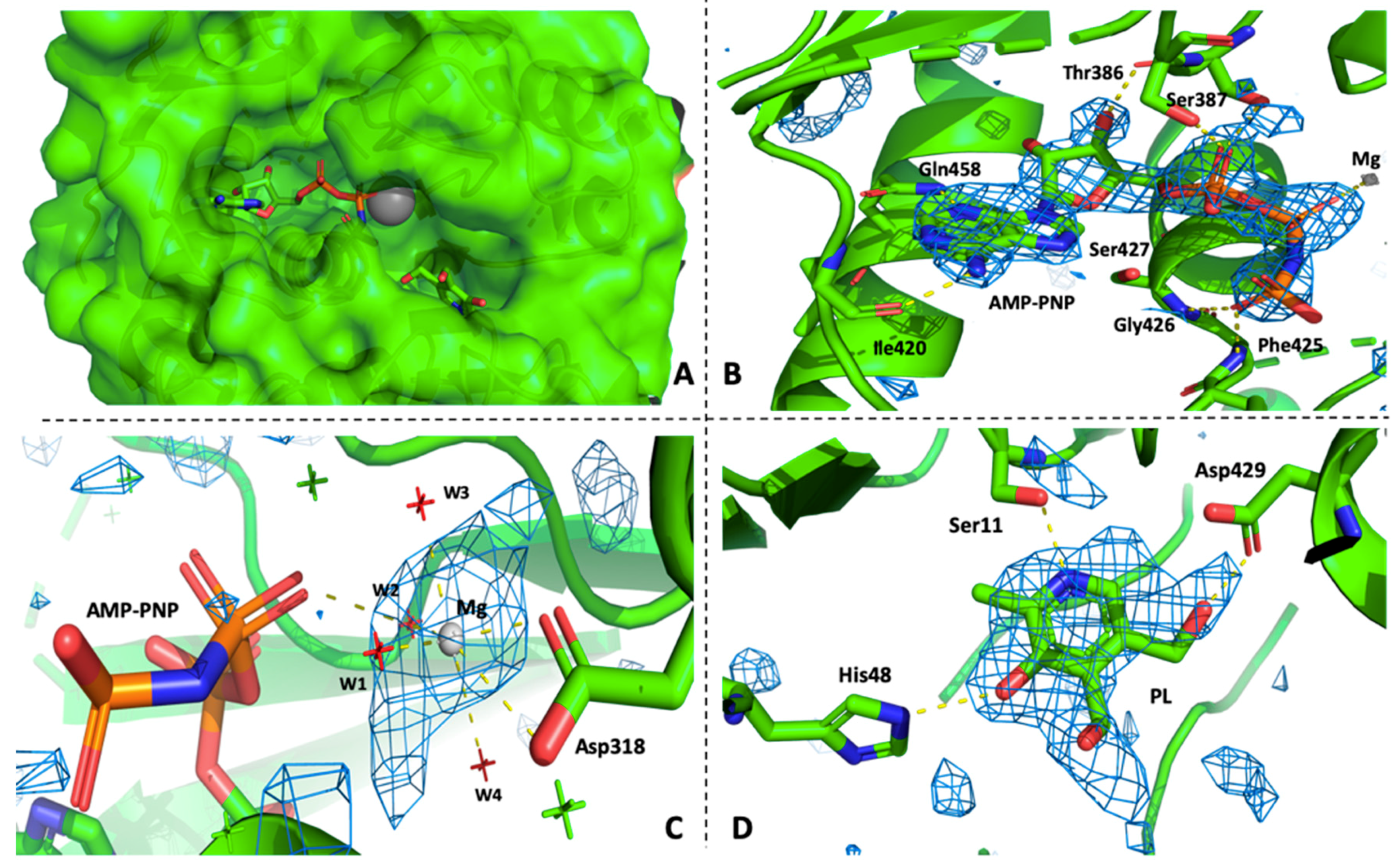
3.2. PfPdxK in Complex with AMP-PNP and PL
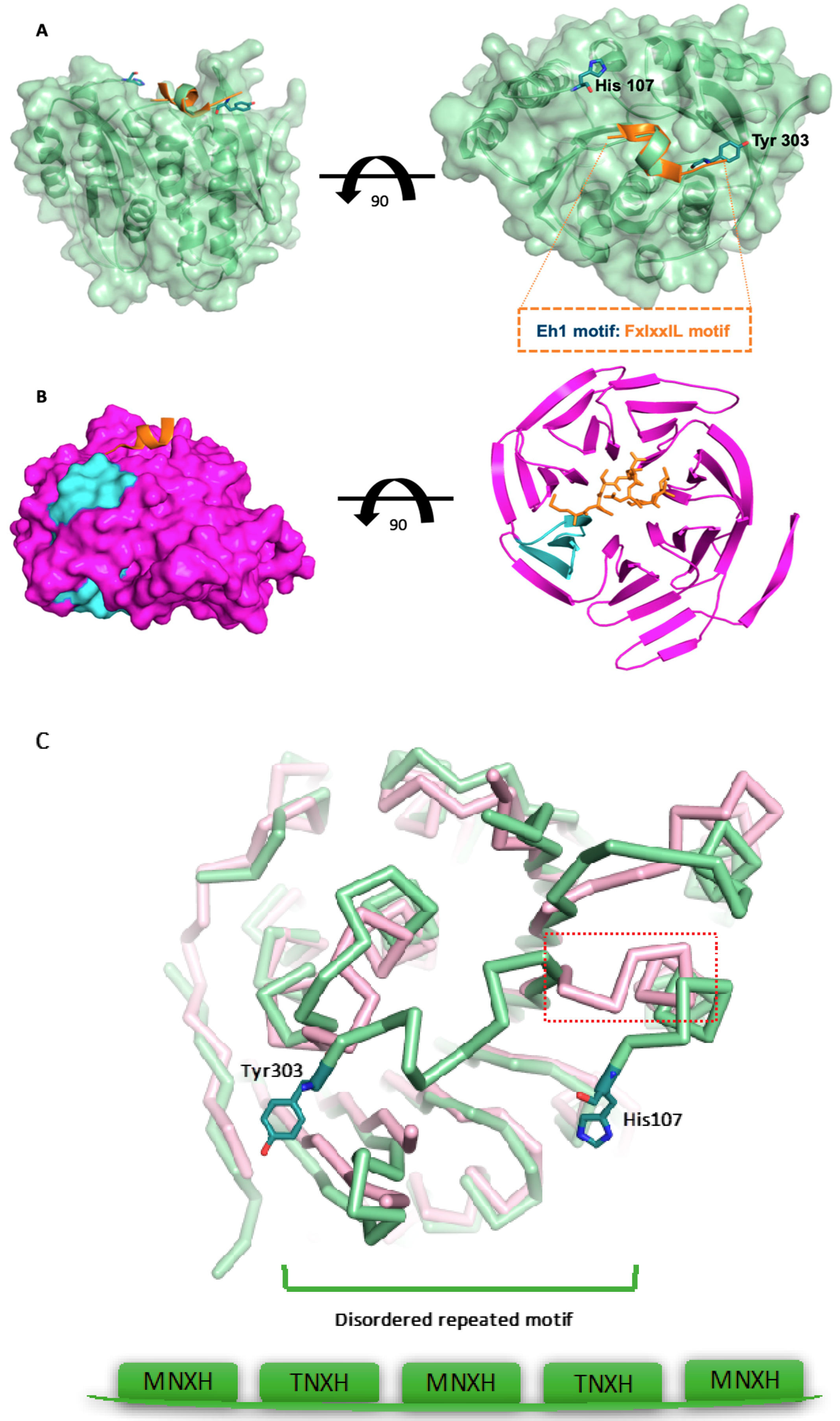
3.3. The Surface-Exposed Conserved FIxxIIxL Motif may Function as a Signal for Degradation
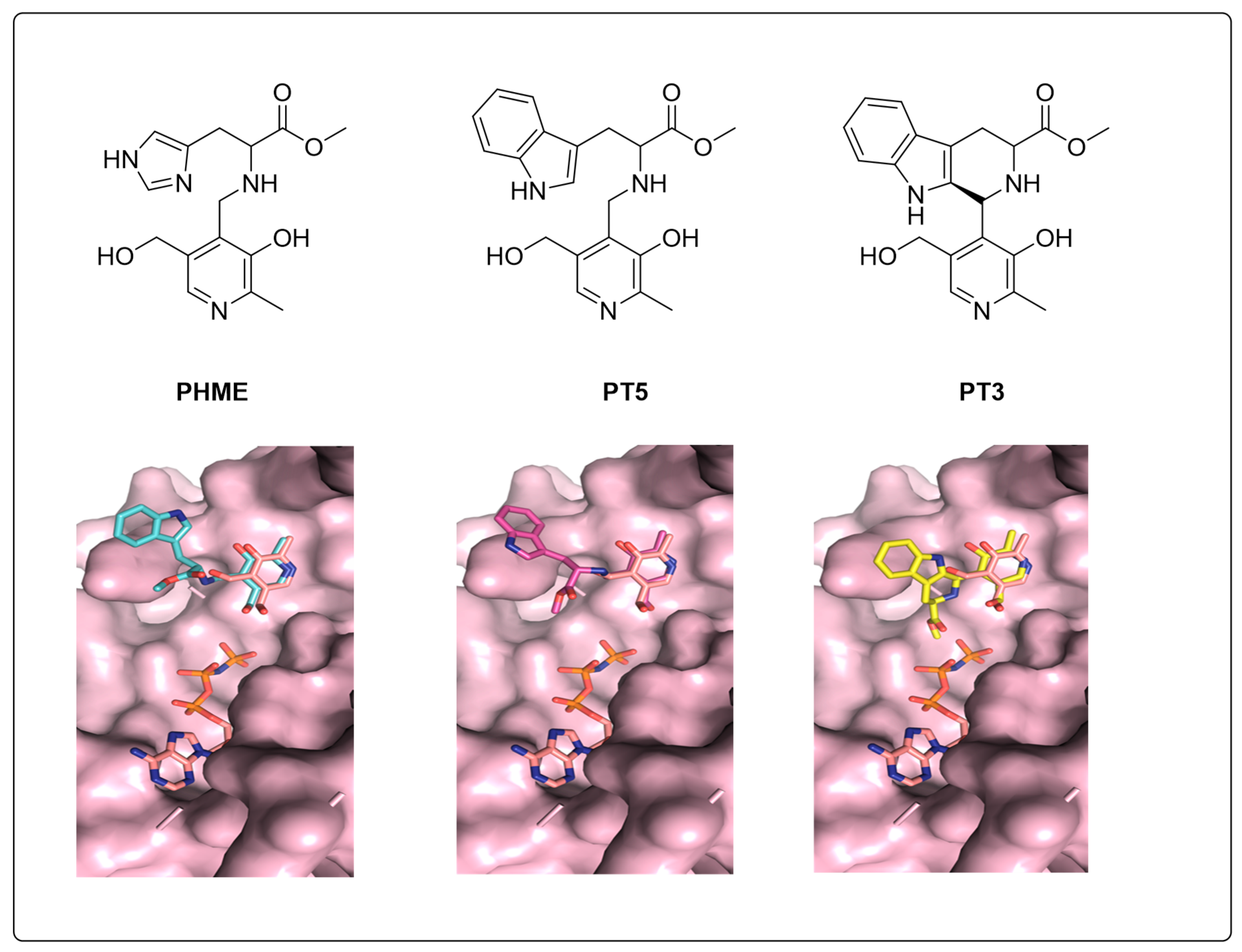
3.4. Molecular Docking Indicates Potential Binding Modes of PT3/PHME/PT5 as Substrates of PfPdxK
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Percudani, R.; Peracchi, A. A genomic overview of pyridoxal-phosphate-dependent enzymes. EMBO Rep. 2003, 4, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, J.; Li, Y.; Banks, J.; Cane, D.E.; Matte, A.; Cygler, M. Crystal Structure of Escherichia Coli PdxA, an Enzyme Involved in the Pyridoxal Phosphate Biosynthesis Pathway. J. Biol. Chem. 2003, 278, 43682–43690. [Google Scholar] [CrossRef] [PubMed]
- Müller, I.B.; Hyde, J.E.; Wrenger, C. Vitamin B Metabolism in Plasmodium falciparum as a Source of Drug Targets. Trends Parasitol. 2010, 26, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhao, G.; Winkler, M.E. Identification of the Pdxk Gene That Encodes Pyridoxine (Vitamin B6) Kinase in Escherichia Coli K-12. FEMS Microbiol. Lett. 1996, 141, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tsui, H.C.; Man, T.K.; Winkler, M.E. Identification and Function of the Pdxy Gene, Which Encodes a Novel Pyridoxal Kinase Involved in the Salvage Pathway of Pyridoxal 5’-Phosphate Biosynthesis in Escherichia Coli K-12. J. Bacteriol. 1998, 180, 1814–1821. [Google Scholar]
- Li, M.H.; Kwok, F.; Chang, W.R.; Liu, S.Q.; Lo, S.C.; Zhang, J.P.; Jiang, T.; Liang, D.C. Conformational Changes in the Reaction of Pyridoxal Kinase. J. Biol. Chem. 2004, 279, 17459–17465. [Google Scholar] [CrossRef]
- Cao, P.; Gong, Y.; Tang, L.; Leung, Y.C.; Jiang, T. Crystal Structure of Human Pyridoxal Kinase. J. Struct. Biol. 2006, 154, 327–332. [Google Scholar] [CrossRef]
- Safo, M.K.; Musayev, F.N.; di Salvo, M.L.; Hunt, S.; Claude, J.B.; Schirch, V. Crystal Structure of Pyridoxal Kinase from the Escherichia Coli Pdxk Gene: Implications for the Classification of Pyridoxal Kinases. J. Bacteriol. 2006, 188, 4542–4552. [Google Scholar] [CrossRef]
- Newman, J.A.; Das, S.K.; Sedelnikova, S.E.; Rice, D.W. The crystal structure of an ADP complex of Bacillus subtilis pyridoxal kinase provides evidence for the parallel emergence of enzyme activity during evolution. J. Mol. Biol. 2006, 363, 520–530. [Google Scholar] [CrossRef]
- Bishop, O.T.; Wells, G.A.; Joubert, F.; Wrenger, C.; Walter, R.D.; Louw, A.I. Progress Towards Determining the Structure of Plasmodium falciparum Pyridoxal Kinase. In Proceedings of the First Southern African Bioinformatics Workshop, Johannesburg, South Africa, 28–30 January 2007; pp. 41–44. [Google Scholar]
- Kronenberger, T.; Lunev, S.; Wrenger, C.; Groves, M.R. Purification, Crystallization and Preliminary X-Ray Diffraction Analysis of Pyridoxal Kinase from Plasmodium falciparum (PfPdxK). Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2014, 70, 1550–1555. [Google Scholar] [CrossRef]
- Wrenger, C.; Eschbach, M.L.; Müller, I.B.; Warnecke, D.; Walter, R.D. Analysis of the Vitamin B6 Biosynthesis Pathway in the Human Malaria Parasite Plasmodium falciparum. J. Biol. Chem. 2005, 280, 5242–5248. [Google Scholar] [CrossRef] [PubMed]
- Reeksting, S.B.; Muller, I.B.; Burger, P.B.; Burgos, E.S.; Salmon, L.; Louw, A.I.; Birkholtz, L.M.; Wrenger, C. Exploring Inhibition of Pdx1, A Component of the PLP Synthase Complex of the Human Malaria Parasite Plasmodium falciparum. Biochem. J. 2013, 449, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Muller, I.B.; Bergmann, B.; Groves, M.R.; Couto, I.; Amaral, L.; Begley, T.P.; Walter, R.D.; Wrenger, C. The Vitamin B1 Metabolism of Staphylococcus aureus Is Controlled at Enzymatic and Transcriptional Levels. PLoS ONE 2009, 4, e7656. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Müller, I.B.; Wu, F.; Bergmann, B.; Knöckel, J.; Walter, R.D.; Gehring, H.; Wrenger, C. Poisoning Pyridoxal 5-Phosphate-Dependent Enzymes: A New Strategy to Target the Malaria Parasite Plasmodium falciparum. PLoS ONE 2009, 4, e4406. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.J.; Hall, N.; Fung, E.; White, O.; Berriman, M.; Hyman, R.W.; Paulsen, I.T.; Carlton, J.M.; Pain, A.; Nelson, K.E.; et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002, 419, 498–511. [Google Scholar] [CrossRef]
- Kabsch, W. Xds. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Cryst. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. Refmac5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. Swissdock, a Protein-Small Molecule Docking Web Service Based on Eadock Dss. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A Novel Method for Fast and Accurate Multiple Sequence Alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering Key Features in Protein Structures with the New Endscript Server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Jennings, B.H.; Pickles, L.M.; Wainwright, S.M.; Roe, S.M.; Pearl, L.H.; Ish-Horowicz, D. Molecular Recognition of Transcriptional Repressor Motifs by the WD Domain of the Groucho/Tle Corepressor. Mol. Cell 2006, 22, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Roberts, R. WD-Repeat Proteins: Structure Characteristics, Biological Function, and Their Involvement in Human Diseases. Cell. Mol. Life Sci. 2001, 58, 2085–2097. [Google Scholar] [CrossRef]
- Neer, E.J.; Schmidt, C.J.; Nambudripad, R.; Smith, T.F. The Ancient Regulatory-Protein Family of WD-Repeat Proteins. Nature 1994, 371, 297–300. [Google Scholar] [CrossRef]
- Xu, C.; Min, J. Structure and Function of WD40 Domain Proteins. Protein Cell 2011, 2, 202–214. [Google Scholar] [CrossRef]
- Copley, R.R. The EH1 motif in metazoan transcription factors. BMC Genom. 2005, 6, 169–177. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set | PfPdxK-AMP-PNP-PL |
|---|---|
| Wavelength | 0.97 Å (12.398 keV) |
| Resolution range | 19.33–2.15 (2.227–2.15) |
| Space group | P 1 21 1 |
| Unit cell | 52.703 62.004 93.712 90 94.988 90 |
| Total reflections | 81052 (8023) |
| Unique reflections | 32284 (3159) |
| Multiplicity | 2.5 (2.6) |
| Completeness (%) | 97.87 (97.17) |
| Mean I/sigma(I) | 15.57 (2.69) |
| Wilson B-factor | 39.31 |
| R-merge | 3.7 (38.61) |
| R-meas | 4.7 (49.31) |
| CC 1/2 | 0.99 (0.88) |
| CC * | 1 (0.967) |
| Reflections used in refinement | 32276 (3159) |
| Reflections used for R-free | 1627 (165) |
| R-work | 0.2094 (0.3002) |
| R-free | 0.2718 (0.3691) |
| Number of non-hydrogen atoms | 4885 |
| macromolecules | 4727 |
| ligands | 89 |
| solvent | 69 |
| Protein residues | 591 |
| RMS(bonds) | 0.008 |
| RMS(angles) | 0.95 |
| Ramachandran favored (%) | 95.62 |
| Ramachandran allowed (%) | 4.20 |
| Ramachandran outliers (%) | 0.18 |
| Rotamer outliers (%) | 0.00 |
| Clashscore | 8.69 |
| Average B-factor | 59.75 |
| macromolecules | 59.58 |
| ligands | 73.01 |
| solvent | 54.41 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, K.; Wang, W.; Kronenberger, T.; Wrenger, C.; Groves, M.R. The Crystal Structure of the Plasmodium falciparum PdxK Provides an Experimental Model for Pro-Drug Activation. Crystals 2019, 9, 534. https://doi.org/10.3390/cryst9100534
Gao K, Wang W, Kronenberger T, Wrenger C, Groves MR. The Crystal Structure of the Plasmodium falciparum PdxK Provides an Experimental Model for Pro-Drug Activation. Crystals. 2019; 9(10):534. https://doi.org/10.3390/cryst9100534
Chicago/Turabian StyleGao, Kai, Wenjia Wang, Thales Kronenberger, Carsten Wrenger, and Matthew R. Groves. 2019. "The Crystal Structure of the Plasmodium falciparum PdxK Provides an Experimental Model for Pro-Drug Activation" Crystals 9, no. 10: 534. https://doi.org/10.3390/cryst9100534
APA StyleGao, K., Wang, W., Kronenberger, T., Wrenger, C., & Groves, M. R. (2019). The Crystal Structure of the Plasmodium falciparum PdxK Provides an Experimental Model for Pro-Drug Activation. Crystals, 9(10), 534. https://doi.org/10.3390/cryst9100534