Synthesis, Single Crystal X-ray Structure, DFT Computations, Hirshfeld Surface Analysis and Molecular Docking Simulations on ({[(1E)-1-(1,3-Benzodioxol-5-yl)-3-(1H-imidazol-1-yl)propylidene]amino}oxy)(furan-2-yl)methanone: A New Antifungal Agent

 , ,
, ,
Abstract
1. Introduction
2. Experimental
2.1. General
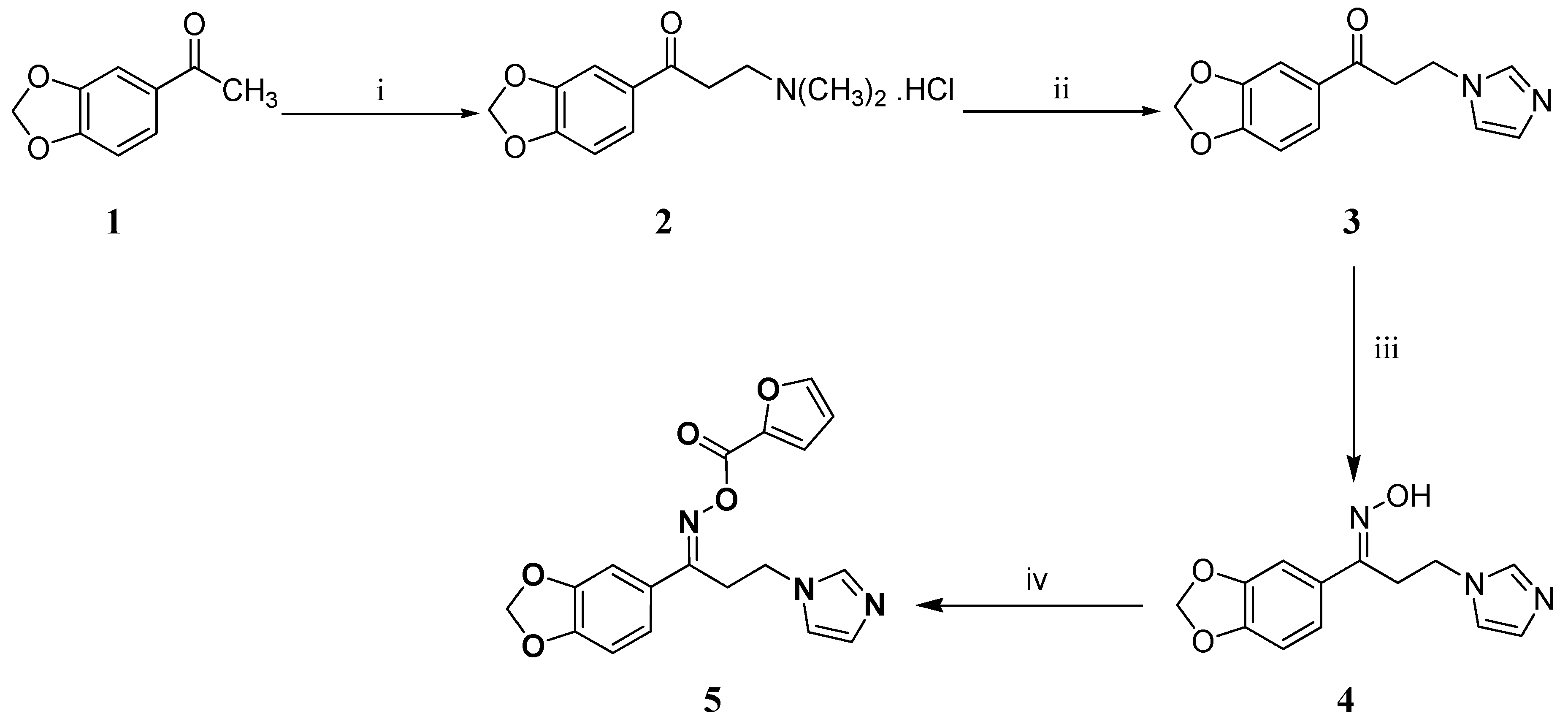
2.2. Synthesis
2.2.1. Synthesis of (1E)-1-(2H-1,3-Benzodioxol-5-yl)-N-hydroxy-3-(1H-imidazol-1-yl)propan-1-imine (4)
2.2.2. Synthesis of ({[(1E)-1-(1,3-Benzodioxol-5-yl)-3-(1H-imidazol-1-yl)propylidene]amino}oxy) (furan-2-yl)methanone (5)
2.3. Crystal Structure Determination
2.4. FT-IR and FT-Raman Measurements
2.5. Quantum Chemical Calculations
2.6. Antifungal Activity
3. Results and Discussion
3.1. Chemistry
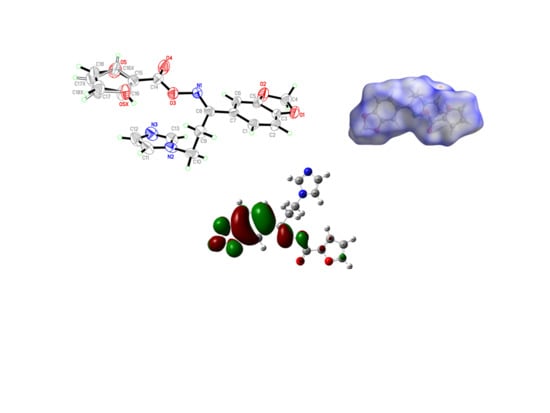
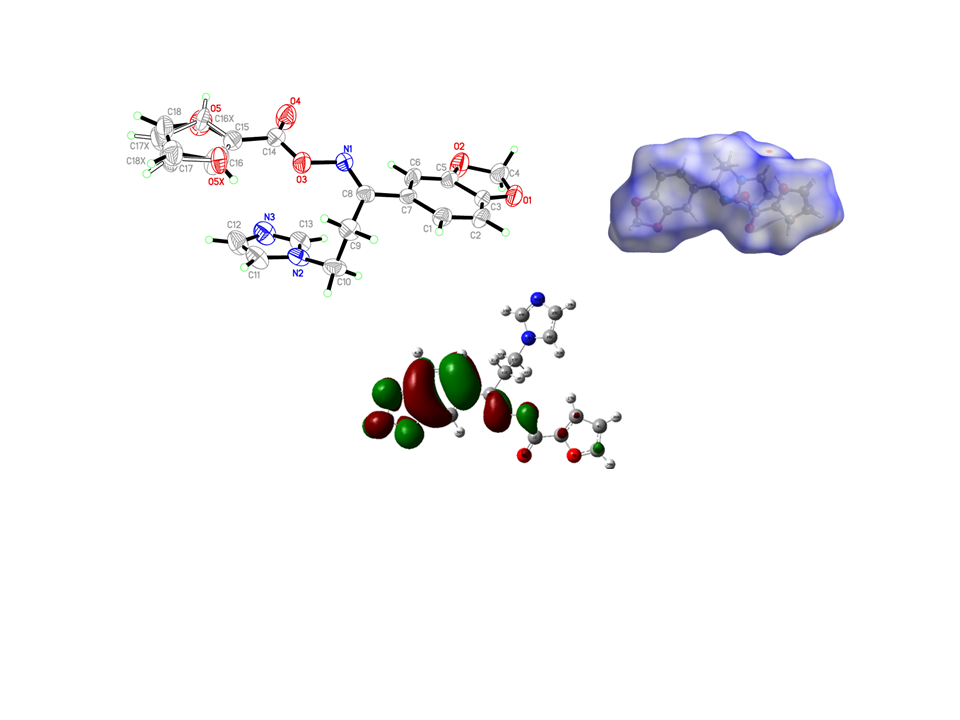
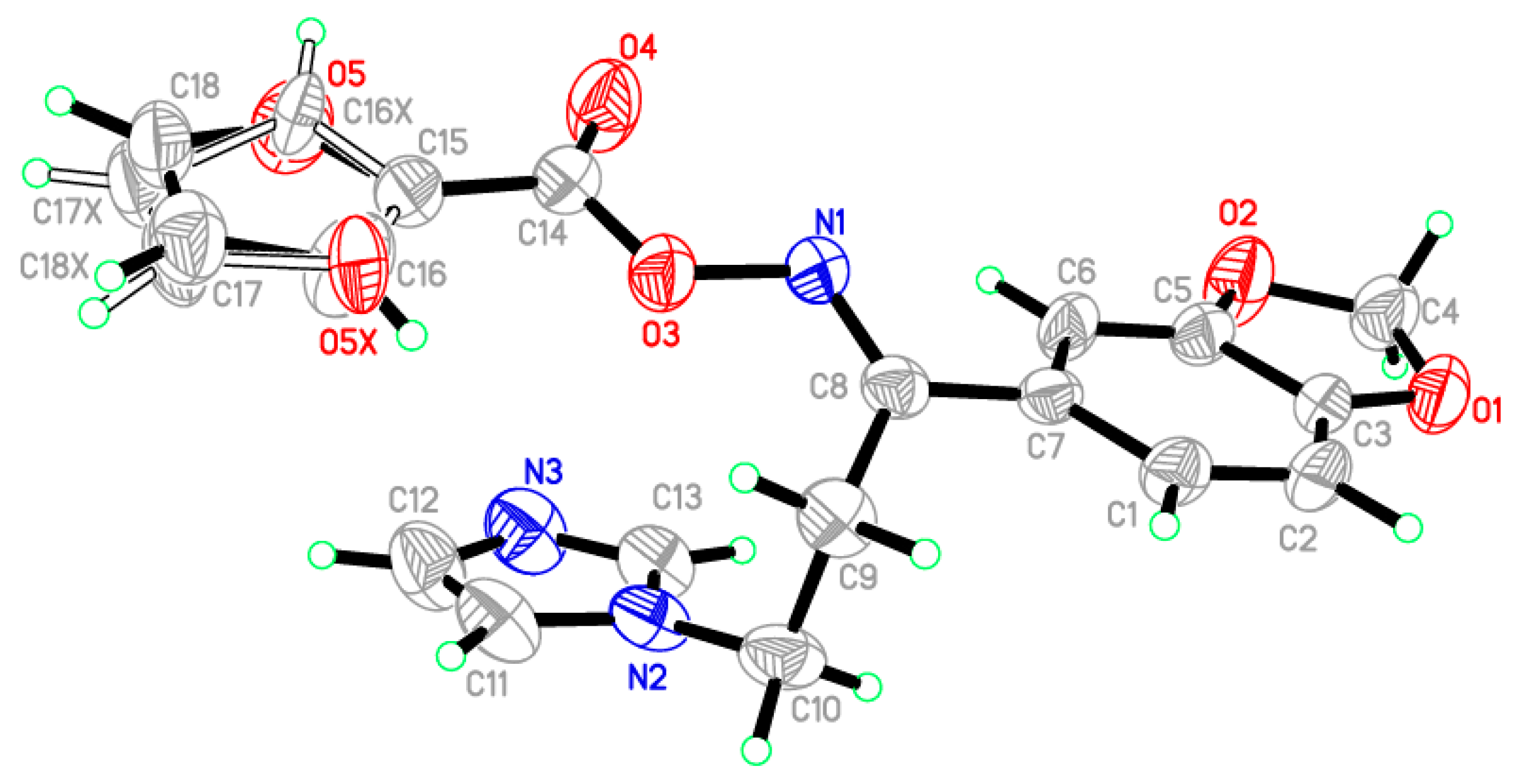
3.2. Crystal Structure of the Target Oximino Ester 5
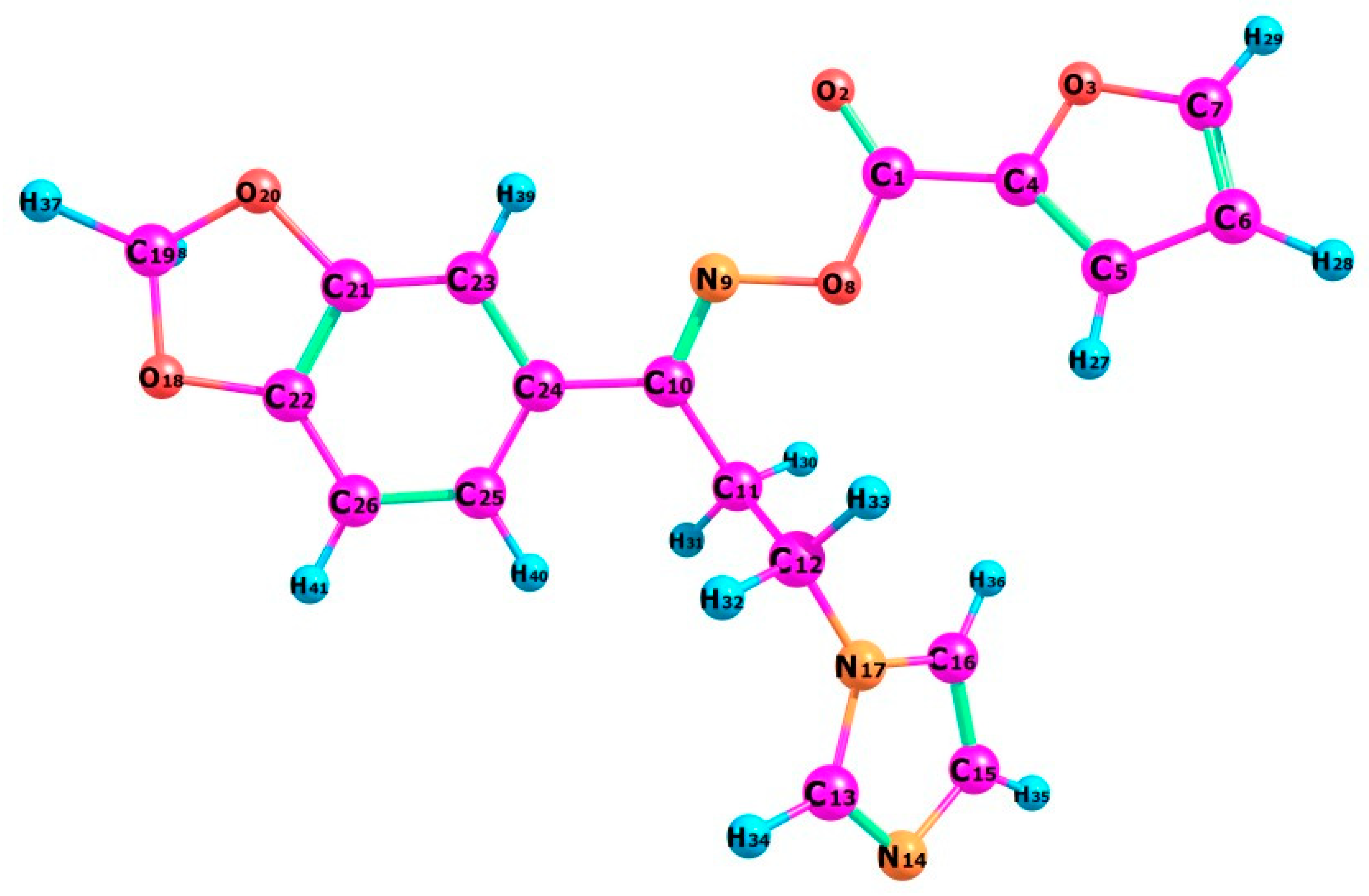
3.3. Structural Geometry Analysis
3.4. Natural Bond Orbital (NBO) Analysis
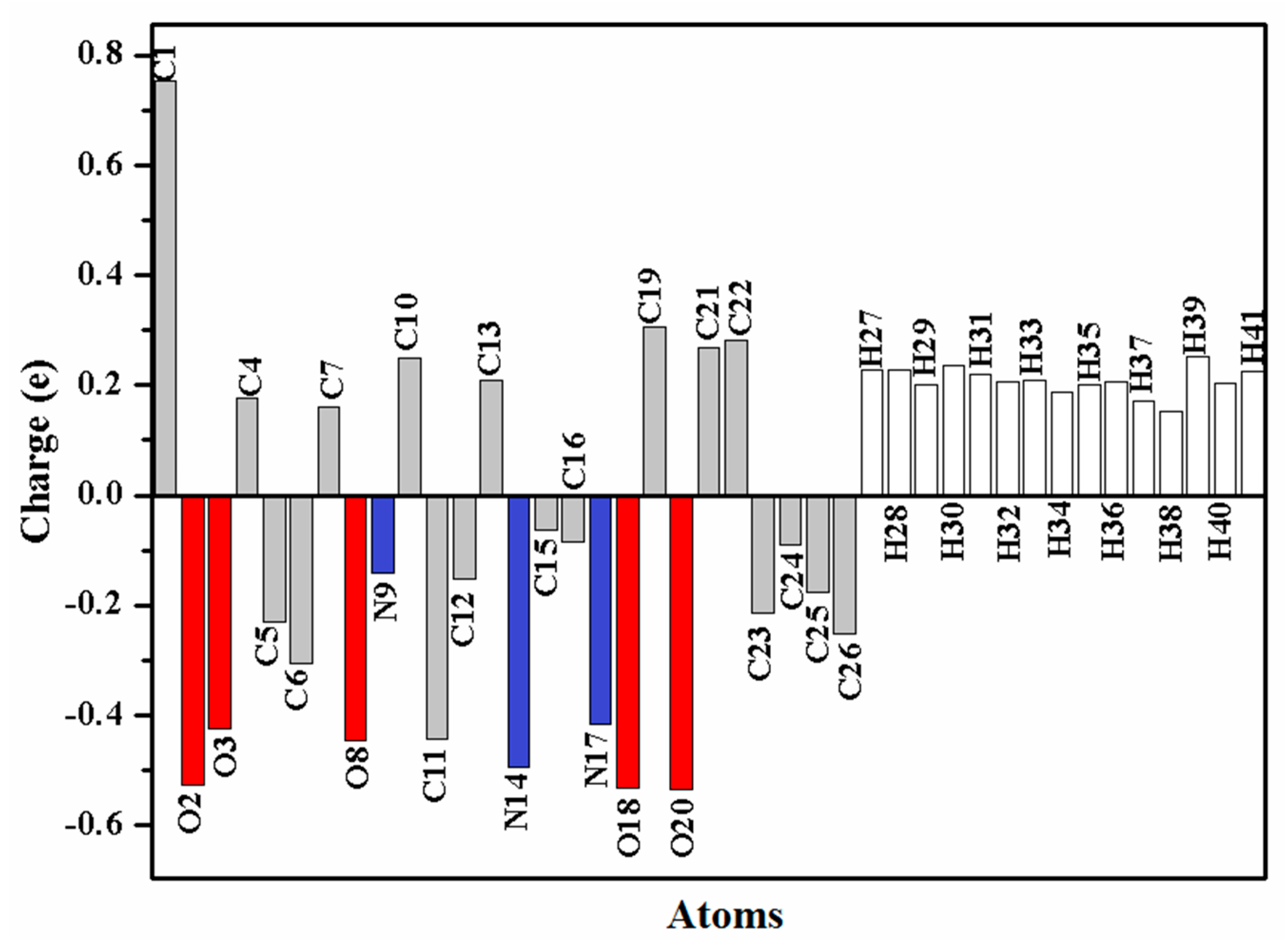
3.5. Natural Population Analysis (NPA)
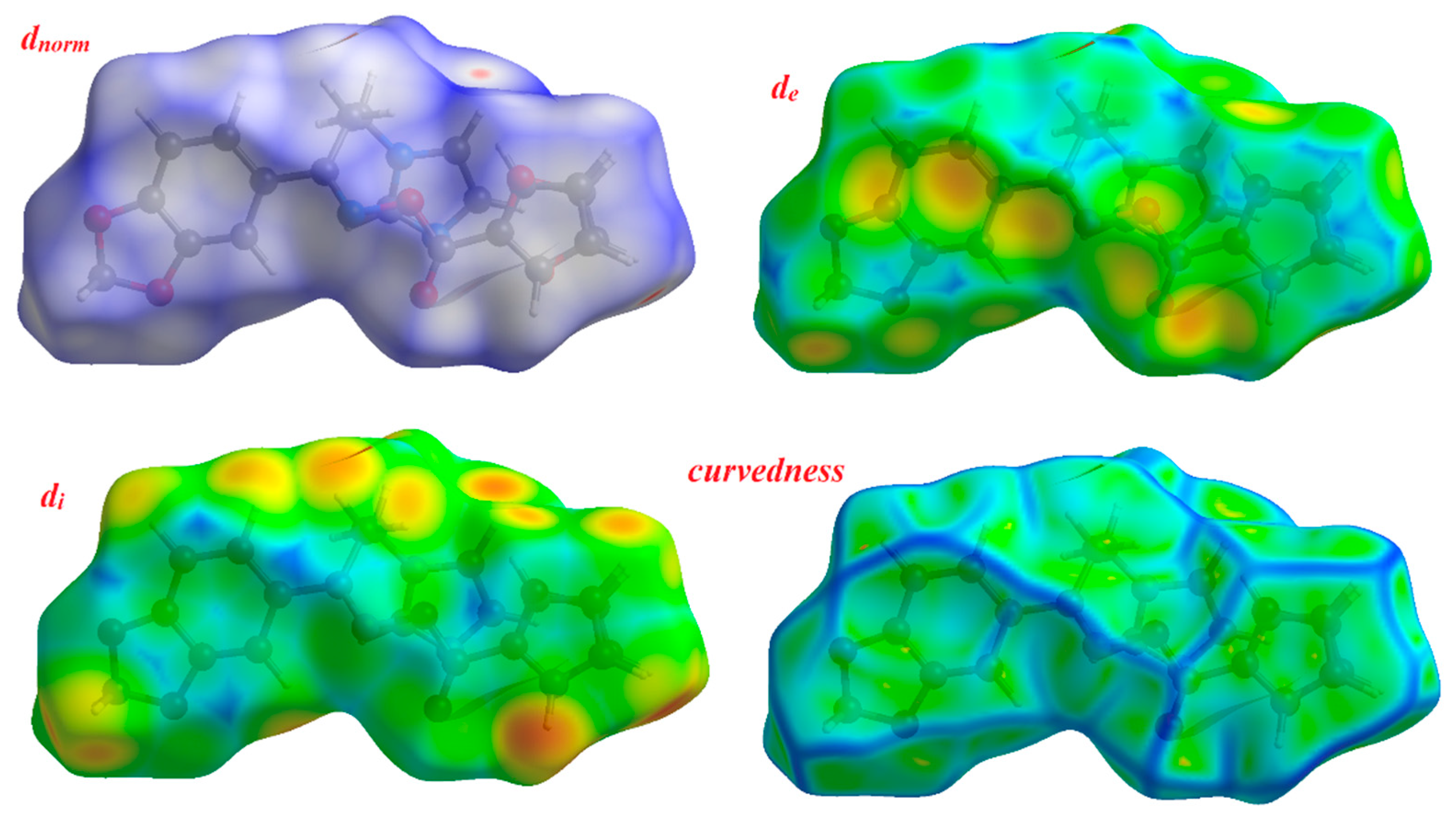
3.6. Hirshfeld Surface Analysis
3.7. Vibrational Spectral Analysis
3.7.1. Imidazole Ring Vibrations
3.7.2. Methylene Group Vibrations
3.7.3. Benzodioxole Ring Vibrations
3.7.4. Side Chain C=N-O-C=O Vibrations
3.7.5. Furan Ring Vibrations
3.7.6. Skeletal Vibrations
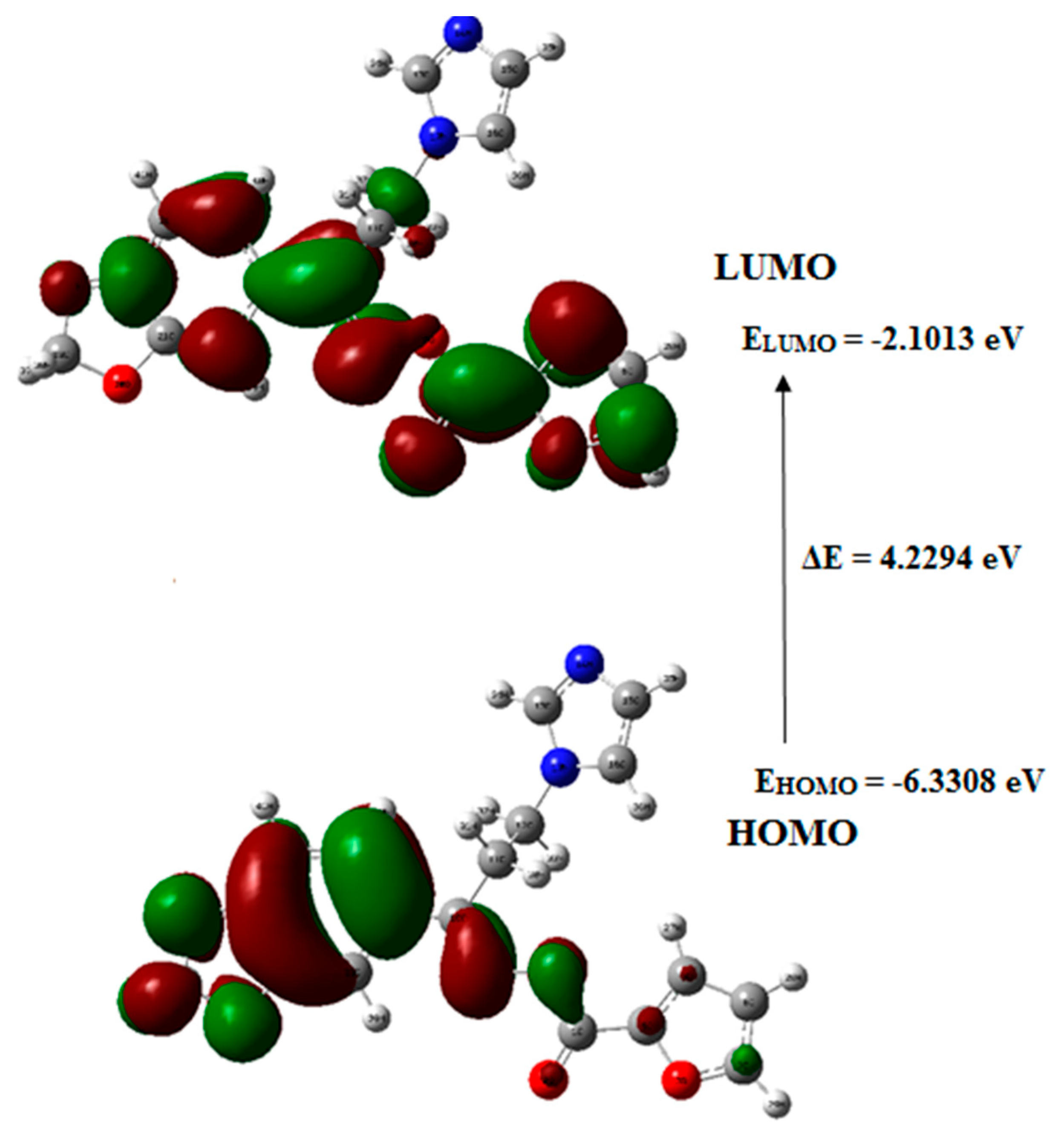
3.8. Frontier Molecular Orbital (FMO) Analysis
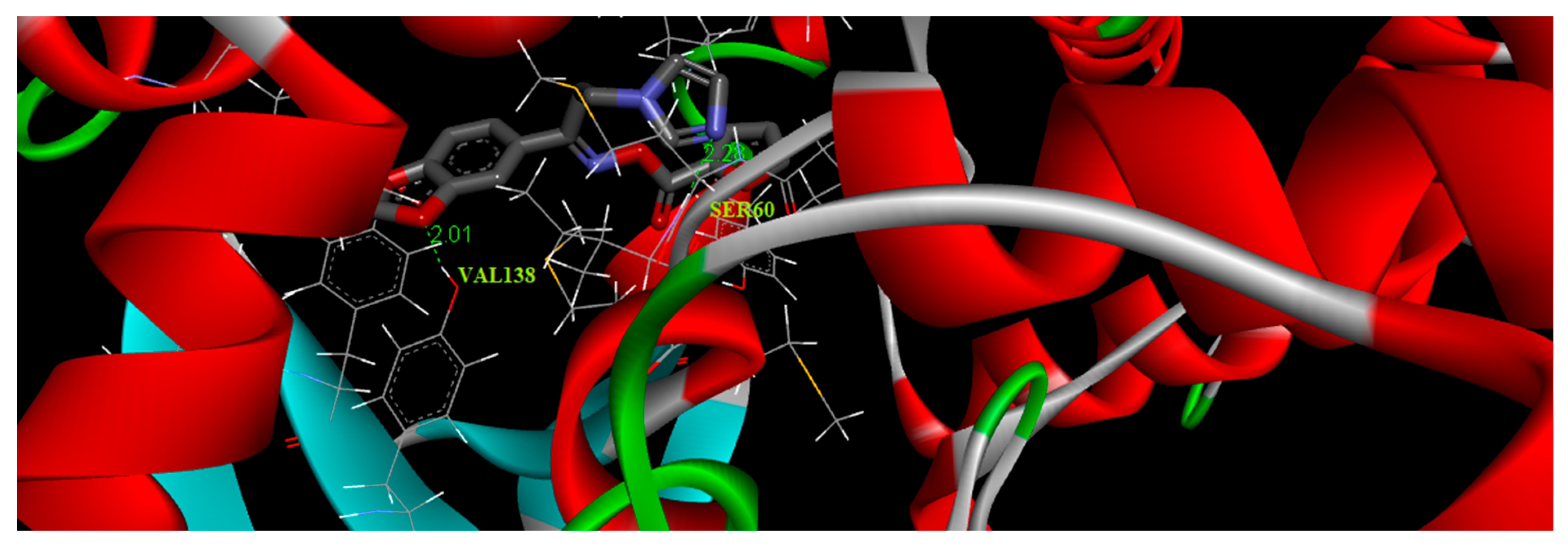
3.9. Molecular Docking Simulations
3.10. Antifungal Activity of the Target Oximino Ester 5
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vandeputte, P.; Ferrari, S.; Coste, A.T. Antifungal resistance and new strategies to control fungal infections. Int. J. Microbiol. 2011, 2012, 713687. [Google Scholar] [CrossRef] [PubMed]
- Groll, A.H.; Lumb, J. New developments in invasive fungal disease. Future Microbiol. 2012, 7, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Cuenca-Estrella, M.; Bernal-Martinez, L.; Buitrago, M.J.; Castelli, M.V.; Gomez-Lopez, A.; Zaragoza, O.; Rodriguez-Tudela, J.L. Update on the epidemiology and diagnosis of invasive fungal infection. Int. J. Antimicrob. Agents 2008, 32, S143–S147. [Google Scholar] [CrossRef]
- Crunkhorn, S. Fungal infection: Protecting from Candida albicans. Nat. Rev. Drug Discov. 2016, 15, 604. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.I.; de Souza, I.G.; Borelli, B.M.; Matos, T.T.S.; Teixeira, I.N.S.; Ramos, J.P.; de Souza Fagundes, E.M.; de Oliveira Fernandes, P.; Maltarollo, V.G.; Johann, S. Synthesis, molecular modeling studies and evaluation of antifungal activity of a novel series of thiazole derivatives. Eur. J. Med. Chem. 2018, 151, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Aperis, G.; Mylonakis, E. Newer triazole antifungal agents: Pharmacology, spectrum, clinical efficacy and limitations. Expert Opin. Investig. Drugs 2006, 15, 579–602. [Google Scholar] [CrossRef] [PubMed]
- Pappas, P.G.; Kauffman, C.A.; Andes, D.R.; Clancy, C.J.; Marr, K.A.; Ostrosky-Zeichner, L.; Reboli, A.C.; Schuster, M.G.; Vazquez, J.A.; Walsh, T.J. Clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2015, 62, 1–50. [Google Scholar] [CrossRef]
- Aoyama, Y.; Yoshida, Y.; Sato, R. Yeast cytochrome P-450 catalyzing lanosterol 14 alpha-demethylation. II. Lanosterol metabolism by purified P-450 (14) DM and by intact microsomes. J. Biol. Chem. 1984, 259, 1661–1666. [Google Scholar]
- Aboul-Enein, M.N.; El-Azzouny, A.A.; Attia, M.I.; Saleh, O.A.; Kansoh, A.L. Synthesis and anti-Candida potential of certain novel 1-[(3-substituted-3-phenyl)propyl]-1H-imidazoles. Arch. Pharm. 2011, 344, 794–801. [Google Scholar] [CrossRef]
- Roman, G.; Mares, M.; Nastasa, V. A novel antifungal agent with broad spectrum: 1-(4-biphenylyl)-3-(1H-imidazol-1-yl)-1-propanone. Arch. Pharm. 2013, 346, 110–118. [Google Scholar] [CrossRef]
- Attia, M.I.; Radwan, A.A.; Zakaria, A.S.; Almutairi, M.S.; Ghoneim, S.W. 1-Aryl-3-(1H-imidazol-1-yl)propan-1-ol esters: Synthesis, anti-Candida potential and molecular modeling studies. Chem. Cent. J. 2013, 7, 168. [Google Scholar] [CrossRef] [PubMed]
- Leite, A.C.L.; da Silva, K.P.; de Souza, I.A.; de Araújo, J.M.; Brondani, D.J. Synthesis, antitumour and antimicrobial activities of new peptidyl derivatives containing the 1,3-benzodioxole system. Eur. J. Med. Chem. 2004, 39, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Cotinguiba, F.; Regasini, L.O.; da Silva Bolzani, V.; Debonsi, H.M.; Passerini, G.D.; Cicarelli, R.M.B.; Kato, M.J.; Furlan, M. Piperamides and their derivatives as potential anti-trypanosomal agents. Med. Chem. Res. 2009, 18, 703–711. [Google Scholar] [CrossRef]
- Al-Wabli, R.I.; Al-Ghamdi, A.R.; Ghabbour, H.A.; Al-Agamy, M.H.; Monicka, J.C.; Joe, I.H.; Attia, M.I. Synthesis, X-ray single crystal structure, molecular docking and DFT computations on N-[(1E)-1-(2H-1,3-benzodioxol-5-yl)-3-(1H-imidazol-1-yl)propylidene]-hydroxylamine: A new potential antifungal agent precursor. Molecules 2017, 22, 373. [Google Scholar] [CrossRef] [PubMed]
- Brucker. APEX2, SAINT and SADABS; Brucker AXS: Madison, WI, USA, 2009. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXTL-PC (Version 7); Siemens Analytical Instruments, Inc.: Madison, WI, USA, 2014. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian-09; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Jamróz, M.H. Vibrational energy distribution analysis (VEDA): Scopes and limitations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 114, 220–230. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Wolff, S.; Grimwood, D.; McKinnon, J.; Jayatilaka, D.; Spackman, M. Crystal Explorer 2.0; University of Western Australia: Perth, Australia, 2007. [Google Scholar]
- Al-Wabli, R.I.; Al-Ghamdi, A.R.; Primsa, I.; Ghabbour, H.A.; Al-Agamy, M.H.; Joe, I.H.; Attia, M.I. (2E)-2-[1-(1,3-benzodioxol-5-yl)-3-(1H-imidazol-1-yl)propylidene]-N-(4-methoxy phenyl)hydrazinecarboxamide: Synthesis, crystal structure, vibrational analysis, DFT computations, molecular docking and antifungal activity. J. Mol. Struct. 2018, 1166, 121–130. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 1987, 2, S1–S19. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Spackman, M.A.; Byrom, P.G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 215–220. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 37, 3814–3816. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Govindarajan, M.; Abdelhameed, A.S.; Al-Saadi, A.A.; Attia, M.I. Experimental and theoretical studies of the vibrational and electronic properties of (2E)-2-[3-(1H-imidazol-1-yl)-1-phenyl-propylidene]-N-phenylhydrazinecarboxamide: An anticonvulsant agent. Appl. Sci. 2015, 5, 955–972. [Google Scholar] [CrossRef]
- Naumov, P.; Ristova, M.; Šoptrajanov, B.; Zugik, M. Vibrational spectra of bis (acetato) tetrakis (imidazole) copper (II). J. Mol. Struct. 2001, 598, 235–243. [Google Scholar] [CrossRef]
- Smith, B.C. Infrared Spectral Interpretation: A Systematic Approach; CRC Press: Boca Raton, FL, USA, 1998. [Google Scholar]
- Bellamy, L. The Infrared Spectra of Complex Molecules; Chapman and Hill: London, UK, 1975. [Google Scholar]
- Fleming, I. Frontier Orbitals and Organic Chemical Reactions; Wiley: Hoboken, NJ, USA, 1977. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comp. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Strushkevich, N.; Usanov, S.; Park, H. Crystal structure of human lanosteroal 14alpha-demethylase (CYP51) in complex with ketoconazole. In Proceedings of the 16th International Conference on Cytochrome P, Graz, Austria, 21–26 June 2009; pp. 21–25. [Google Scholar]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.F., Jr.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The protein data bank: A computer-based archival file for macromolecular structures. Arch. Biochem. Biophys. 1978, 185, 584–591. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comp. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Data | |
| Molecular formula | C18H15N3O5 |
| Mr | 353.33 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 296 (2) |
| a, b, c (Å) | 10.4067 (5), 6.8534 (3), 23.2437 (12) |
| β (°) | 94.627 (2) |
| V (Å3) | 1652.37 (14) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.11 |
| Crystal size (mm) | 0.42 × 0.36 × 0.22 |
| Data Collection | |
| Diffractometer | Bruker APEX-II D8 venture diffractometer |
| Absorption correction | Multiscan SADABS Bruker 2014 |
| Tmin, Tmax | 0.957, 0.977 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 46277, 6612, 3174 |
| Rint | 0.068 |
| (sin θ/λ)max (Å−1) | 0.782 |
| Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.067, 0.181, 1.02 |
| No. of reflections | 6612 |
| No. of parameters | 272 |
| No. of restraints | 106 |
| H-atom treatment | H atoms parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.21, −0.24 |
| O1-C3 | 1.372 (2) | O5-C18 | 1.382 (3) |
| O1-C4 | 1.418 (3) | N1-C8 | 1.282 (2) |
| O2-C4 | 1.417 (3) | N2-C10 | 1.454 (3) |
| O2-C5 | 1.370 (3) | N2-C11 | 1.355 (3) |
| O3-N1 | 1.437 (2) | N2-C13 | 1.345 (3) |
| O3-C14 | 1.355 (2) | N3-C12 | 1.362 (4) |
| O4-C14 | 1.190 (2) | N3-C13 | 1.315 (3) |
| O5-C15 | 1.326 (3) | ||
| C3-O1-C4 | 105.87 (16) | O2-C5-C6 | 128.18 (19) |
| C4-O2-C5 | 106.04 (16) | N1-C8-C7 | 113.73 (18) |
| N1-O3-C14 | 112.68 (14) | N1-C8-C9 | 125.53 (17) |
| C15-O5-C18 | 106.7 (2) | N2-C10-C9 | 112.46 (17) |
| O3-N1-C8 | 109.89 (14) | N2-C11-C12 | 106.4 (2) |
| C10-N2-C11 | 126.76 (16) | N3-C12-C11 | 110.5 (2) |
| C10-N2-C13 | 126.55 (18) | N2-C13-N3 | 112.17 (19) |
| C11-N2-C13 | 106.44 (17) | O3-C14-O4 | 125.31 (18) |
| C12-N3-C13 | 104.5 (2) | O3-C14-C15 | 108.30 (17) |
| O1-C3-C2 | 128.97 (18) | O4-C14-C15 | 126.40 (19) |
| O1-C3-C5 | 109.62 (17) | O5-C15-C14 | 120.68 (19) |
| O1-C4-O2 | 108.72 (16) | O5-C15-C16 | 109.76 (19) |
| O2-C5-C3 | 109.60 (17) | O5-C18-C17 | 108.6 (2) |
| Bond Length | Value (Å) | Exp. | Angle | DFT Value (°) | Exp. Value (°) | Dihedral Angle | Value (°) | Exp. |
|---|---|---|---|---|---|---|---|---|
| C10-C24 | 1.4843 | 1.503 | O8-N9-C10 | 111.55 | 120.00 | O8-C1-C4-O3 | 173.68 | 180.00 |
| C11-C12 | 1.5428 | 1.523 | N9-C10-C11 | 123.56 | 116.26 | O8-C1-C4-C5 | 7.57 | 180.00 |
| C13-N14 | 1.3135 | 1.260 | N9-C10-C24 | 115.28 | 121.17 | C1-O8-N9-C10 | 164.71 | 120.00 |
| C13-N17 | 1.3684 | 1.462 | C11-C10-C24 | 121.16 | 122.56 | O8-N9-C10-C11 | 1.19 | 0.55 |
| C13-H34 | 1.0803 | 1.100 | C10-C11-C12 | 111.99 | 109.50 | O8-N9-C10-C24 | 178.35 | −180.00 |
| N14-C15 | 1.3747 | 1.805 | C13-N14-C15 | 105.23 | 105.88 | N9-C10-C11-C12 | 81.97 | 0.00 |
| C15-C16 | 1.3718 | 1.337 | N14-C15-C16 | 110.49 | 101.12 | N9-C10-C11-H30 | 38.35 | 119.91 |
| C15-H35 | 1.079 | 1.100 | C19-O18-C22 | 105.40 | 101.53 | C24-C10-C11-C12 | 97.54 | −179.45 |
| C16-N17 | 1.3817 | 1.462 | O18-C19-O20 | 107.25 | 111.96 | C9-C10-C24-C23 | 10.75 | −40.00 |
| C21-C22 | 1.3956 | 1.420 | C21-C22-C26 | 121.52 | 120.00 | C9-C10-C24-C25 | 168.60 | 139.43 |
| C21-C23 | 1.3704 | 1.420 | C21-C23-C24 | 117.34 | 120.00 | C11-C10-C24-C23 | 169.70 | 139.42 |
| C23-C24 | 1.4186 | 1.420 | C10-C24-C23 | 119.31 | 120.00 | C11-C10-C24-C25 | 10.94 | −41.15 |
| C24-C25 | 1.4004 | 1.420 | C10-C24-C25 | 121.01 | 120.00 | C11-C12-N17-C13 | 111.27 | −60.00 |
| C25-C26 | 1.404 | 1.420 | C23-C24-C25 | 119.68 | 120.00 | C11-C12-N17-C16 | 67.36 | 119.42 |
| H39…N9 | 2.429 | - | C24-C25-C26 | 122.14 | 119.99 | |||
| C22-C26-C25 | 116.88 | 120.00 | ||||||
| C22-C26-H41 | 121.56 | 120.01 |
| Calculated Wavenumber (cm−1) | Experimental Wavenumber (cm−1) | Assignments with PED (%) a | |
|---|---|---|---|
| IR | Raman | ||
| 3159 | 3269 m | νC5-H27(84), νC7-H29(11)f | |
| 3106 | 3106 vw | νC25-H40(37), νC26-H41(62), νC15-H35(16)f | |
| 3023 | 3141 m | νasyH38-C19-H37(97), νasy H32-C12-H33(10), νsymH30-C11-H31(30)m | |
| 2999 | 3090 m | νasy H32-C12-H33(35)m | |
| 2979 | 3036 vw | 2998 m | νsym H30-C11-H31(36)m |
| 2961 | 2993 vw | 2951 m | νsym H33-C12-H32(42), νC12-H33(49)m |
| 2916 | 2930 m | 2801 vs | νC19-H38(92), νsymH38-C19-H37(97)m |
| 1764 | 2874 vw | 1814 vs | νO2=C1(86)si |
| 1604 | 1772s | 1774 vs | νN9-C10 (24), νC21-C23(33)b |
| 1603 | 1730 w | νN9-C10 (23), νC22-C26(15), νC24-C25(18)b | |
| 1579 | 1611 vw | 1619 w | νN9-C10 (30), νC22-C26(26)b, νC10=N9 (10)s |
| 1546 | 1587 vw | 1584 vw | νC6-C7(25), νC4-C5(43) |
| 1489 | 1553 vw | νC16=C15(31)im | |
| 1440 | 1477 w | 1471 vw | νC13=N14(55)im |
| 1422 | 1441 m | νC21-C23(14), νC25-C26(11)b | |
| 1382 | 1429 w | 1433 w | νC15-C16(10)im |
| 1345 | 1388 vw | 1399 m | νC10-C11(20)sk |
| 1298 | 1343 vw | 1349 m | νC10-C24(22)sk |
| 1264 | 1302 m | 1308 m | νC10-C11(20)sk |
| 1237 | νC25-C26(15)b | ||
| 1219 | 1220 m | νN17-C12(10)sk | |
| 1124 | 1176 vs | νC25-C26(11)b | |
| 1103 | νN14-C15(55)im | ||
| 1102 | 1139 s | νC10-C11(20)sk | |
| 1075 | 1161 vw | νC6-C7(13) νO3-C7(40) β H41-C26-C22(15)b | |
| 1039 | 1044 s | 1079 m | νO8-C1(23)si |
| 1023 | 1019 vs | νO20-C1(40)νO18-C19(19) | |
| 1009 | 1002 m | 1049 vs | νN17-C13(15)im |
| 990 | 993 m | νC10-C11(20)sk | |
| 924 | 1009 vs | νC4-C5(10), νO3-C4(14), νO8-C1(23)f | |
| 886 | 918 m | 938 w | Ring deformation b O8-C1 (18)si |
| 872 | 894 vw | γC25-C24-C23-H39(52)b | |
| 775 | 805 vw | 808 w | γC25-C24-C26-H41(58)b |
| 747 | 799 vw | Ring breathing b(34) | |
| 739 | 751 vw | ωC13-N14-C15(55)f | |
| 695 | 704 vw | 719 vw | νC10-C11(11), νN17-C12(11)sk |
| 478 | 479 vw | 481 vw | νC1-C4(14)sk |
| 409 | 423 vw | νC1-C4(10)sk | |
| 396 | 410 vw | νC1-C4(13)sk | |
| 356 | 362 vw | ||
| 222 | 223 w | ||
| 220 | 203 w | ||
| Compound No. | MIC (µmol/mL) | |||
|---|---|---|---|---|
| C. albicans | C. tropicalis | C. parapsilosis | Aspergillus niger | |
| 5 | 0.181 | 0.724 | 0.181 | 0.724 |
| Fluconazole | 0.051 | 0.045 | 0.047 | ND |
| Ketoconazole | ND | ND | ND | 0.02 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Wabli, R.I.; Al-Ghamdi, A.R.; Aswathy, S.V.; Ghabbour, H.A.; Al-Agamy, M.H.; Hubert Joe, I.; Attia, M.I. Synthesis, Single Crystal X-ray Structure, DFT Computations, Hirshfeld Surface Analysis and Molecular Docking Simulations on ({[(1E)-1-(1,3-Benzodioxol-5-yl)-3-(1H-imidazol-1-yl)propylidene]amino}oxy)(furan-2-yl)methanone: A New Antifungal Agent. Crystals 2019, 9, 25. https://doi.org/10.3390/cryst9010025
Al-Wabli RI, Al-Ghamdi AR, Aswathy SV, Ghabbour HA, Al-Agamy MH, Hubert Joe I, Attia MI. Synthesis, Single Crystal X-ray Structure, DFT Computations, Hirshfeld Surface Analysis and Molecular Docking Simulations on ({[(1E)-1-(1,3-Benzodioxol-5-yl)-3-(1H-imidazol-1-yl)propylidene]amino}oxy)(furan-2-yl)methanone: A New Antifungal Agent. Crystals. 2019; 9(1):25. https://doi.org/10.3390/cryst9010025
Chicago/Turabian StyleAl-Wabli, Reem I., Alwah R. Al-Ghamdi, S. V. Aswathy, Hazem A. Ghabbour, Mohamed H. Al-Agamy, I. Hubert Joe, and Mohamed I. Attia. 2019. "Synthesis, Single Crystal X-ray Structure, DFT Computations, Hirshfeld Surface Analysis and Molecular Docking Simulations on ({[(1E)-1-(1,3-Benzodioxol-5-yl)-3-(1H-imidazol-1-yl)propylidene]amino}oxy)(furan-2-yl)methanone: A New Antifungal Agent" Crystals 9, no. 1: 25. https://doi.org/10.3390/cryst9010025
APA StyleAl-Wabli, R. I., Al-Ghamdi, A. R., Aswathy, S. V., Ghabbour, H. A., Al-Agamy, M. H., Hubert Joe, I., & Attia, M. I. (2019). Synthesis, Single Crystal X-ray Structure, DFT Computations, Hirshfeld Surface Analysis and Molecular Docking Simulations on ({[(1E)-1-(1,3-Benzodioxol-5-yl)-3-(1H-imidazol-1-yl)propylidene]amino}oxy)(furan-2-yl)methanone: A New Antifungal Agent. Crystals, 9(1), 25. https://doi.org/10.3390/cryst9010025