Anthracene-Based Lanthanide Metal-Organic Frameworks: Synthesis, Structure, Photoluminescence, and Radioluminescence Properties


Abstract
:1. Introduction
2. Experimental Method
2.1. Synthesis
2.2. Characterization
2.3. Photoluminescence and Radioluminescence
3. Results and Discussion
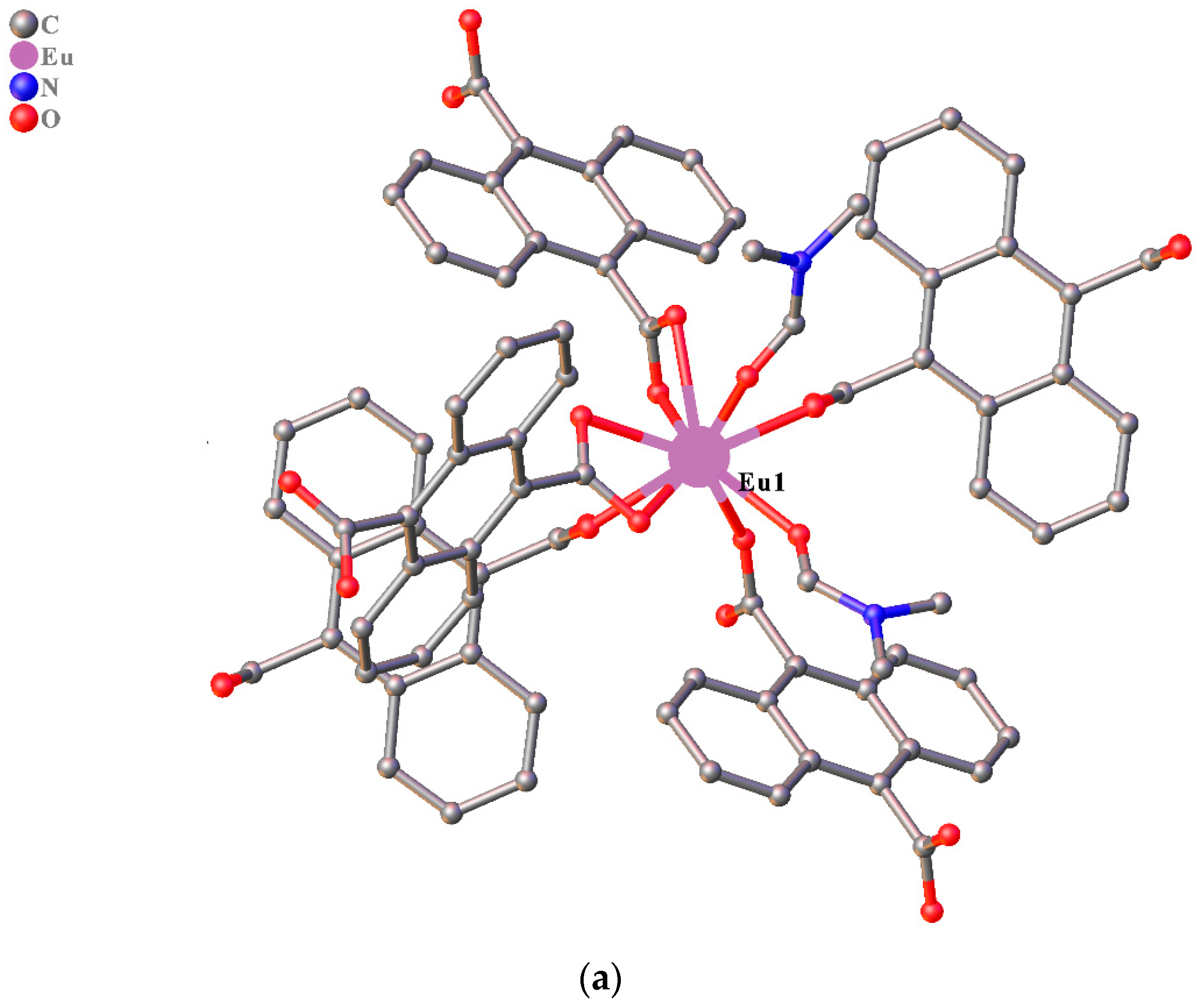
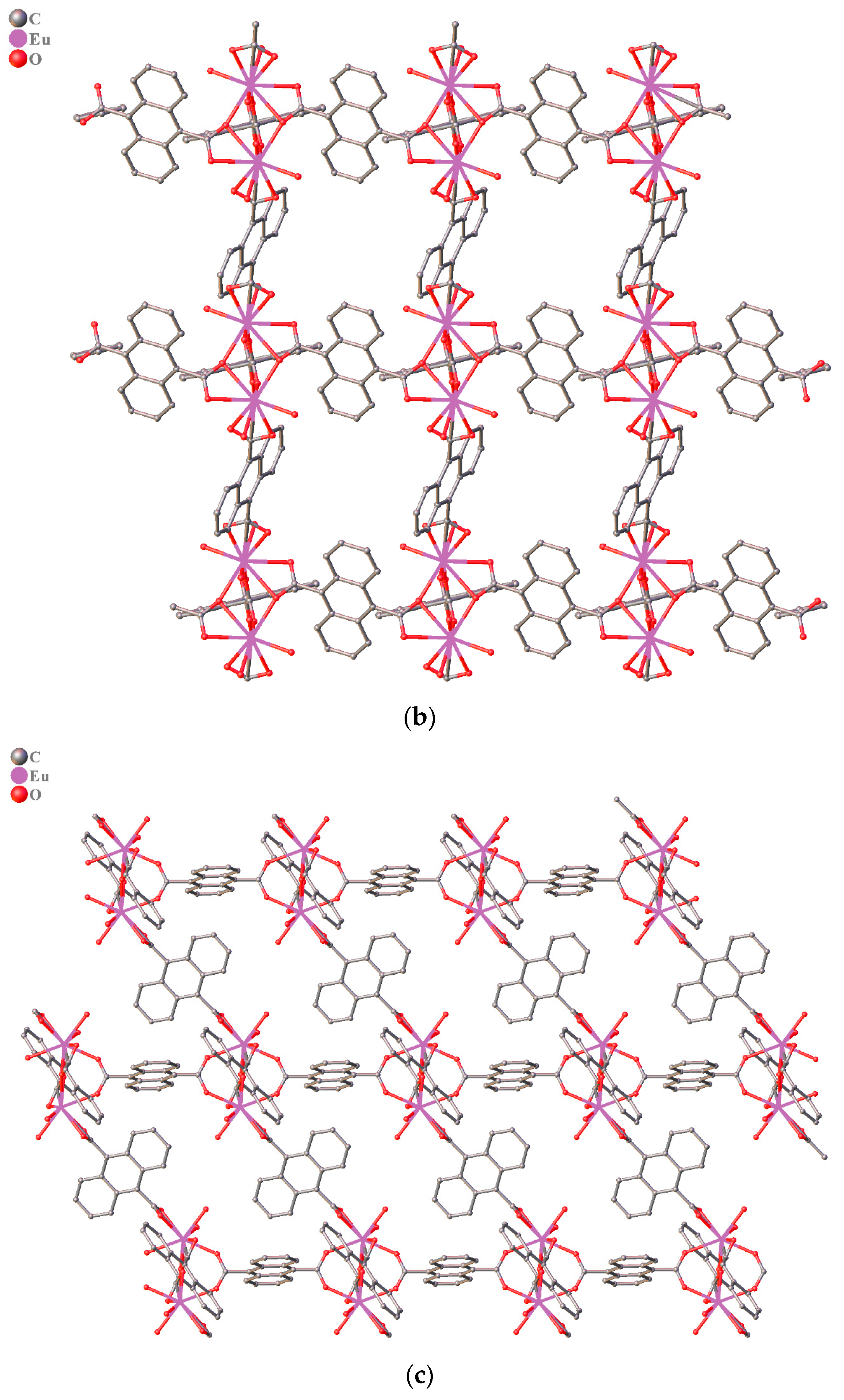
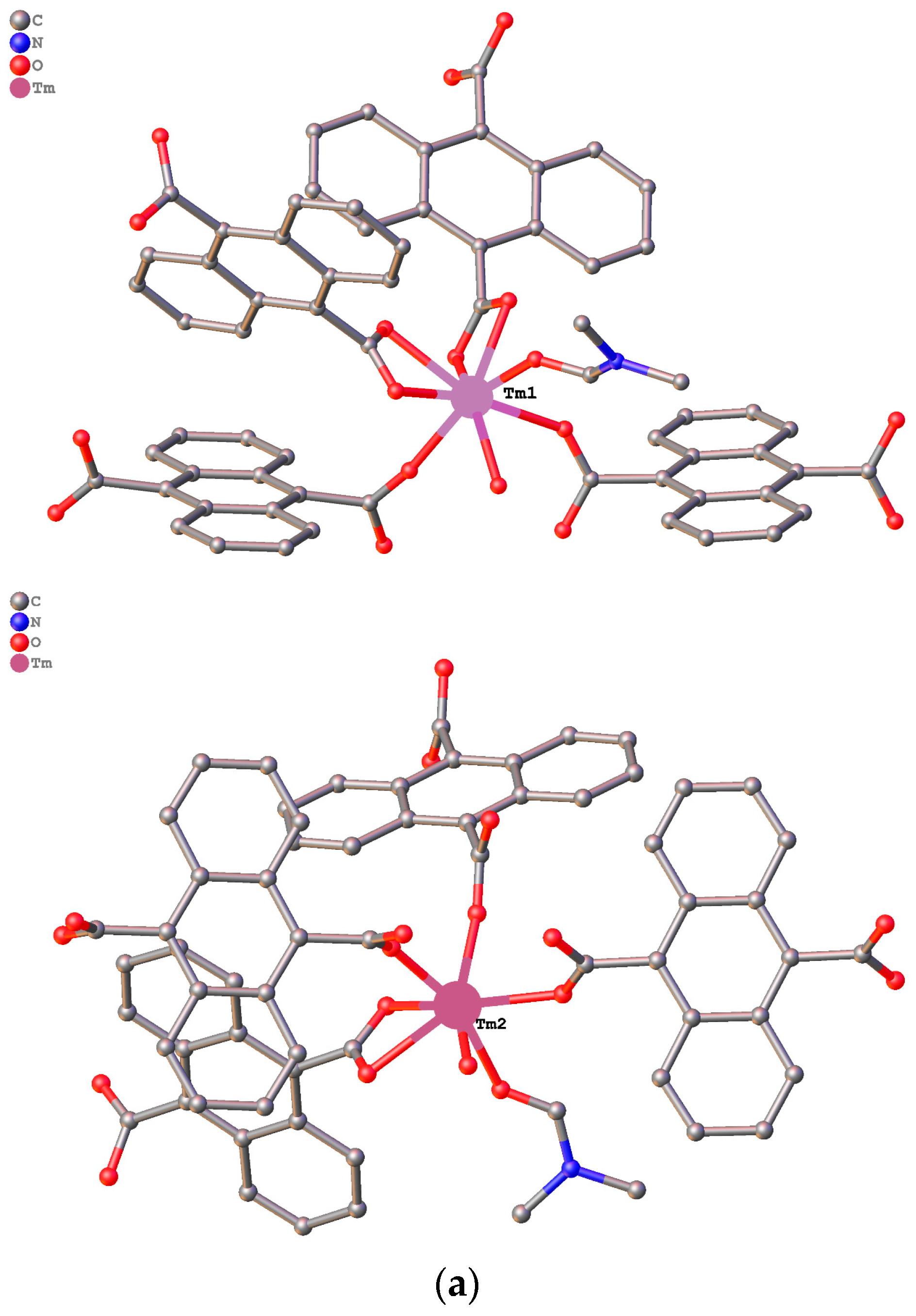
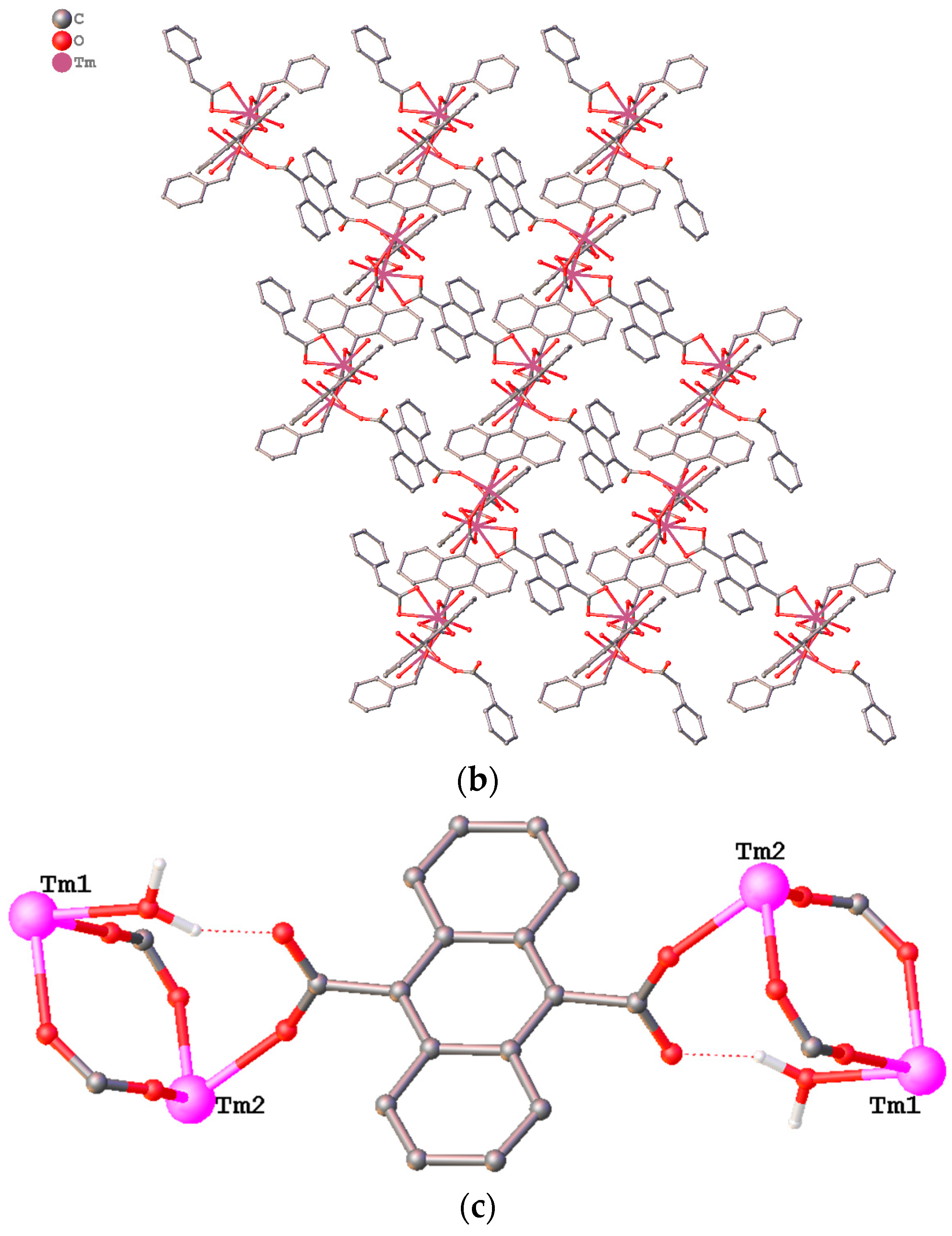
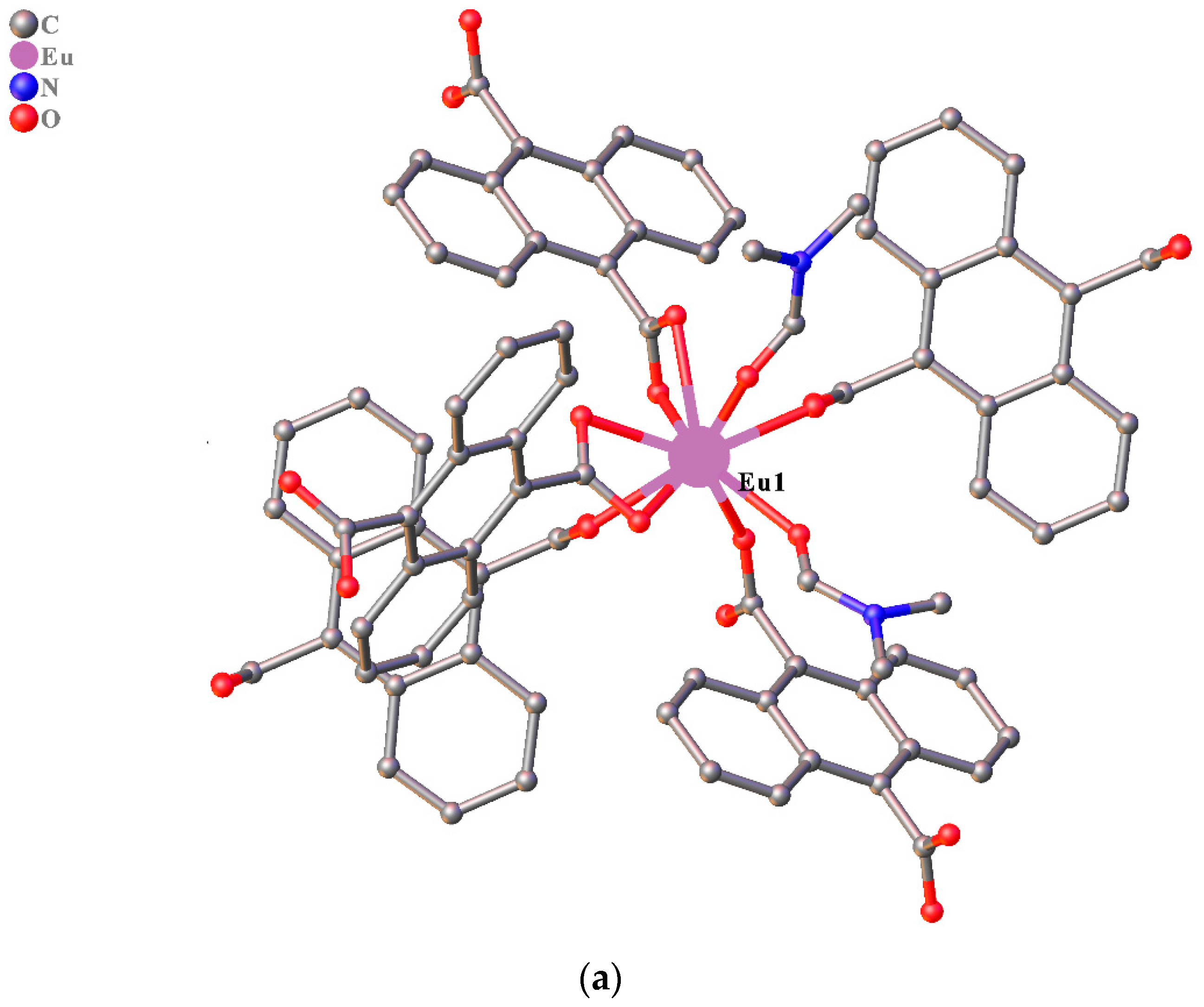
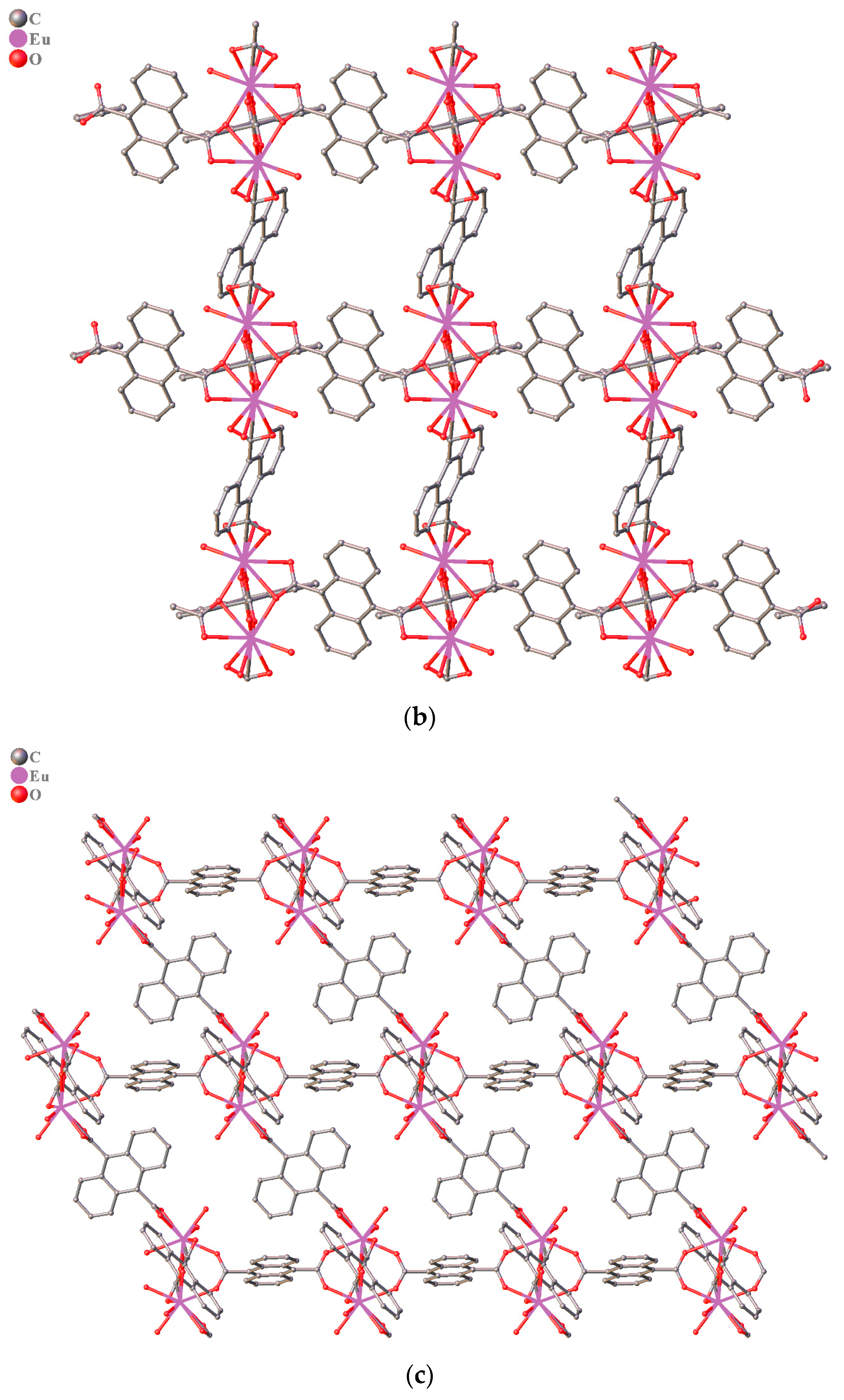
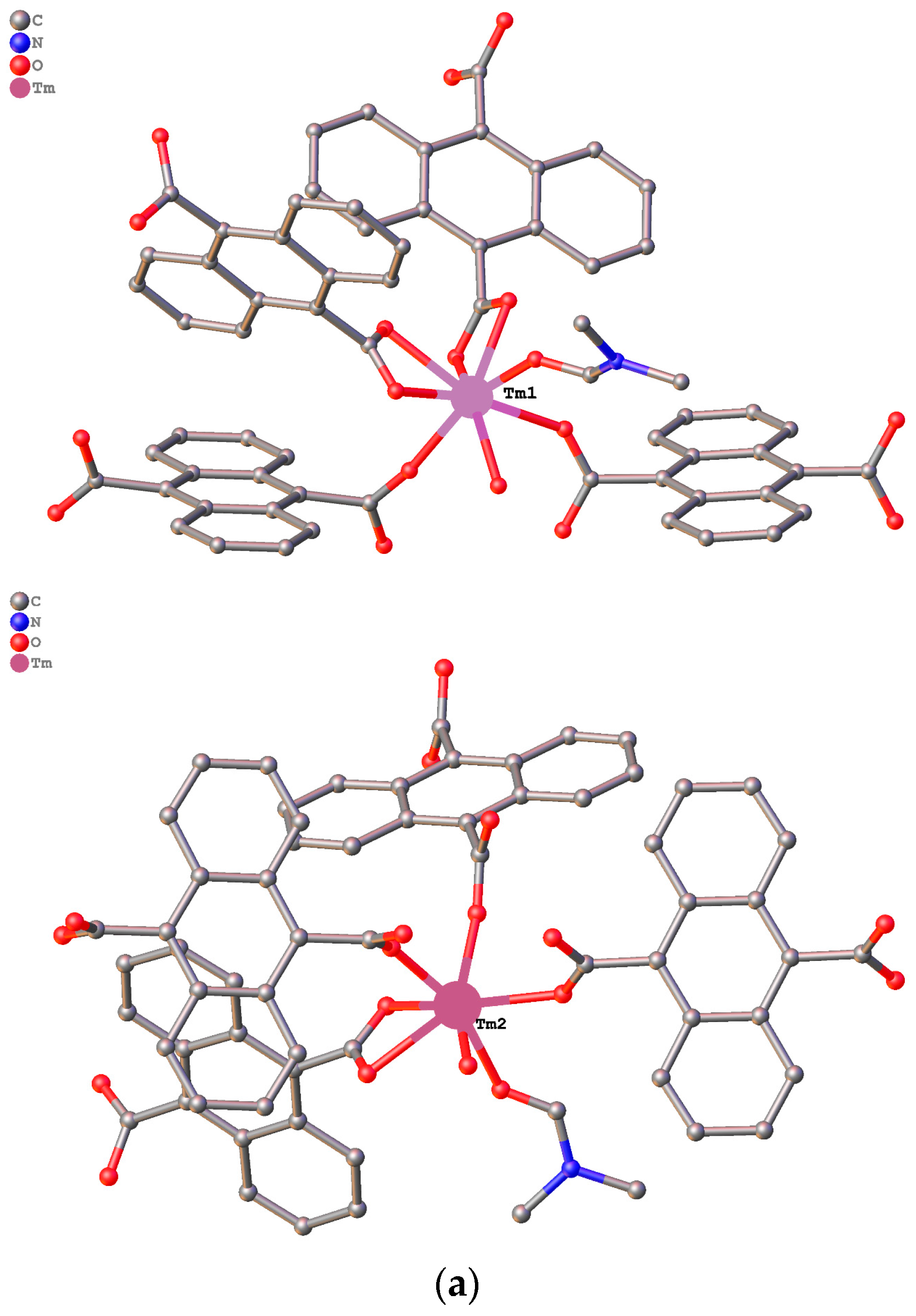
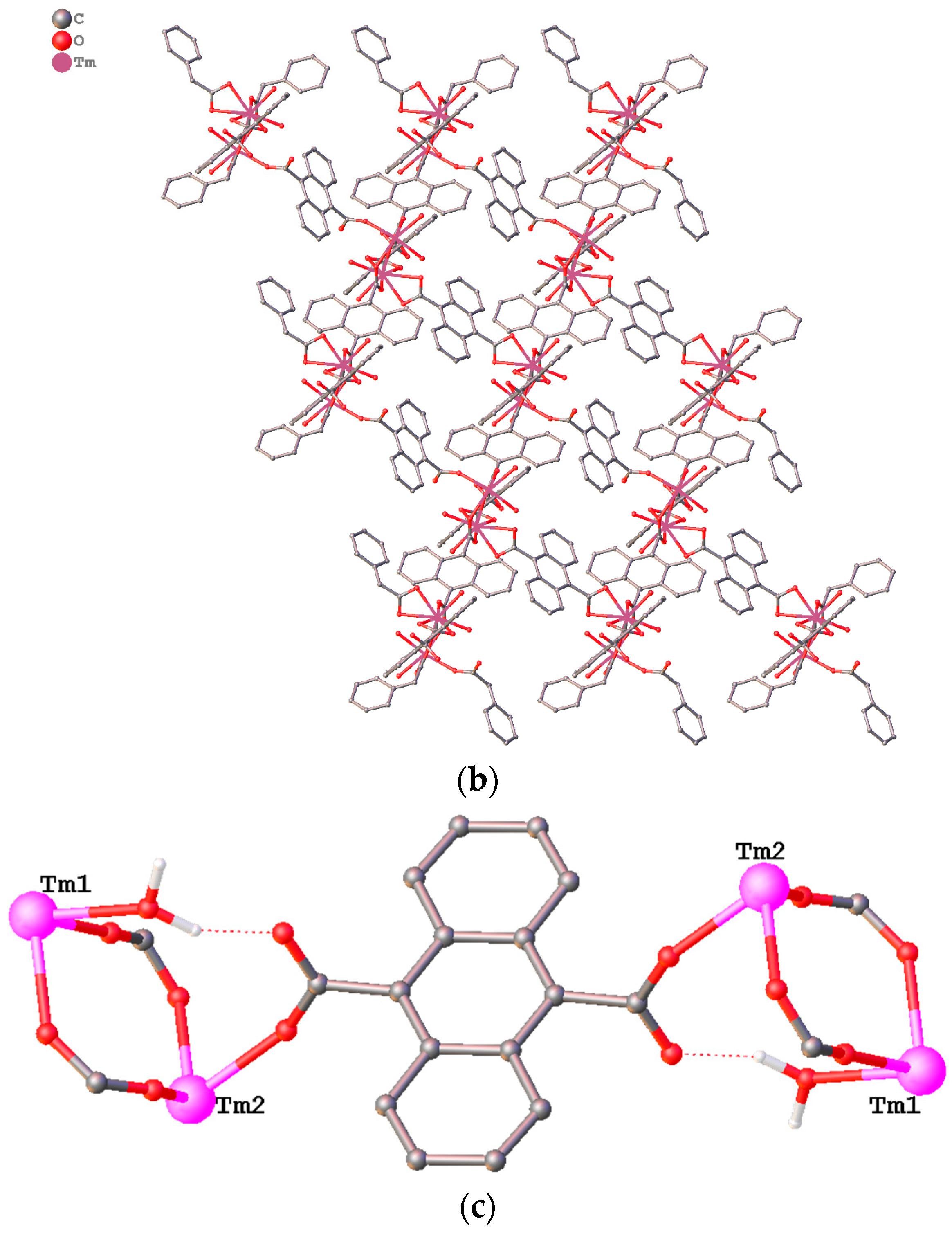
3.1. Structure Description
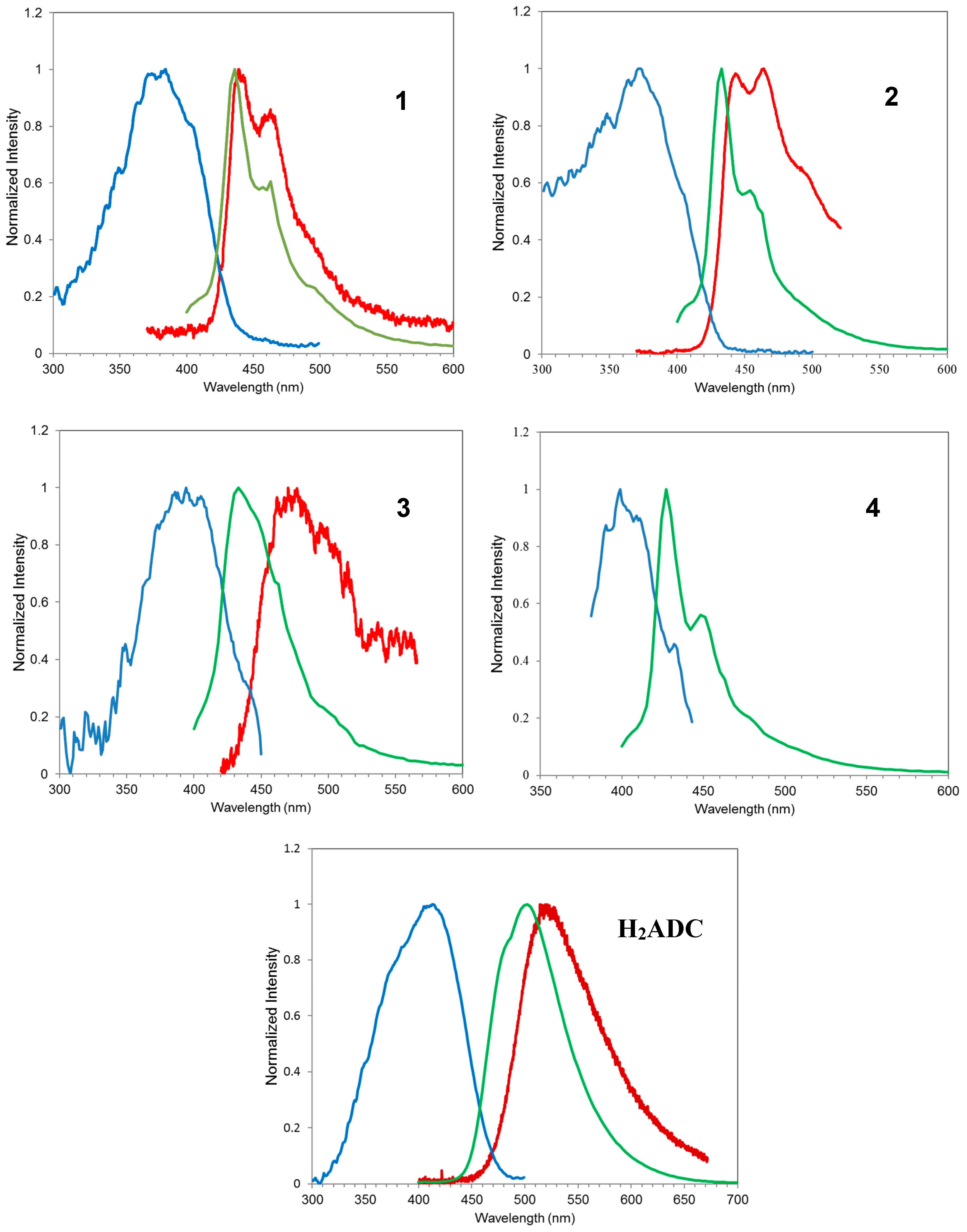
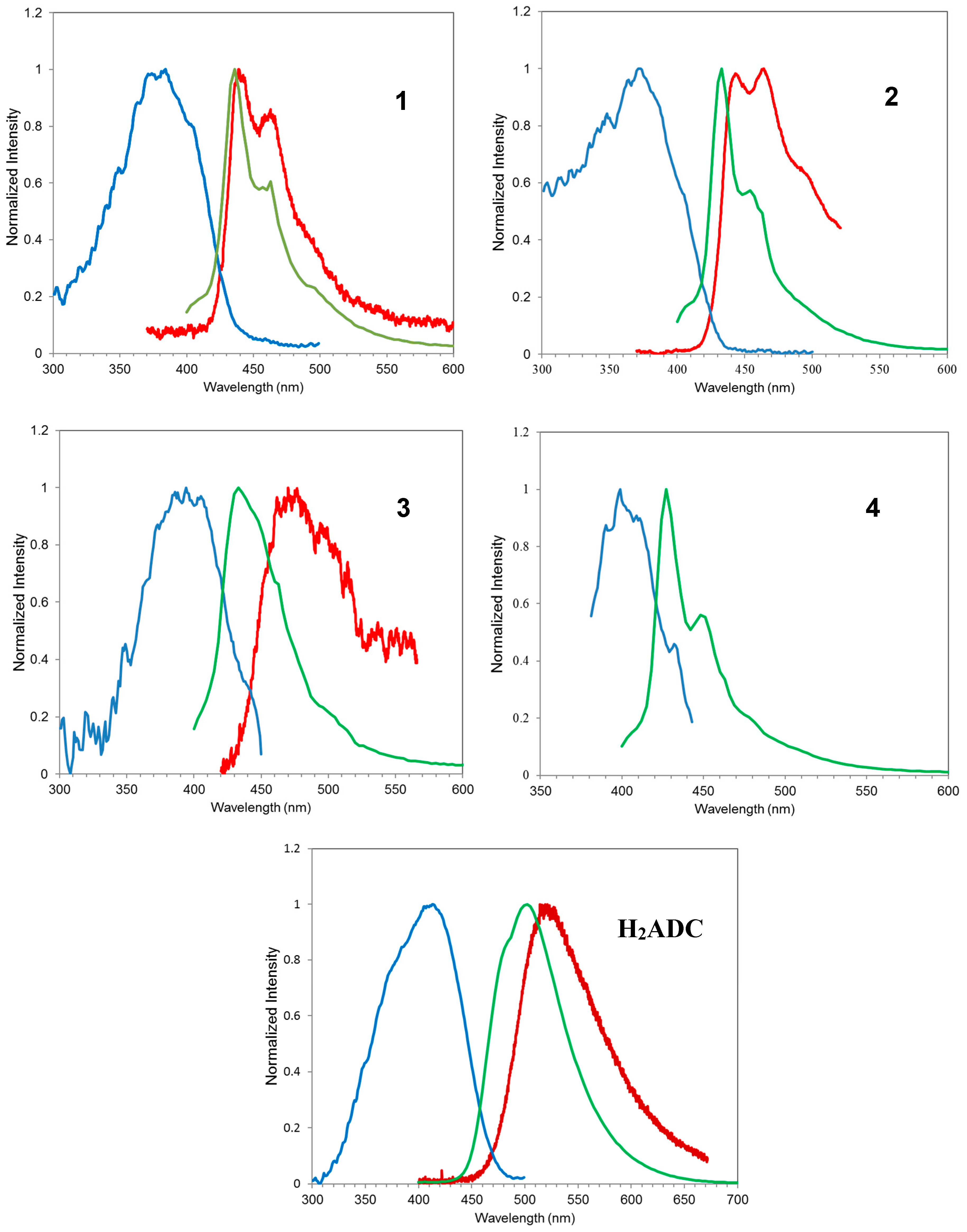
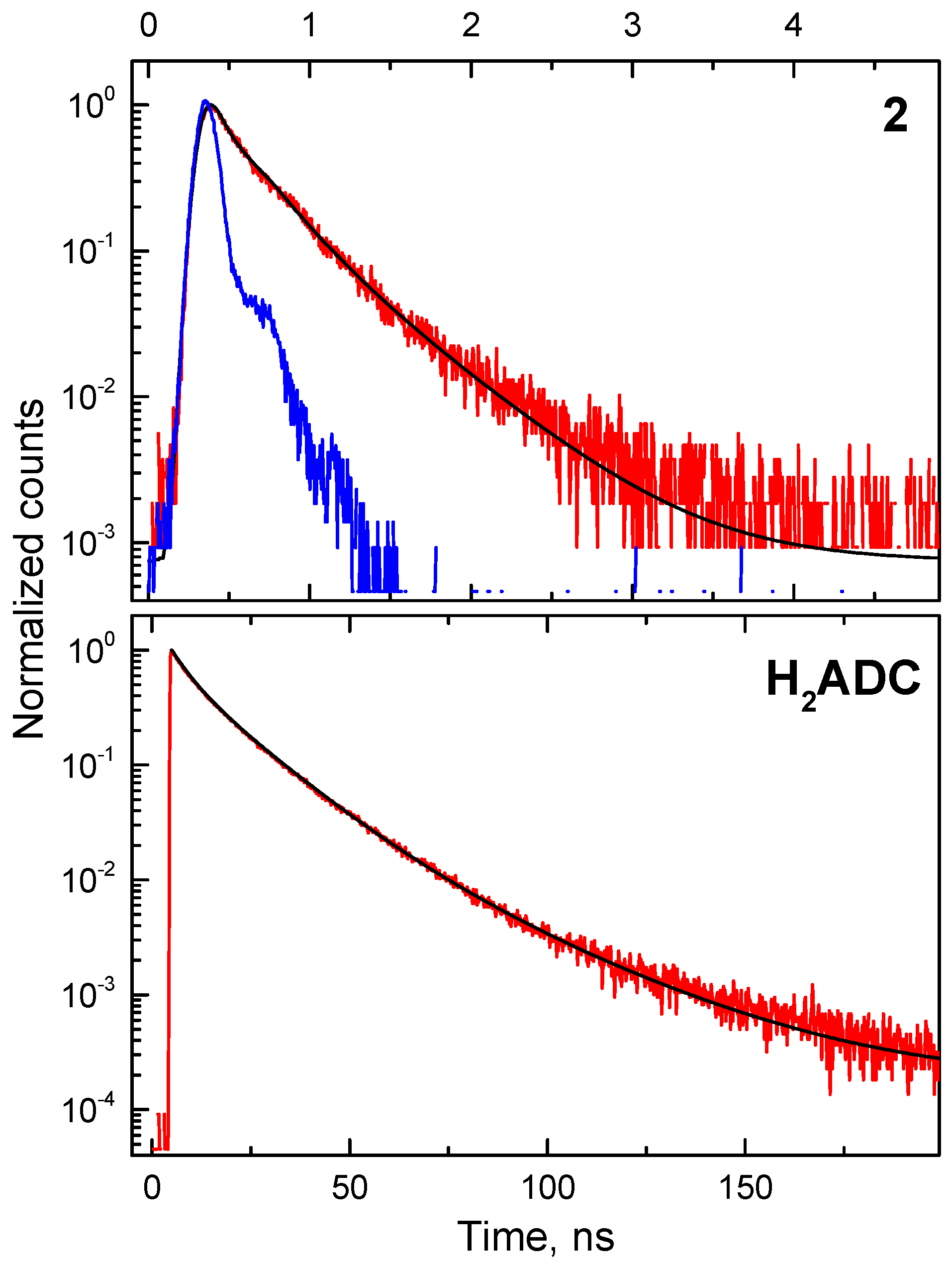
3.2. Photoluminescence
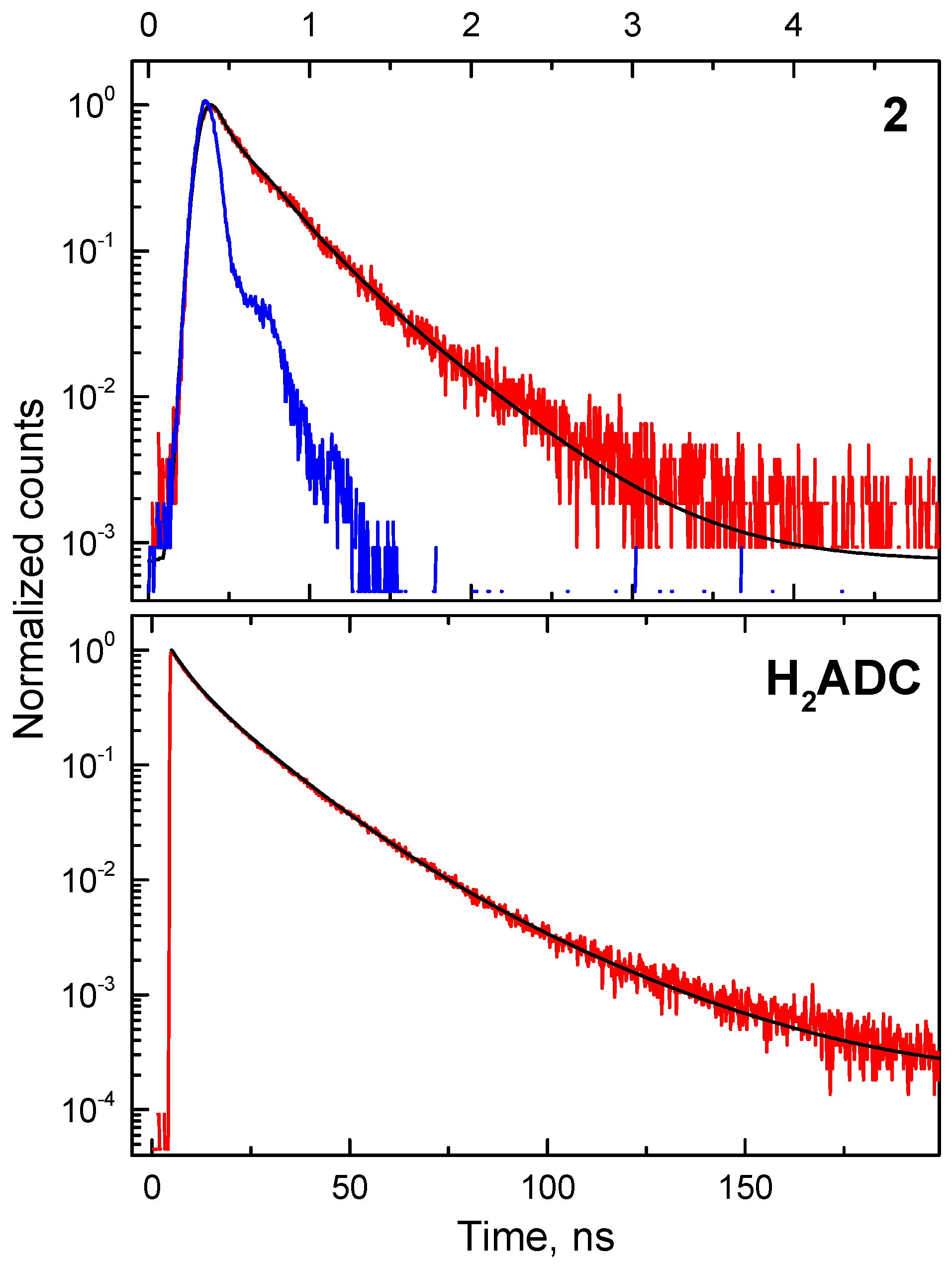
4. Radioluminescence
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Allendorf, M.D.; Bauer, C.A.; Bhakta, R.K.; Houk, R.J.T. Luminescent metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1330–1352. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.J., IV; Feng, P.L.; Meek, S.T.; Leong, K.; Doty, F.P.; Allendorf, M.D. Connecting structure with function in metal-organic frameworks to design novel photo- and radioluminescent materials. J. Mater. Chem. 2012, 22, 10235–10248. [Google Scholar] [CrossRef]
- Kreno, L.E.; Leong, K.; Farha, O.K.; Allendorf, M.D.; Van Duyne, R.P.; Hupp, J.T. Metal-organic framework materials as chemical sensors. Chem. Rev. 2012, 112, 1105–1125. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.; Carlos, L.D.; Almeida Paz, F.A.; Ananias, D. Luminescent multifunctional lanthanides-based metal–organic frameworks. Chem. Soc. Rev. 2011, 40, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.G.; Corma, A.; Garciá, H. Metal-organic frameworks as semiconductors. J. Mater. Chem. 2010, 20, 3141–3156. [Google Scholar] [CrossRef]
- Cui, Y.; Yue, Y.; Qian, G.; Chen, B. Luminescent functional metal-organic frameworks. Chem. Rev. 2012, 112, 1126–1162. [Google Scholar] [CrossRef] [PubMed]
- Knoll, G.F. Radiation Detection and Measurement, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2000; pp. 219–263. [Google Scholar]
- Liu, J.Q. Crystal engineering of Cd(II) metal-organic frameworks bridged by dicarboxylates and N-donor coligands. J. Coord. Chem. 2011, 64, 1503–1512. [Google Scholar] [CrossRef]
- Doty, F.; Bauer, C.; Skulan, A.; Grant, P.; Allendorf, M.D. Scintillating metal-organic frameworks: A new class of radiation detection materials. Adv. Mater. 2009, 21, 95–101. [Google Scholar] [CrossRef]
- Bauer, C.A.; Timofeeva, T.V.; Settersten, T.B.; Patterson, B.D.; Liu, V.H.; Simmons, B.A.; Allendorf, M.D. Influence of connectivity and porosity on ligand-based luminescence in zinc metal-organic frameworks. J. Am. Chem. Soc. 2007, 129, 7136–7144. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.L.; Branson, J.V.; Hattar, K.; Vizkelethy, G.; Allendorf, M.D.; Doty, F.P. Designing metal-organic framework for radiation dectection. Nucl. Inst. Methods Phys. Res. A 2011, 652, 295–298. [Google Scholar] [CrossRef]
- Mathis, S.R., II; Golafale, S.T.; Bacsa, J.; Steiner, A.; Ingram, C.W.; Doty, F.P.; Auden, E.; Hattar, K. Mesoporous stilbene-based lanthanide metal organic frameworks: Synthesis, photoluminescence and radioluminescence characteristics. Dalton Trans. 2017, 46, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hu, T.; Bu, X. Cadmium(II) and zinc(II) metal-organic frameworks with anthracene-based dicarboxylic ligands: Solvothermal synthesis, crystal structures, and luminescent properties. CrystEngComm 2011, 13, 5152–5161. [Google Scholar] [CrossRef]
- Li, X.; Yang, L.; Zhao, L.; Wang, X.-L.; Shao, K.Z.; Su, Z.-M. Luminescent metal-organic frameworks with anthracene chromophores: Small-molecule sensing and highly selective sensing for nitro explosives. Cryst. Growth Des. 2016, 16, 4374–4382. [Google Scholar] [CrossRef]
- Wang, J.; Liu, C.; Hu, T.; Chang, Z.; Li, C.; Yan, L.; Chen, P.; Bu, X.; Wu, Q.; Zhao, L.; et al. Zinc(II) coordination architectures with two bulky anthracene-based carboxylic ligands: Crystal structures and luminescent properties. CrystEngComm 2008, 10, 681–692. [Google Scholar] [CrossRef]
- Liu, C.; Wang, J.; Chang, Z.; Yan, L.; Bu, X. Cadmium(II) coordination polymers based on a bulky anthracene-based dicarboxylate ligand: Crystal structures and luminescent properties. CrystEngComm 2010, 12, 1833–1841. [Google Scholar] [CrossRef]
- Ma, S.; Wang, X.-S.; Collier, C.D.; Manis, E.S.; Zhou, H.-C. Ultramicroporous metal-organic framework based on 9,10-anthracenedicarboxylate for selective gas adsorption. Inorg. Chem. 2007, 46, 8499–8501. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Zhang, A.-S.; Hiu, T.-L.; Bu, X.-H. ZnII coordination poylmers based on 2,3,6,7-anthracenetetracarboxylic acid: Synthesis, structures, and luminescence properties. Cryst. Growth Des. 2009, 9, 4840–4846. [Google Scholar] [CrossRef]
- Zhuang, J.; Friedel, J.; Terfort, A. The oriented and patterned growth of fluorescent metal-organic frameworks onto functionalized surfaces. Belstein J. Nanotechnol. 2012, 3, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Kojtari, A.; Carroll, P.; Ji, H. Metal organic framework (MOF) micro/nanopillars. CrystEngComm 2014, 16, 2885–2888. [Google Scholar] [CrossRef]
- Gao, Q.; Xie, Y.-B.; Li, J.-R.; Yuan, D.-Q.; Yakovenko, A.A.; Sun, J.-H.; Zhou, H.-C. Tuning the formations of metal-organic frameworks by modification of ratio of reactant, acidity of reaction system, and use of a secondary ligand. Cryst. Growth Des. 2012, 12, 281–288. [Google Scholar] [CrossRef]
- Ma, S.; Simmons, J.M.; Yuan, D.; Li, J.-R.; Weng, W.; Liu, D.-J.; Zhou, H.-C. A. Nanotubular metal-organic framework with permanent porosity: Structure analysis and gas sorption studies. Chem. Commun. 2009, 27, 4049–4051. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Zhu, B.; Li, D.; Guo, D.; Zhao, J.; Ma, L. A new highly-connected 3D [Co4(μ3-OH)2] cluster-based framework from different dicarboxylates and N-donor co-ligands: Synthesis, structure, and magnetic propert. Inorg. Chem. Commun. 2013, 33, 86–89. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, L.; Wang, R.; Sun, J.; Yang, J.; Chen, Z.; Wang, X.; Sun, D. Five MOFs with different topologies based on anthracene functionalized tetracarboxylic acid: Syntheses, structures, and properties. CrysEngComm 2014, 16, 2917–2928. [Google Scholar] [CrossRef]
- Liu, C.; Chang, Z.; Wang, J.; Yan, L.; Bu, X.; Batten, S. A photoluminescent 3D silver(I) coordination polymer with mixed ligands anthracene-9,10-dicarboxylate and hexamethylenetetramine, showing binodal 4-connected (43·63)2(42·62·82)3 topology. Inorg. Chem. Commun. 2008, 11, 889–892. [Google Scholar] [CrossRef]
- Chen, J.; Fan, Q.; Kitagawa, S. Synthesis, structures, and adsorption properties of two new magnesium coordination polymers. Solid State Sci. 2013, 16, 29–33. [Google Scholar] [CrossRef]
- Calahorro, A.J.; Fernández, B.; Oyarzabal, I.; Seco, J.M.; Tian, T.; Fairen-Jimenez, D.; Colacio, E.; Rodríguez-Diéguez, A. Rare earth anthracenedicarboxylate metal-organic frameworks: Slow relaxation of magnetization of Nd3+, Gd3+, Dy3+, Er3+ and Yb3+ based materials. Dalton Trans. 2016, 45, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Cryst. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2—A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 339–341. [Google Scholar]
- APEX2; Bruker AXS Inc.: Madison, WI, USA, 2012.
- SAINT; Bruker AXS Inc.: Madison, WI, USA, 2013.
- Wang, J.; Wang, J.; Li, Y.; Jiang, M.; Zhang, L.; Wu, P. A europium(III)-based metal–organic framework as a naked-eye and fast response luminescence sensor for acetone and ferric iron. New J. Chem. 2016, 40, 8600–8606. [Google Scholar] [CrossRef]
- Wen, G.-X.; Wu, Y.-P.; Dong, W.-W.; Zhao, J.; Li, D.-S.; Zhang, J. An Ultrastable europium(III)-organic framework with the capacity of discriminating Fe2+/Fe3+ ions in various solutions. Inorg. Chem. 2016, 55, 10114–10117. [Google Scholar] [CrossRef] [PubMed]
- Werner, T.C.; Hercules, D.M. Fluorescence of 9-anthroic acid and its esters. Environmental effects on excited-state behavior. J. Phys. Chem. 1969, 73, 2005–2011. [Google Scholar] [CrossRef]
- Dametto, P.; Siqueira, A.; Carvalho, C.; Ionashiro, M. Synthesis, characterization and thermal studies on solid state 3-methoxybenzoate of lighter trivalent lanthanides. Eclética Química 2007, 32, 17–21. [Google Scholar] [CrossRef]
- Clarke, H.B.; Northrop, D.C.; Simpson, O. The Scintillation Phenomenon in Anthracene I. Radiation Damage. Prog. Phys. Soc. 1962, 79, 366–372. [Google Scholar] [CrossRef]
- Nag, A.; Kutty, T. The light induced valence change of europium in Sr2SiO4: Eu involving transient crystal structure. J. Mater. Chem. 2004, 14, 1598–1604. [Google Scholar] [CrossRef]
- Hinoue, T.; Shigenoi, Y.; Sugino, M.; Mizobe, Y.; Hisaki, I.; Miyata, M.; Tohnai, N. Regulation of π-stacked anthracene arrangement for fluorescence modulation of organic solid from monomer to excited oligomer emission. Chem. Eur. J. 2012, 18, 4634–4643. [Google Scholar] [CrossRef] [PubMed]
- Birks, J.B. The Theory and Practice of Scintillation Counting, 1st ed.; Pargamon: Oxford, UK, 1964. [Google Scholar]
- King, T.A.; Voltz, R. The time dependence of scintillation intensity in aromatic materials. Proc. R. Soc. Lond. Ser. A 1966, 269, 424–439. [Google Scholar] [CrossRef]
- Birks, J.B. Energy Transfer in organic phosphorsm. Phys. Rev. 1954, 94, 1567. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Formula | C63H59N5O17Eu2 | C63H59N5O17Tb2 | C60H58N4O19Er2 | C60H58N4O19Tm2 |
| Density g cm−3 | 1.645 | 1.681 | 1.697 | 1.703 |
| μ/mm−1 | 2.183 | 2.484 | 2.969 | 3.141 |
| Formula Weight | 1462.07 | 1475.99 | 1473.62 | 1476.96 |
| Crystal System | triclinic | triclinic | triclinic | Triclinic |
| Space Group | P-1 | P-1 | P-1 | P-1 |
| a/Å | 10.4144(5) | 10.3706(9) | 12.6026(14) | 12.5949(3) |
| b/Å | 11.4340(5) | 11.3600(9) | 14.9605(16) | 14.9329(4) |
| c/Å | 12.9875(6) | 12.9850(11) | 17.1024(19) | 17.1174(5) |
| α/° | 72.8203(11) | 72.5410(10) | 87.5905(14) | 87.538(2) |
| β/° | 89.9297(11) | 89.7340(10) | 69.7644(15) | 69.7810(10) |
| γ/° | 87.1572(12) | 87.4110(10) | 72.8494(14) | 72.8160(10) |
| V/Å3 | 1475.56(12) | 1457.7(2) | 2884.5(5) | 2879.8(8) |
| Z | 1 | 1 | 4 | 2 |
| Z’ | 0.5 | 0.5 | 2 | 1 |
| Θmin/° | 1.867 | 2.551 | 1.938 | 1.430 |
| Θmax/° | 31.000 | 30.058 | 30.046 | 30.522 |
| Measured Refl. | 27,686 | 38,805 | 28,992 | 47,108 |
| Independent Refl. | 9326 | 8544 | 15,975 | 17,387 |
| Refl. Used | 8571 | 8036 | 11,489 | 13,514 |
| Rint | 0.0338 | 0.0338 | 0.0450 | 0.0527 |
| Parameters | 458 | 392 | 803 | 803 |
| Restraints | 95 | 24 | 98 | 65 |
| Largest Peak | 1.346 | 1.700 | 2.080 | 2.372 |
| Deepest Hole | −0.999 | −0.978 | −1.169 | −1.057 |
| GooF | 1.102 | 1.053 | 0.967 | 1.048 |
| wR2(all data) | 0.0753 | 0.0673 | 0.1202 | 0.0906 |
| wR2 | 0.0680 | 0.660 | 0.1040 | 0.0814 |
| R1(all data) | 0.0322 | 0.0285 | 0.0750 | 0.0554 |
| R1 | 0.0280 | 0.0264 | 0.0470 | 0.0374 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathis, S.R., II; Golafale, S.T.; Solntsev, K.M.; Ingram, C.W. Anthracene-Based Lanthanide Metal-Organic Frameworks: Synthesis, Structure, Photoluminescence, and Radioluminescence Properties. Crystals 2018, 8, 53. https://doi.org/10.3390/cryst8010053
Mathis SR II, Golafale ST, Solntsev KM, Ingram CW. Anthracene-Based Lanthanide Metal-Organic Frameworks: Synthesis, Structure, Photoluminescence, and Radioluminescence Properties. Crystals. 2018; 8(1):53. https://doi.org/10.3390/cryst8010053
Chicago/Turabian StyleMathis, Stephan R., II, Saki T. Golafale, Kyril M. Solntsev, and Conrad W. Ingram. 2018. "Anthracene-Based Lanthanide Metal-Organic Frameworks: Synthesis, Structure, Photoluminescence, and Radioluminescence Properties" Crystals 8, no. 1: 53. https://doi.org/10.3390/cryst8010053
APA StyleMathis, S. R., II, Golafale, S. T., Solntsev, K. M., & Ingram, C. W. (2018). Anthracene-Based Lanthanide Metal-Organic Frameworks: Synthesis, Structure, Photoluminescence, and Radioluminescence Properties. Crystals, 8(1), 53. https://doi.org/10.3390/cryst8010053