A 12-Fold ThSi2 Interpenetrated Network Utilizing a Glycine-Based Pseudopeptidic Ligand



Abstract
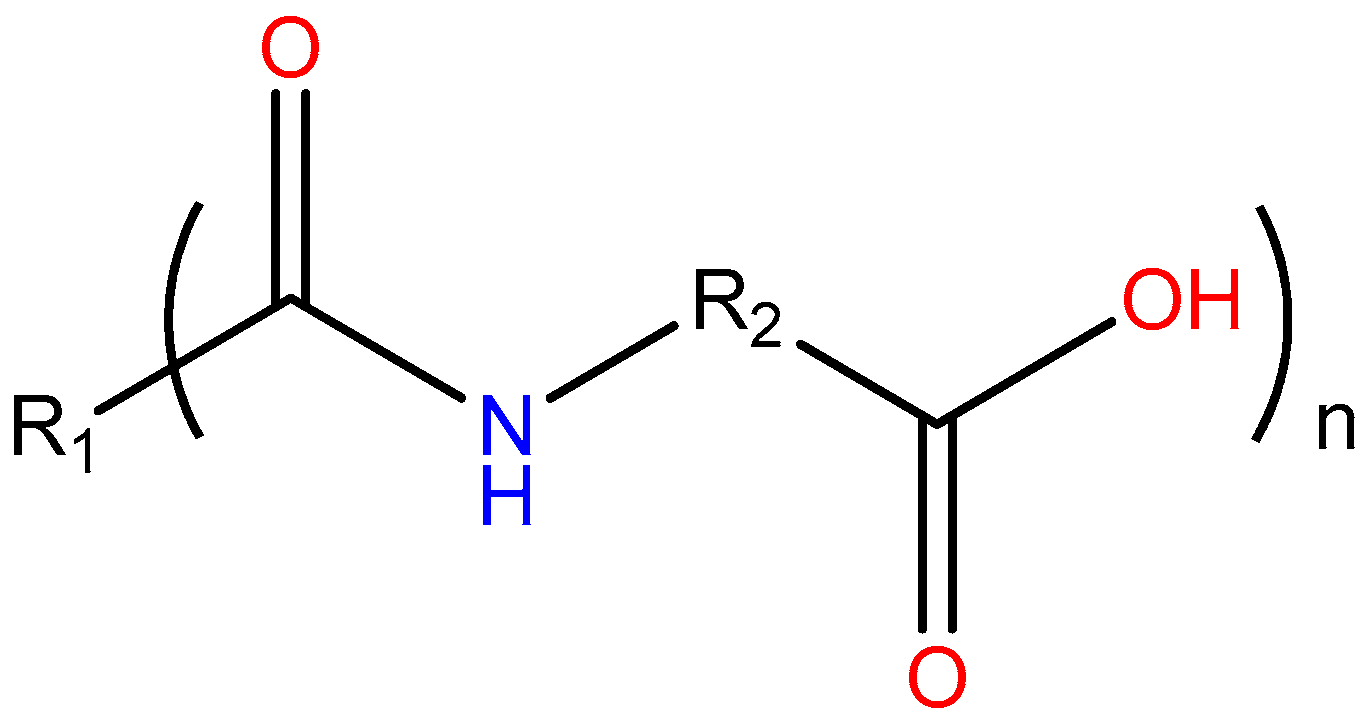
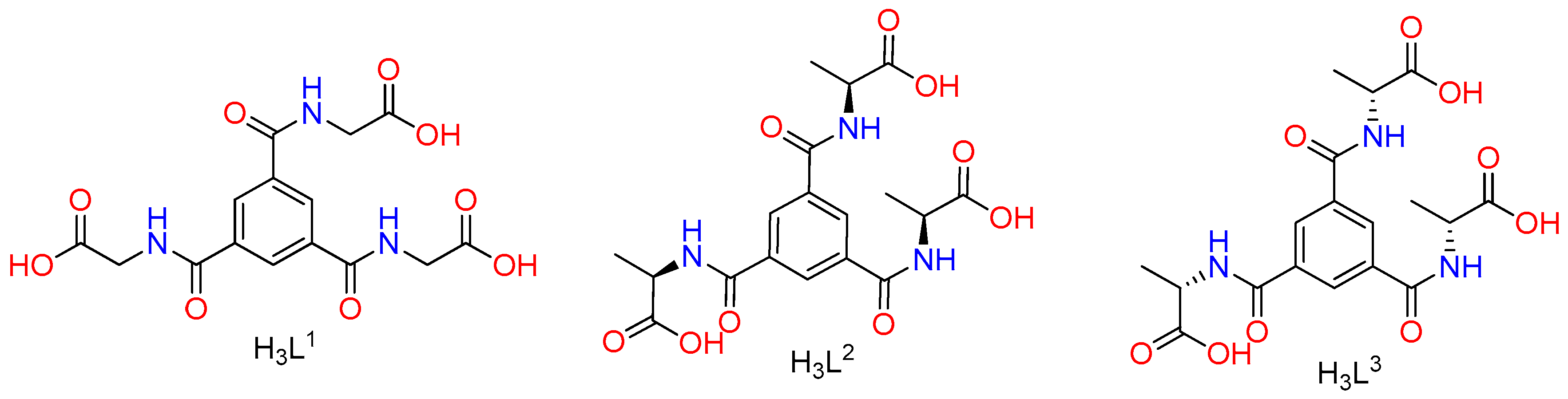
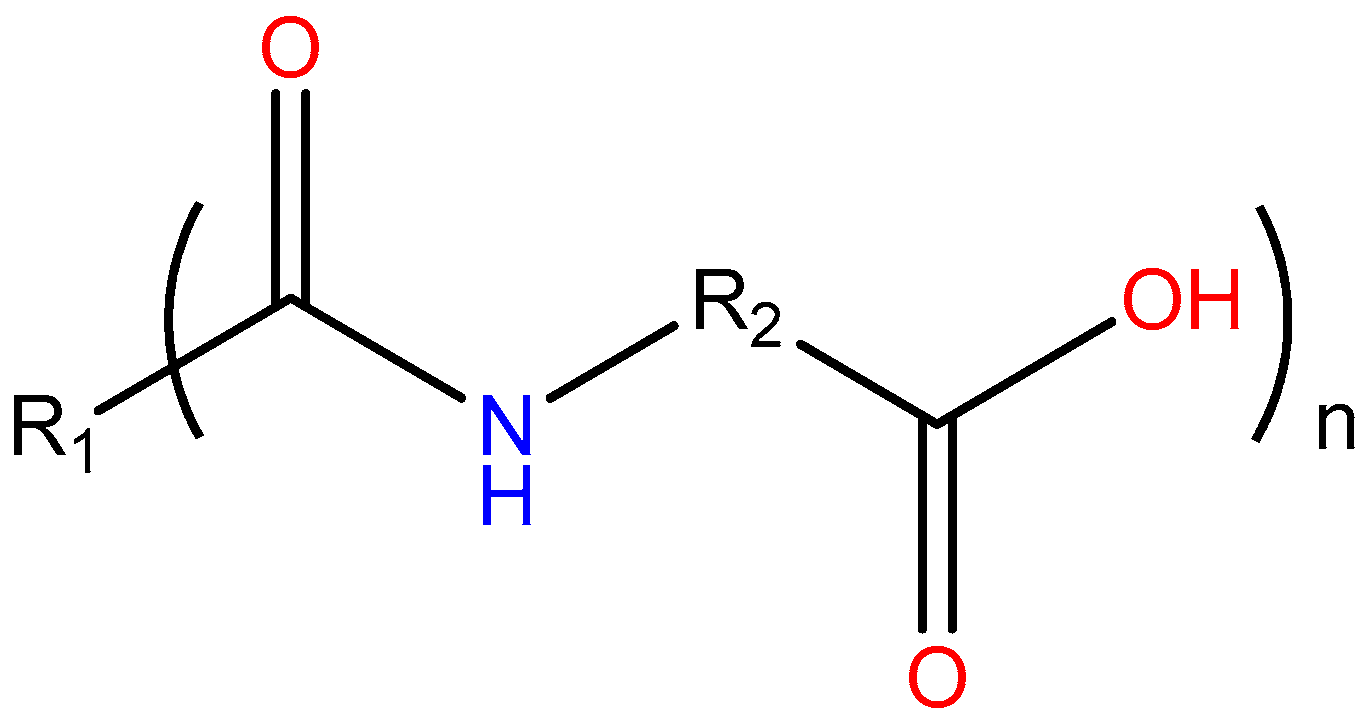
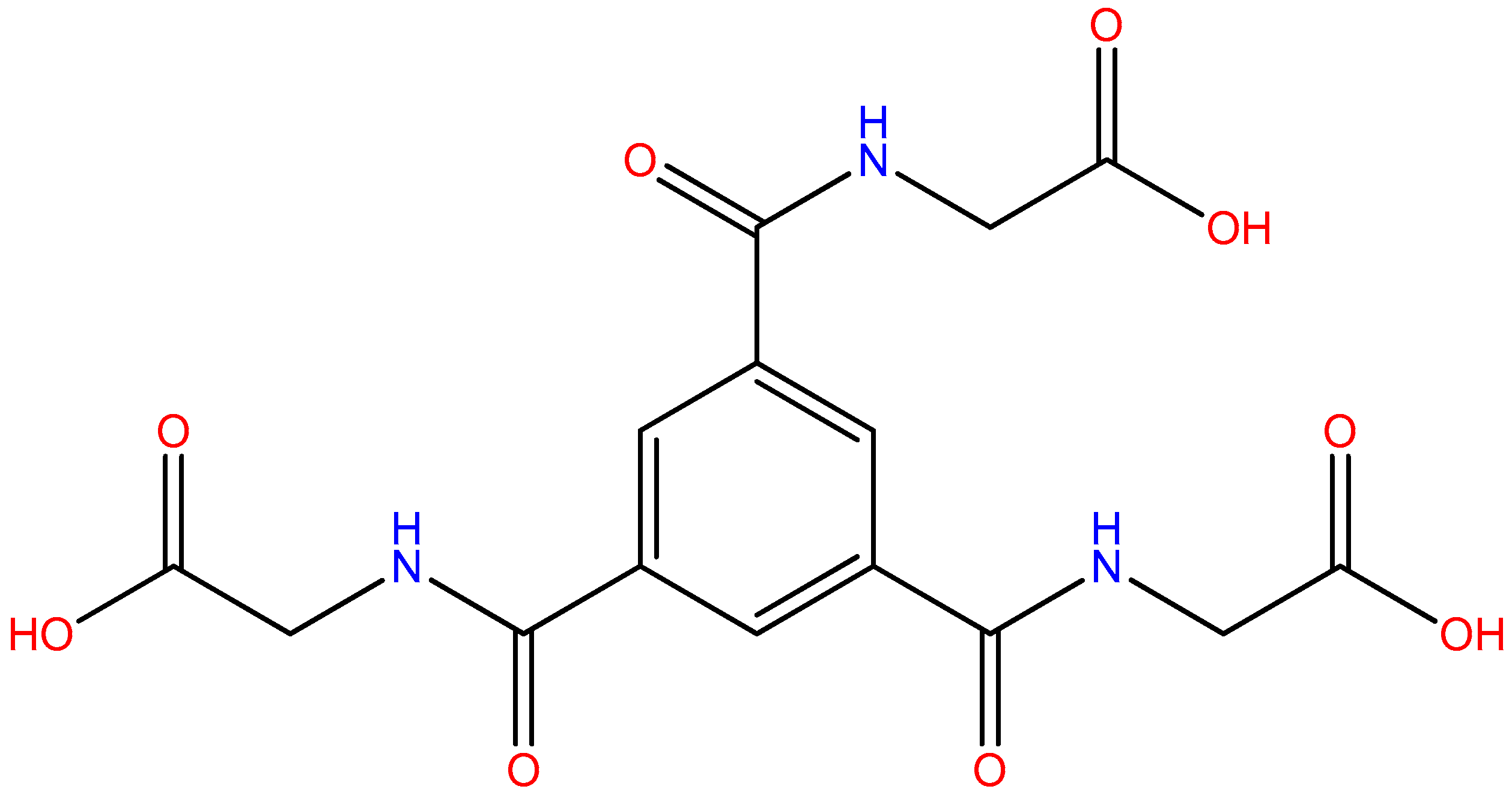
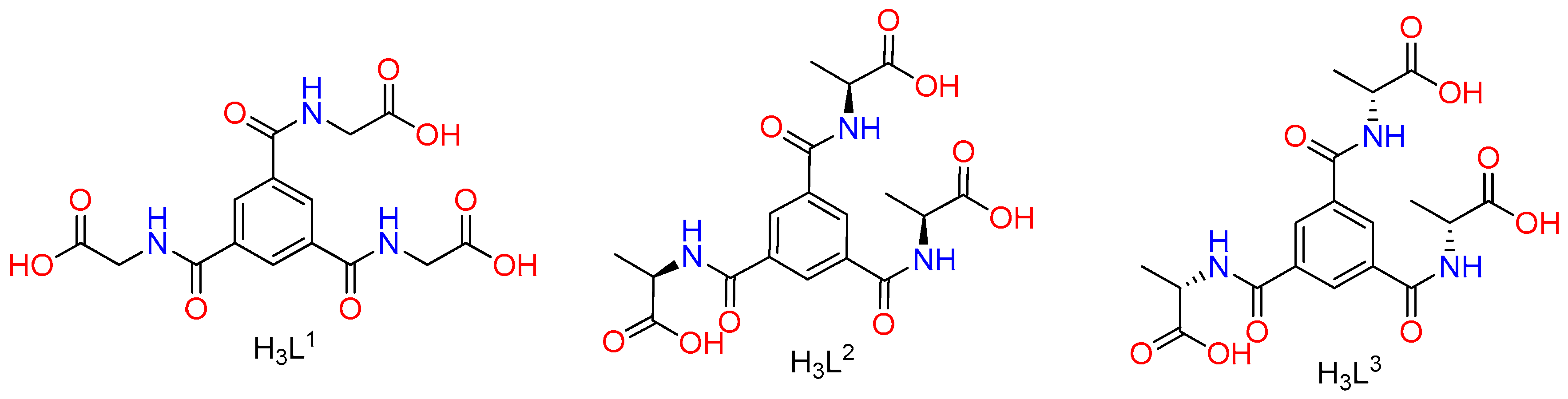
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Instrumentation
2.3. X-ray Crystallography
2.4. Synthetic Procedures
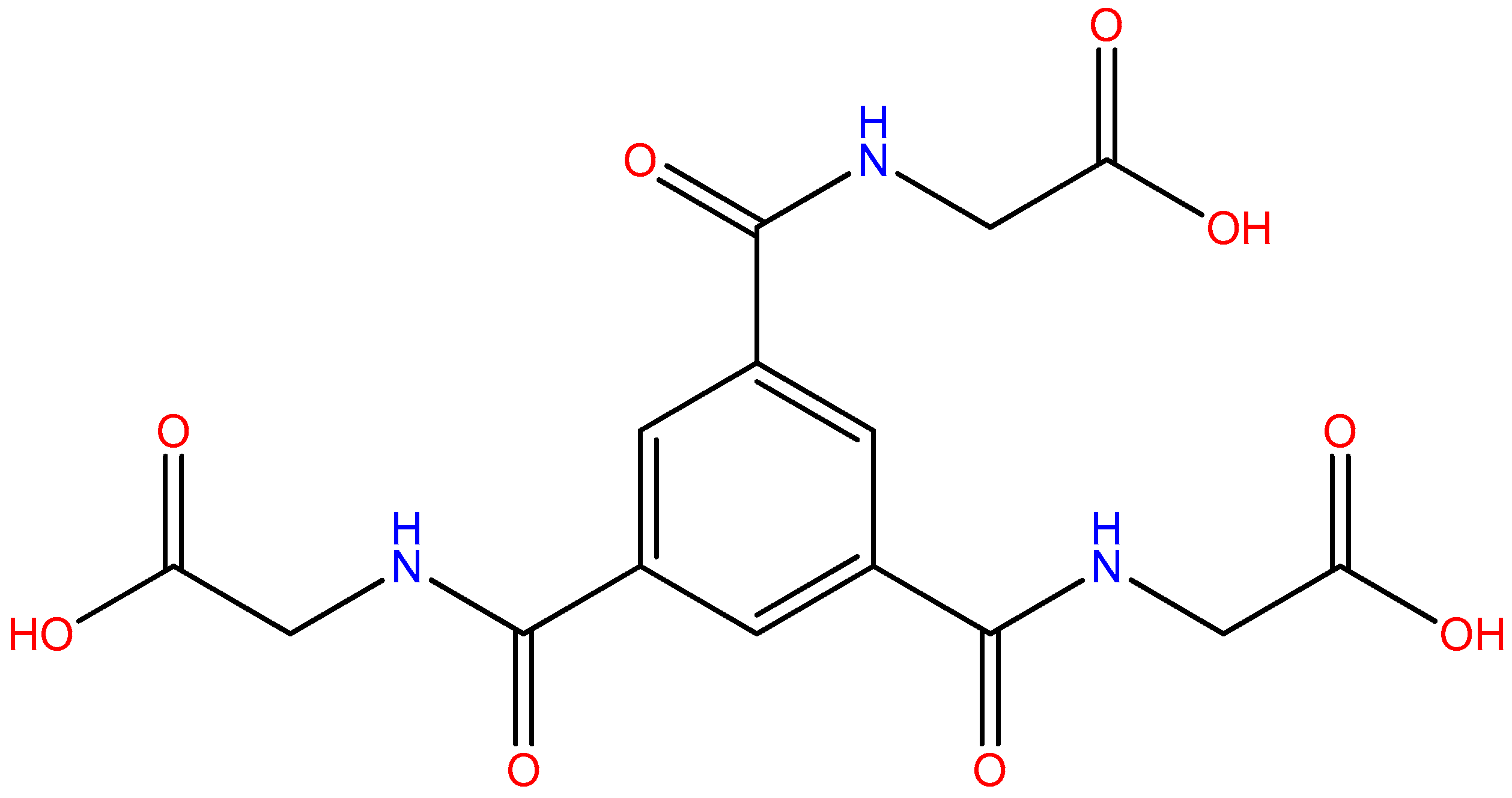
2.4.1. Synthesis of [Cu3(L1)2(H2O)8]·8H2O (1)
2.4.2. General Catalytic Protocol for A3 Coupling
3. Results
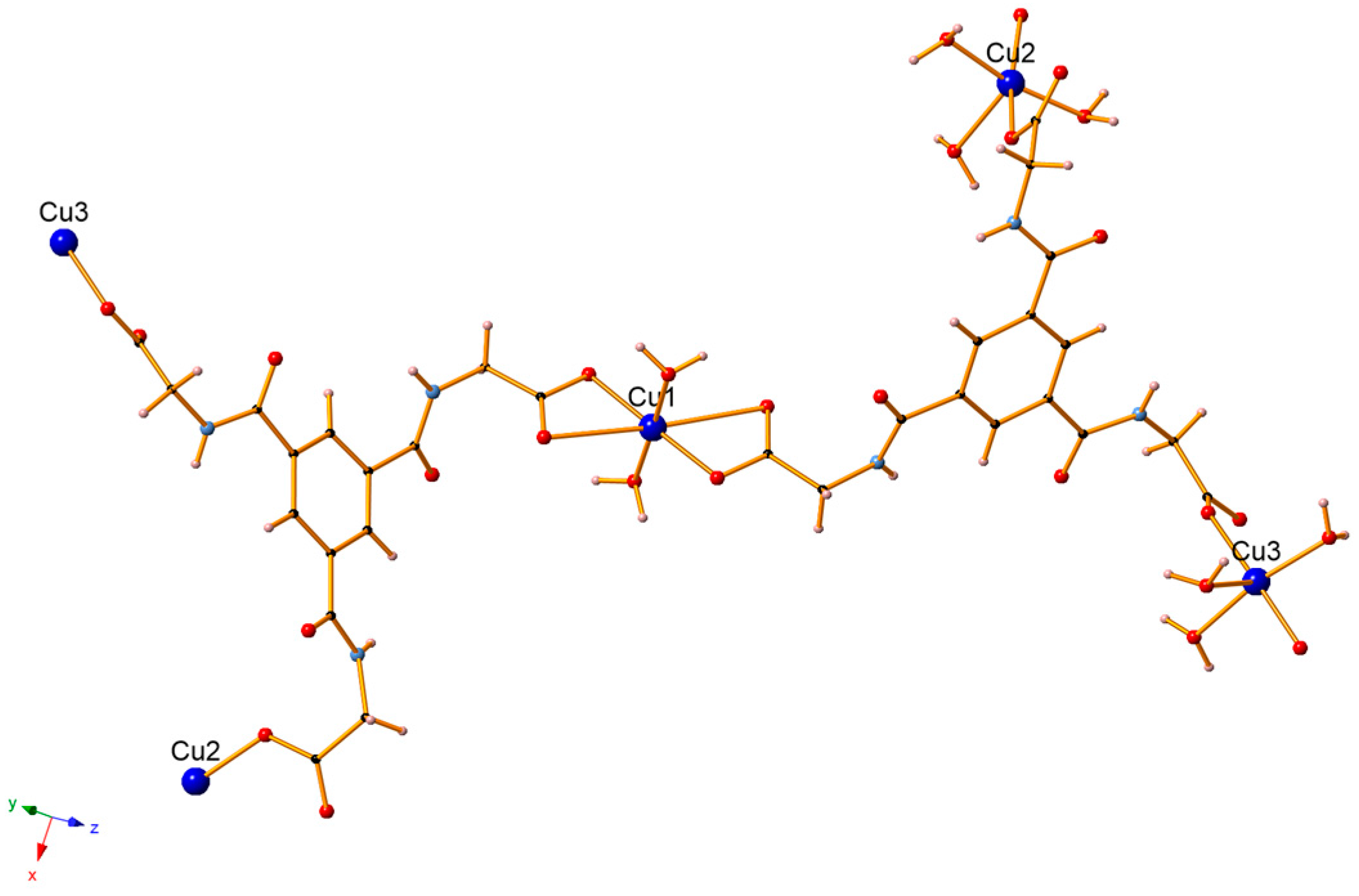
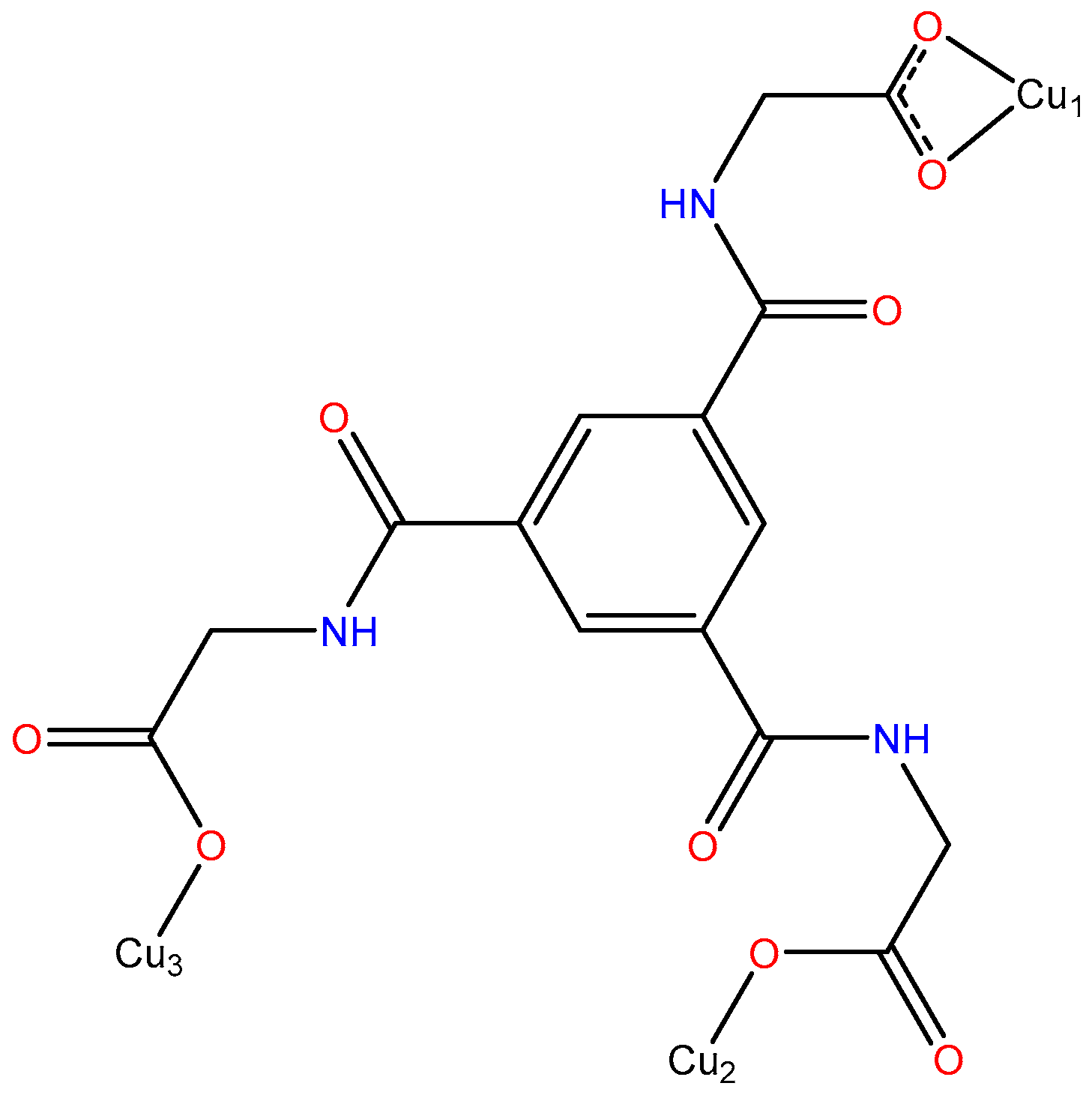
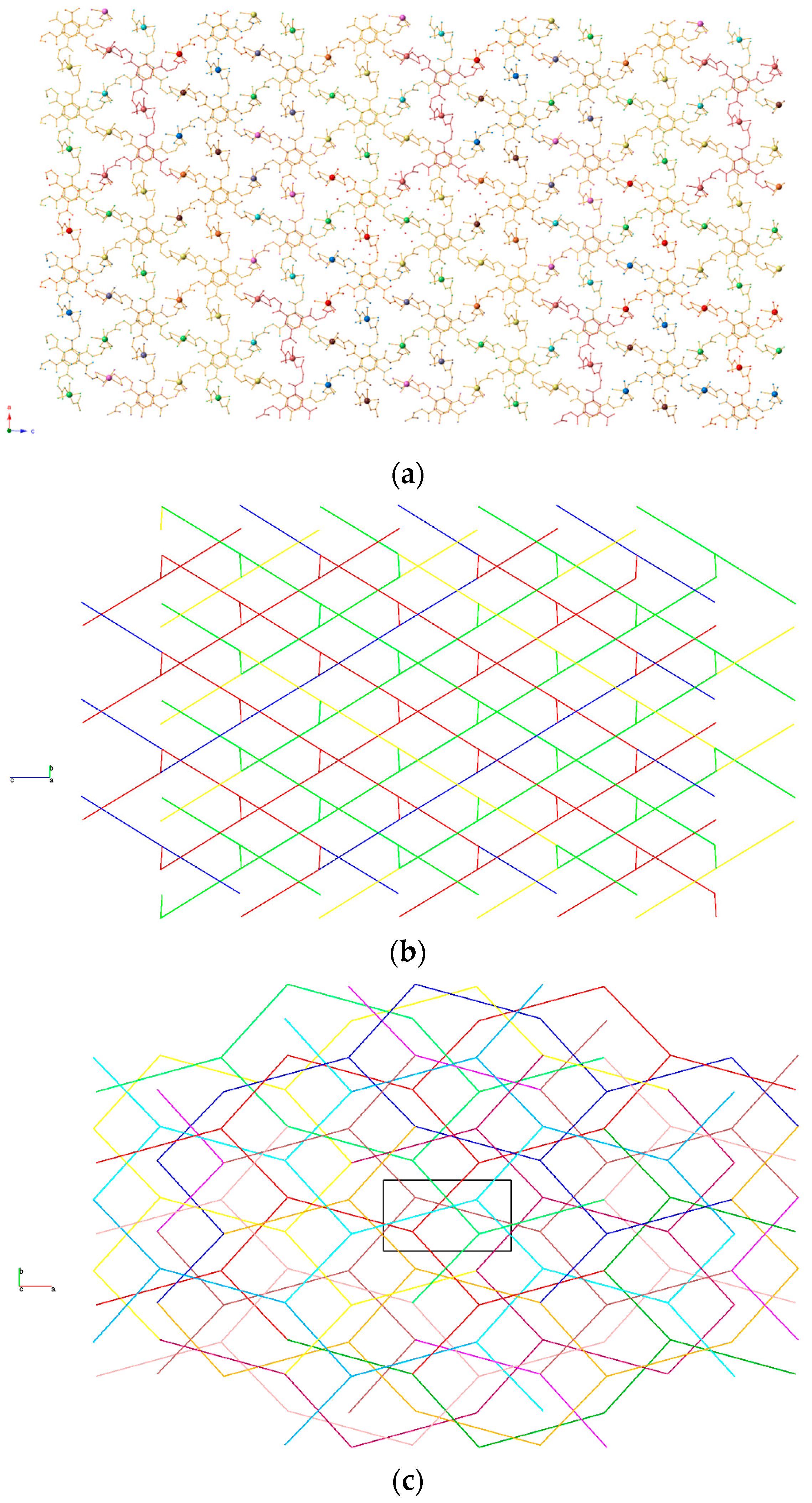
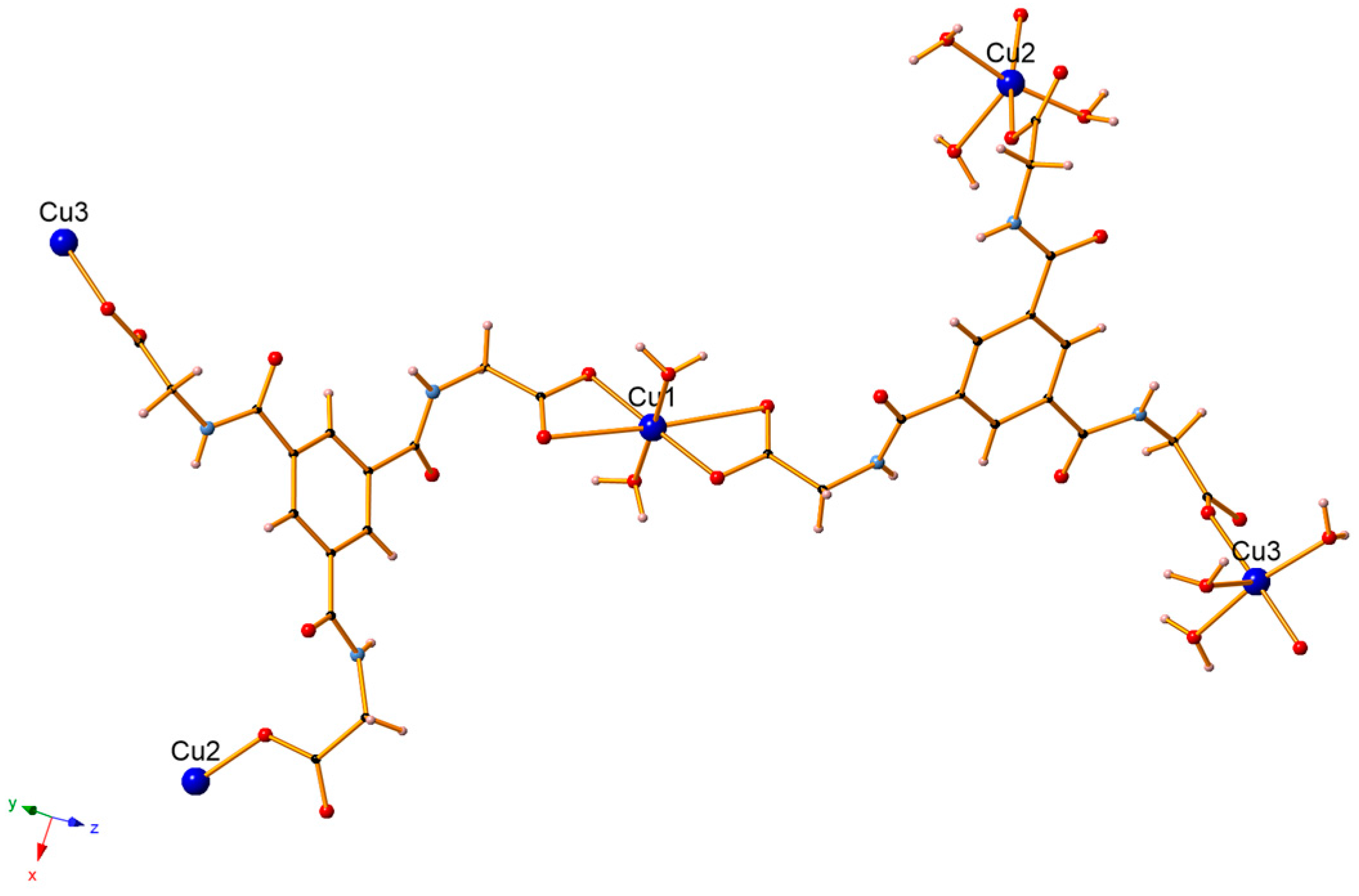
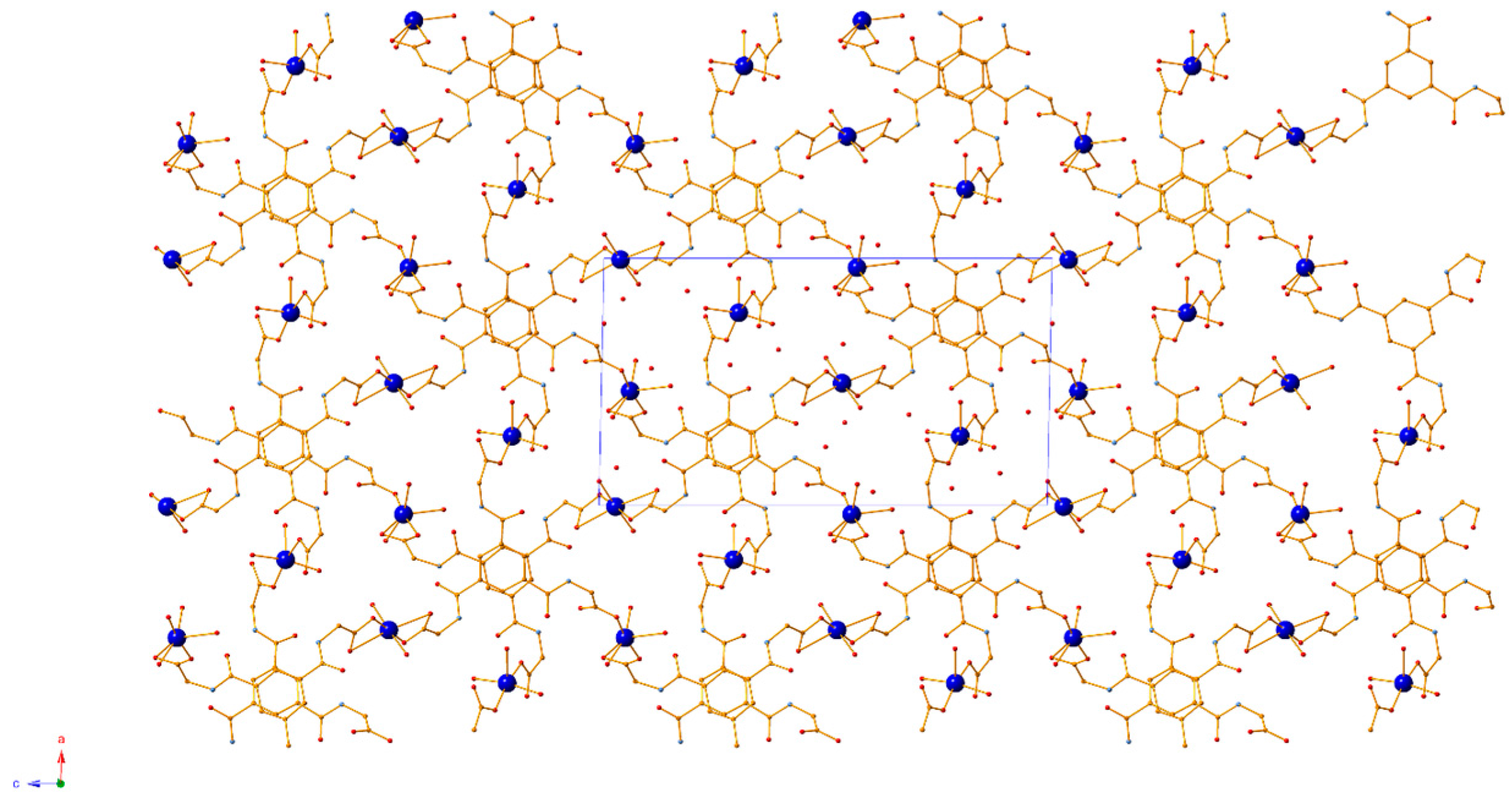
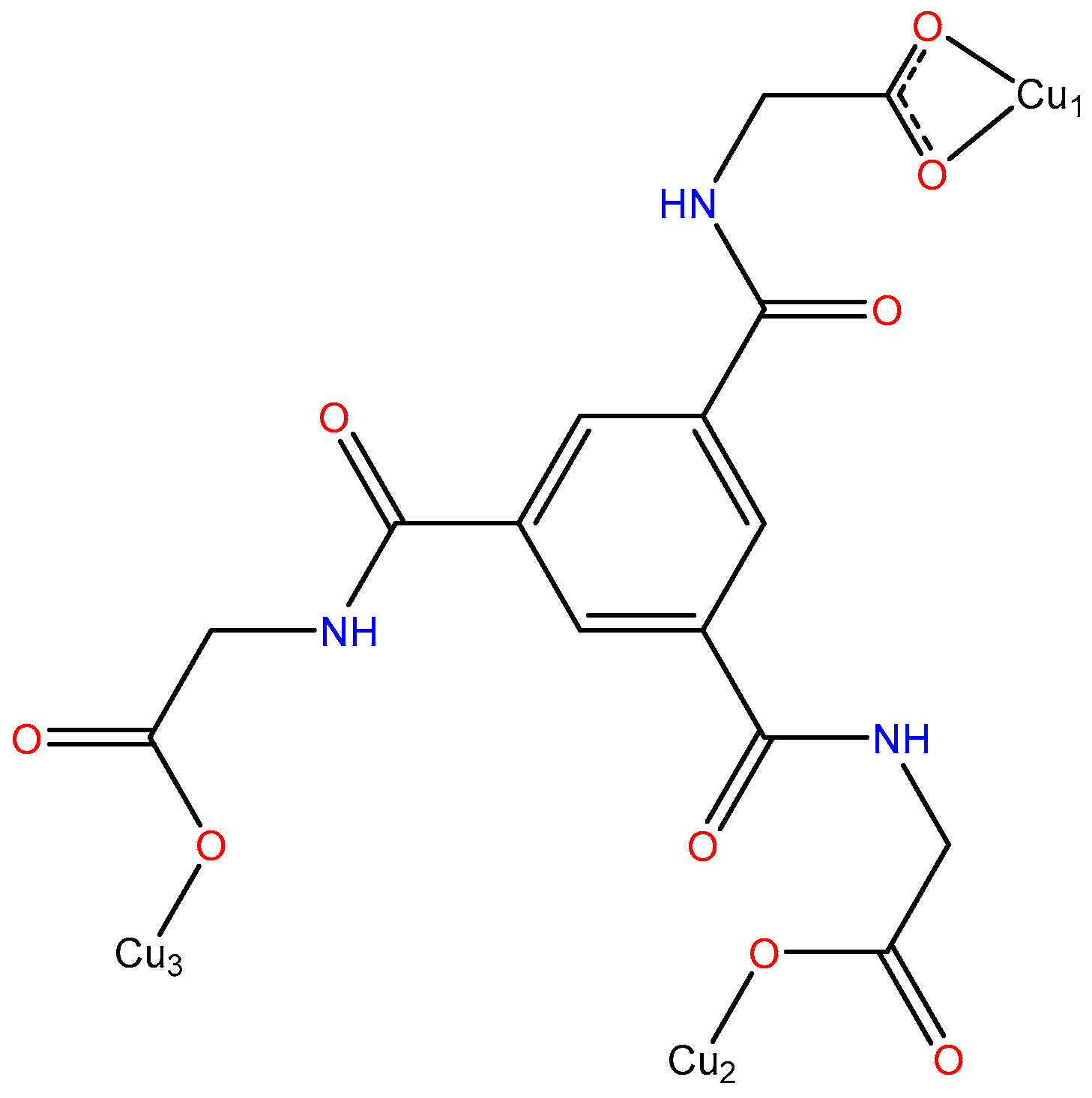
3.1. Crystal Structure Description
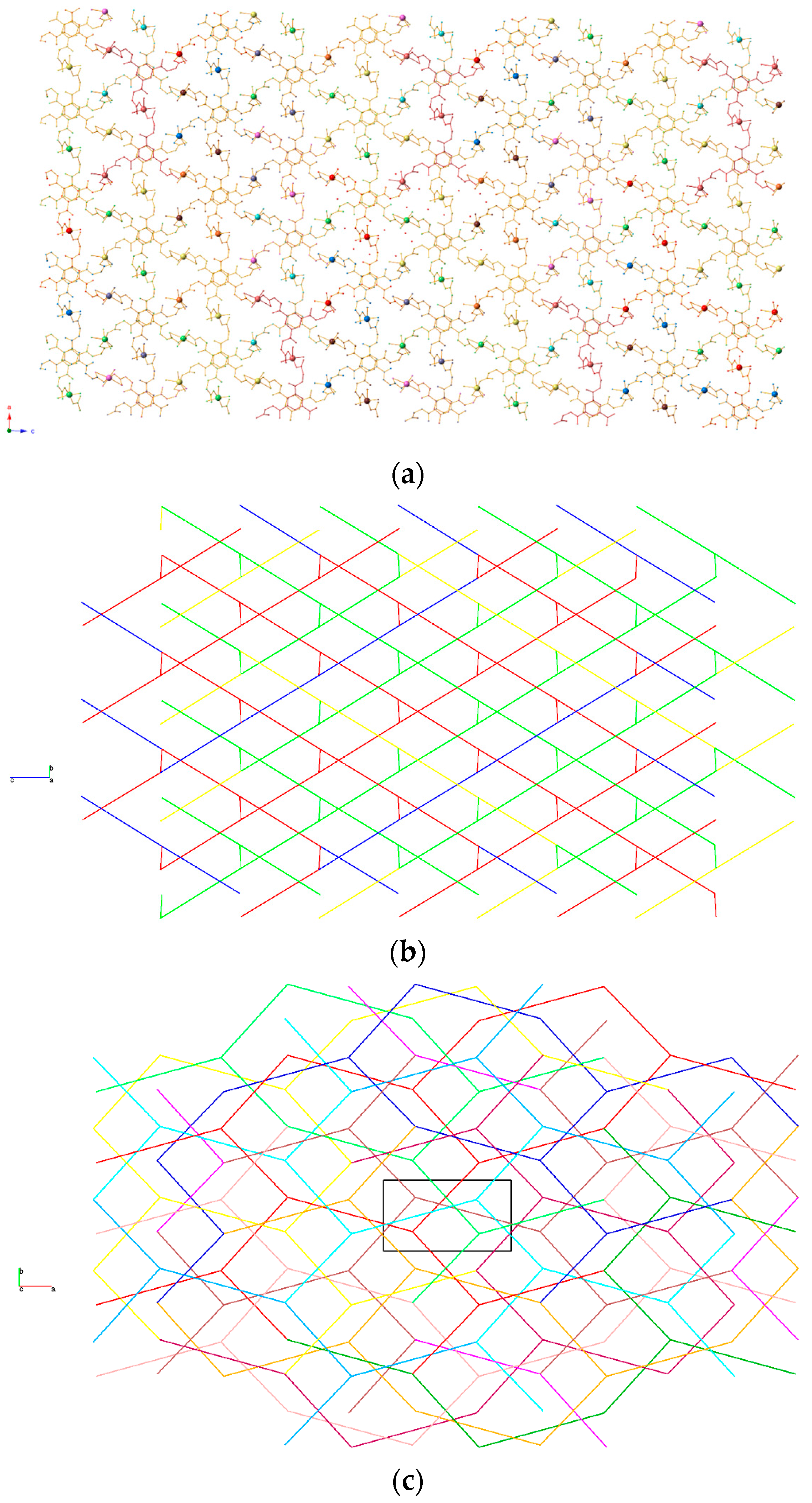
3.2. Topological Analysis
3.3. TGA and IR Studies
3.4. Synthetic Aspects
3.5. Catalytic Studies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The Chemistry and Applications of Metal-Organic Frameworks. Science 2013, 341, 1230444. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wen, H.M.; Cui, Y.; Zhou, W.; Qian, G.; Chen, B. Emerging Multifunctional Metal–Organic Framework Materials. Adv. Mater. 2016, 28, 8819–8860. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liu, X.Q.; Jiang, H.L.; Sun, L.B. Metal-Organic Frameworks for Heterogeneous Basic Catalysis. Chem. Rev. 2017, 117, 8129–8176. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Xie, L.-H.; Li, J.-R.; Ma, Y.; Seminario, J.M.; Balbuena, P.B. CO2 Capture and Separations Using MOFs: Computational and Experimental Studies. Chem. Rev. 2017, 117, 9674–9754. [Google Scholar] [CrossRef] [PubMed]
- Kirillov, A.M.; Karabach, Y.Y.; Kirillova, M.V.; Haukka, M.; Pombeiro, A.J.L. Topologically unique 2D heterometallic Cu II/Mg coordination polymer: Synthesis, structural features, and catalytic use in alkane hydrocarboxylation. Cryst. Growth Des. 2012, 12, 1069–1074. [Google Scholar] [CrossRef]
- Liu, B. Metal–organic framework-based devices: Separation and sensors. J. Mater. Chem. 2012, 22, 10094. [Google Scholar] [CrossRef]
- Wu, M.X.; Yang, Y.W. Metal–Organic Framework (MOF)-Based Drug/Cargo Delivery and Cancer Therapy. Adv. Mater. 2017, 29, 1606134. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Chen, B.; Qian, G. Lanthanide metal-organic frameworks for luminescent sensing and light-emitting applications. Coord. Chem. Rev. 2014, 273–274, 76–86. [Google Scholar] [CrossRef]
- Zhang, M.; Bosch, M.; Gentle, T., III; Zhou, H.-C. Rational design of metal–organic frameworks with anticipated porosities and functionalities. CrystEngComm 2014, 16, 4069. [Google Scholar] [CrossRef]
- Burneo, I.; Stylianou, K.; Imaz, I.; Maspoch, D. The influence of the enantiomeric ratio of an organic ligand on the structure and chirality of Metal-Organic Frameworks. Chem. Commun. 2014, 50, 13829–13832. [Google Scholar] [CrossRef] [PubMed]
- Loukopoulos, E.; Chilton, N.F.; Abdul-Sada, A.; Kostakis, G.E. Exploring the Coordination Capabilities of a Family of Flexible Benzotriazole-Based Ligands Using Cobalt(II) Sources. Cryst. Growth Des. 2017, 17, 2718–2729. [Google Scholar] [CrossRef]
- Lu, W.; Wei, Z.; Gu, Z.-Y.; Liu, T.-F.; Park, J.; Park, J.; Tian, J.; Zhang, M.; Zhang, Q.; Gentle, T.; et al. Tuning the structure and function of metal-organic frameworks via linker design. Chem. Soc. Rev. 2014, 43, 5561–5593. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.L.; Stylianou, K.C. Biologically derived metal organic frameworks. Coord. Chem. Rev. 2017, 349, 102–128. [Google Scholar] [CrossRef]
- An, J.; Fiorella, R.P.; Geib, S.J.; Rosi, N.L. Synthesis, structure, assembly, and modulation of the CO2 adsorption properties of a zinc-adeninate macrocycle. J. Am. Chem. Soc. 2009, 131, 8401–8403. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.X.; He, Y.P.; Zhang, J. Serine-based homochiral nanoporous frameworks for selective CO2 uptake. Inorg. Chem. 2011, 50, 11527–11531. [Google Scholar] [CrossRef] [PubMed]
- Martí-Gastaldo, C.; Warren, J.E.; Stylianou, K.C.; Flack, N.L.O.; Rosseinsky, M.J. Enhanced stability in rigid peptide-based porous materials. Angew. Chem. Int. Ed. 2012, 51, 11044–11048. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.D.; Hu, A.; Zhang, L.; Lin, W. A homochiral porous metal-organic framework for highly enantioselective heterogeneous asymmetric catalysis. J. Am. Chem. Soc. 2005, 127, 8940–8941. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, H.-B.; Xu, Z.-X.; Zhang, J. Asymmetric induction in homochiral MOFs: From interweaving double helices to single helices. Chem. Commun. 2015, 51, 16331–16333. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zheng, M.; Lin, W. Asymmetric catalysis with chiral porous metal-organic frameworks: Critical issues. J. Phys. Chem. Lett. 2011, 2, 1701–1709. [Google Scholar] [CrossRef]
- An, J.; Shade, C.M.; Chengelis-Czegan, D.A.; Petoud, S.; Rosi, N.L. Zinc-adeninate metal-organic framework for aqueous encapsulation and sensitization of near-infrared and visible emitting lanthanide cations. J. Am. Chem. Soc. 2011, 133, 1220–1223. [Google Scholar] [CrossRef] [PubMed]
- McKinlay, A.C.; Morris, R.E.; Horcajada, P.; Férey, G.; Gref, R.; Couvreur, P.; Serre, C. BioMOFs: Metal-organic frameworks for biological and medical applications. Angew. Chem. Int. Ed. 2010, 49, 6260–6266. [Google Scholar] [CrossRef] [PubMed]
- Dokorou, V.N.; Milios, C.J.; Tsipis, A.C.; Haukka, M.; Weidler, P.G.; Powell, A.K.; Kostakis, G.E. Pseudopeptidic ligands: Exploring the self-assembly of isophthaloylbisglycine (H2IBG) and divalent metal ions. Dalton Trans. 2012, 41, 12501–12513. [Google Scholar] [CrossRef] [PubMed]
- Dokorou, V.N.; Powell, A.K.; Kostakis, G.E. Two pseudopolymorphs derived from alkaline earth metals and the pseudopeptidic ligand trimesoyl-tris-glycine. Polyhedron 2013, 52, 538–544. [Google Scholar] [CrossRef]
- Morrison, C.N.; Powell, A.K.; Kostakis, G.E. Influence of Metal Ion on Structural Motif in Coordination Polymers of the Pseudopeptidic Ligand Terephthaloyl-bis-beta-alaninate. Cryst. Growth Des. 2011, 11, 3653–3662. [Google Scholar] [CrossRef]
- Lymperopoulou, S.; Dokorou, V.N.; Tsipis, A.C.; Weidler, P.G.; Plakatouras, J.C.; Powell, A.K.; Kostakis, G.E. Influence of the metal salt on the self-assembly of isophthaloylbis-β-alanine and Cu(II) ion. Polyhedron 2015, 89, 313–321. [Google Scholar] [CrossRef]
- Kostakis, G.E.; Casella, L.; Boudalis, A.K.; Monzani, E.; Plakatouras, J.C. Structural variation from 1D chains to 3D networks: A systematic study of coordination number effect on the construction of coordination polymers using the terepthaloylbisglycinate ligand. New J. Chem. 2011, 35, 1060–1071. [Google Scholar] [CrossRef]
- Kostakis, G.E.; Casella, L.; Hadjiliadis, N.; Monzani, E.; Kourkoumelis, N.; Plakatouras, J.C. Interpenetrated networks from a novel nanometer-sized pseudopeptidic ligand, bridging water, and transition metal ions with cds topology. Chem. Commun. 2005, 987, 3859–3861. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Zheng, B.; Bai, J.; Zhang, Q.; Zuo, C. Metal-dependent dimensionality in coordination polymers of a semi-rigid dicarboxylate ligand with additional amide groups: Syntheses, structures and luminescent properties. Inorganica Chim. Acta 2010, 363, 3172–3177. [Google Scholar] [CrossRef]
- Wisser, B.; Chamayou, A.-C.; Miller, R.; Scherer, W.; Janiak, C. A chiral C3-symmetric hexanuclear triangular-prismatic copper(ii) cluster derived from a highly modular dipeptidic N,N′-terephthaloyl-bis(S-aminocarboxylato) ligand. CrystEngComm 2008, 10, 461–464. [Google Scholar] [CrossRef]
- Sun, R.; Li, Y.Z.; Bai, J.; Pan, Y. Synthesis, structure, water-induced reversible crystal-to-amorphous transformation, and luminescence properties of novel cationic spacer-filled 3D transition metal supramolecular frameworks from N,N′,N″-tris(carboxymethyl)-1,3,5-benzenetricarboxamide. Cryst. Growth Des. 2007, 7, 890–894. [Google Scholar] [CrossRef]
- Sun, R.; Wang, S.; Xing, H.; Bai, J.; Li, Y.; Pan, Y.; You, X. Unprecedented 4264 topological 2-D rare-earth coordination polymers from a flexible tripodal acid with additional amide groups. Inorg. Chem. 2007, 46, 8451–8453. [Google Scholar] [CrossRef] [PubMed]
- Loukopoulos, E.; Griffiths, K.; Akien, G.; Kourkoumelis, N.; Abdul-Sada, A.; Kostakis, G. Dinuclear Lanthanide (III) Coordination Polymers in a Domino Reaction. Inorganics 2015, 3, 448–466. [Google Scholar] [CrossRef]
- Kallitsakis, M.; Loukopoulos, E.; Abdul-Sada, A.; Tizzard, G.J.; Coles, S.J.; Kostakis, G.E.; Lykakis, I.N. A Copper-Benzotriazole-Based Coordination Polymer Catalyzes the Efficient One-Pot Synthesis of (N′-Substituted)-hydrazo-4-aryl-1,4-dihydropyridines from Azines. Adv. Synth. Catal. 2017, 359, 138–145. [Google Scholar] [CrossRef]
- Loukopoulos, E.; Kallitsakis, M.; Tsoureas, N.; Abdul-Sada, A.; Chilton, N.F.; Lykakis, I.N.; Kostakis, G.E. Cu(II) Coordination Polymers as Vehicles in the A3 Coupling. Inorg. Chem. 2017, 56, 4898–4910. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, C.; Meijer, E.W.; Palmans, A.R.A. Cooperativity Scale: A Structure–Mechanism Correlation in the Self-Assembly of Benzene-1,3,5-tricarboxamides. Acc. Chem. Res. 2017, 50, 1928–1936. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Blake, A.J.; Champness, N.R.; Schröder, M. OLEX: New software for visualization and analysis of extended crystal structures. J. Appl. Crystallogr. 2003, 36, 1283–1284. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXS97, Program for the Solution of Crystal Structures. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Van De Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Stiefel, E.I.; Brown, G.F. On the Detailed Nature of the Six-Coordinate Polyhedra in Tris(bidentate ligand) Complexes. Inorg. Chem. 1972, 11, 434–436. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen-sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane] copper(II) perchlorate. J. Chem. Soc. Dalton. Trans. 1984, 1349. [Google Scholar] [CrossRef]
- Blatov, V.A.; Shevchenko, A.P.; Proserpio, D.M. Applied Topological Analysis of Crystal Structures with the Program Package ToposPro. Cryst. Growth Des. 2014, 14, 3576–3586. [Google Scholar] [CrossRef]
- Hao, Y.; Wu, B.; Li, S.; Jia, C.; Huang, X.; Yang, X.-J. Coordination polymers derived from a flexible bis(pyridylurea) ligand: Conformational change of the ligand and structural diversity of the complexes. CrystEngComm 2011, 13, 215–222. [Google Scholar] [CrossRef]
- Hsu, Y.F.; Lin, C.H.; Chen, J.D.; Wang, J.C. A novel interpenetrating diamondoid network from self-assembly of N,N′-di(4-pyridyl)adipoamide and copper sulfate: An unusual 12-fold, [6 + 6] mode. Cryst. Growth Des. 2008, 8, 1094–1096. [Google Scholar] [CrossRef]
- Blatov, V.A.; Carlucci, L.; Ciani, G.; Proserpio, D.M. Interpenetrating metal organic and inorganic 3D networks: A computer-aided systematic investigation. Part I. Analysis of the Cambridge structural database. CrystEngComm 2004, 6, 378. [Google Scholar] [CrossRef]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Lu, Z.; Xing, H.; Sun, R.; Bai, J.; Zheng, B.; Li, Y. Water stable metal-organic framework evolutionally formed from a flexible multidentate ligand with acylamide groups for selective CO2 adsorption. Cryst. Growth Des. 2012, 12, 1081–1084. [Google Scholar] [CrossRef]
- Min, T.; Zheng, B.; Bai, J.; Sun, R.; Li, Y.; Zhang, Z. Topology diversity and reversible crystal-to-amorphous transformation properties of 3D cobalt coordination polymers from a series of 1D rodlike dipyridyl-containing building blocks and a flexible tripodal acid with additional amide groups. CrystEngComm 2010, 12, 70–72. [Google Scholar] [CrossRef]
- Zuo, C.; Bai, J.; Sun, R.; Li, Y. Synthesis, structure, novel topology and reversible crystal-to-amorphous transformation of calcium coordination polymers from a flexible tripodal acid with additional amide groups. Inorg. Chim. Acta 2012, 383, 305–311. [Google Scholar] [CrossRef]
- Karmakar, A.; Oliver, C.L.; Roy, S.; Öhrström, L. The synthesis, structure, topology and catalytic application of a novel cubane-based copper(ii) metal-organic framework derived from a flexible amido tripodal acid. Dalton Trans. 2015, 44, 10156–10165. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, X.; Wu, A.; Liang, Y.; Wang, X.; Liang, F. Synthesis, structure and properties of an octahedral dinuclear-based Cu 12 nanocage of trimesoyltri(l-alanine). RSC Adv. 2016, 6, 9911–9915. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, C.; Liu, X.; Zhang, Z.; Liang, F. Synthesis, structure, and properties of a chiral zinc(II) metal-organic framework featuring linear trinuclear secondary building blocks. Aust. J. Chem. 2012, 65, 1662–1666. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, X.; Zhang, C.; Zhang, Z.; Liang, F. Structure, adsorption and magnetic properties of chiral metal–organic frameworks bearing linear trinuclear secondary building blocks. Dalton Trans. 2011, 40, 1911. [Google Scholar] [CrossRef] [PubMed]
- Peshkov, V.A.; Pereshivko, O.P.; Van der Eycken, E.V. A walk around the A3-coupling. Chem. Soc. Rev. 2012, 41, 3790. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Yuan, W.; Ma, S. Unexpected E-stereoselective reductive A(3)-coupling reaction of terminal alkynes with aldehydes and amines. Nat. Commun. 2014, 5, 3884. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Kumar, M.; Bhalla, V. Aggregates of perylene bisimide stabilized superparamagnetic Fe3O4 nanoparticles: An efficient catalyst for the preparation of propargylamines and quinolines via C–H activation. Chem. Commun. 2015, 51, 16327–16330. [Google Scholar] [CrossRef] [PubMed]
- Paioti, P.H.S.; Abboud, K.A.; Aponick, A. Catalytic Enantioselective Synthesis of Amino Skipped Diynes. J. Am. Chem. Soc. 2016, 138, 2150–2153. [Google Scholar] [CrossRef] [PubMed]
- De, D.; Pal, T.K.; Neogi, S.; Senthilkumar, S.; Das, D.; Gupta, S.S.; Bharadwaj, P.K. A Versatile CuII Metal-Organic Framework Exhibiting High Gas Storage Capacity with Selectivity for CO2: Conversion of CO2 to Cyclic Carbonate and Other Catalytic Abilities. Chem. Eur. J. 2016, 22, 3387–3396. [Google Scholar] [CrossRef] [PubMed]
- Arcadi, A.; Cacchi, S.; Cascia, L.; Fabrizi, G.; Marinelli, F. Preparation of 2,5-disubstituted oxazoles from N-propargylamides. Org. Lett. 2001, 3, 2501–2504. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, B.M.; Hacksell, U. Base-Catalyzed cyclization of N-propargylamides to oxazoles. J. Heterocycl. Chem. 1989, 26, 269–275. [Google Scholar] [CrossRef]
- Chauhan, D.P.; Varma, S.J.; Vijeta, A.; Banerjee, P.; Talukdar, P. A 1,3-amino group migration route to form acrylamidines. Chem. Commun. 2014, 50, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Hayashi, H.; Saigoku, T.; Nishiyama, H. Domino coupling relay approach to polycyclic pyrrole-2-carboxylates. J. Am. Chem. Soc. 2005, 127, 10804–10805. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhou, X.; Okamura, T.-A.; Chen, M.; Lu, Y.; Sun, W.-Y.; Yu, J.-Q. Silver supramolecule catalyzed multicomponent reactions under mild conditions. Dalton Trans. 2012, 41, 5889–5896. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.J.; Xi, F.G.; Pan, W.L.; Gao, E.Q. MIL-101(Cr)-SO3Ag: An efficient catalyst for solvent-free A3 coupling reactions. J. Mol. Catal. A Chem. 2016, 430, 36–42. [Google Scholar] [CrossRef]
- Jayaramulu, K.; Datta, K.K.R.; Suresh, M.V.; Kumari, G.; Datta, R.; Narayana, C.; Eswaramoorthy, M.; Maji, T.K. Honeycomb porous framework of zinc(II): Effective host for palladium nanoparticles for efficient three-component (A3) coupling and selective gas storage. Chempluschem 2012, 77, 743–747. [Google Scholar] [CrossRef]
- Lili, L.; Xin, Z.; Jinsen, G.; Chunming, X. Engineering metal–organic frameworks immobilize gold catalysts for highly efficient one-pot synthesis of propargylamines. Green Chem. 2012, 14, 1710. [Google Scholar] [CrossRef]
- Prat, D.; Hayler, J.; Wells, A. A survey of solvent selection guides. Green Chem. 2014, 16, 4546–4551. [Google Scholar] [CrossRef]

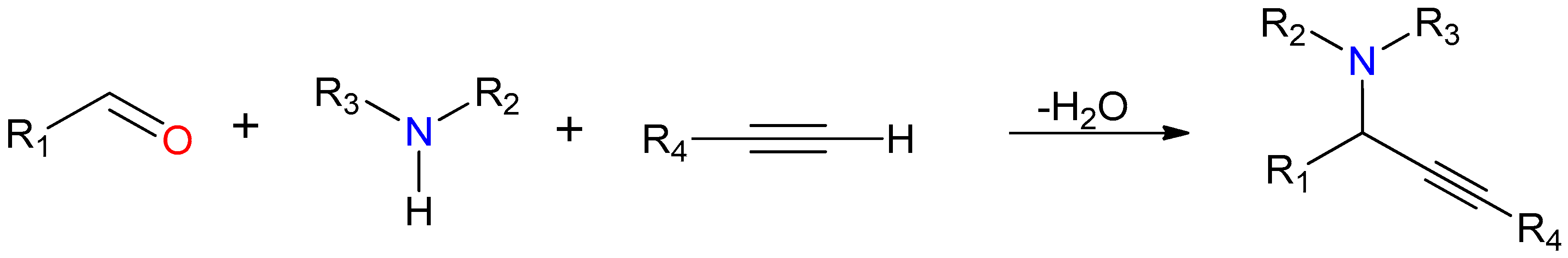

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
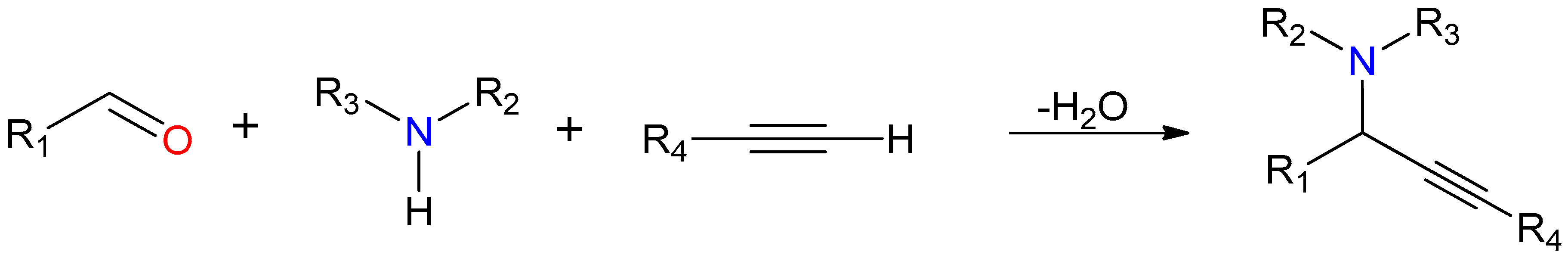{kind=link}
| Bond | Å |
|---|---|
| Cu1–O1 | 1.932(11) |
| Cu1–O2 | 2.668(11) |
| Cu1–O10 | 1.964(10) |
| Cu1–O11 | 2.611(11) |
| Cu1–O19 | 1.938(11) |
| Cu1–O20 | 1.931(10) |
| Cu2–O8 | 1.942(11) |
| Cu2–O28 | 1.991(9) |
| Cu2–O29 | 2.191(11) |
| Cu2–O30 | 1.959(10) |
| Cu2–O14 1 | 1.927(11) |
| Cu3–O5 | 1.928(10) |
| Cu3–O22 | 1.925(12) |
| Cu3–O23 | 2.005(11) |
| Cu3–O24 | 2.343(11) |
| Cu3–O18 2 | 1.943(10) |
| Cu1–Cu2 | 11.400(3) |
| Cu1–Cu3 | 14.176(3) |
| Cu2–Cu3 | 12.554(3) |
| Entry | Metal Salt | L | Additive a | Conditions | Formula | Ref. |
|---|---|---|---|---|---|---|
| 1 | Cu(NO3)2·2.5H2O | H3L1 | Et3N | rt/H2O/MeOH (10:1)/Me2CO | [Cu3(L1)2(H2O)8]·8H2O | b |
| 2 | CuCl2·2H2O | H3L1 | None | 100 °C/3 h/H2O/DMF (1:2) | [Cu(L1)(H2O)3]2[Cu(H2O)6]·(H2O)3 | [50] |
| 3 | CuCl2·2H2O | H3L1 | py c | 100 °C/24 h/H2O/DMF (1:1) | [Cu3(L1)2(H2O)3]·2H2O | [50] |
| 4 | CuCl2·2H2O | H3L1 | py | 90 °C/40 h/H2O/DMF (1:1) | [Cu2(L1)(Py)2(μ3-OH)]·(H2O)2 | [50] |
| 5 | CuCl2·2H2O | H3L1 | bpp d | 100 °C/48 h/H2O/MeOH (1:1) | [Cu2(L1)(bpp)(μ3-OH)]·6H2O | [50] |
| 6 | Zn(NO3)2·2.5H2O | H3L1 | None | 100 °C/48 h/H2O/MeOH (10:1) | [Zn(L1)(H2O)3]2[Zn(H2O)6]·(H2O)3 | [30] |
| 7 | Ni(NO3)2·2.5H2O | H3L1 | None | 100 °C/48 h/H2O/MeOH (10:1) | [Ni(L1)(H2O)3]2[Ni(H2O)6]·(H2O)3 | [30] |
| 8 | Mn(OAc)2·4H2O | H3L1 | None | 100 °C/48 h/H2O/MeOH (10:1) | [Mn(L1)(H2O)3]2[Mn(H2O)6]·(H2O)3 | [30] |
| 9 | Co(NO3)2·6H2O | H3L1 | None | 100 °C/48 h/H2O/MeOH (10:1) | [Co(L1)(H2O)3]2[Co(H2O)6]·(H2O)3 | [30] |
| 10 | Co(NO3)2·6H2O | H3L1 | bpy e | 100 °C/48 h/H2O | [Co1.5(L1)(bpy)1.5(H2O)3]·(H2O)5 | [51] |
| 11 | Co(NO3)2·6H2O | H3L1 | bpe f | 100 °C/48 h/H2O | [Co1.5(L1)(bpe)1.5(H2O)2] | [51] |
| 12 | Co(NO3)2·6H2O | H3L1 | bpp | 100 °C/48 h/H2O | [Co2(L1)(bpp)2(NO3)(μ2-H2O)2]·(H2O)2 | [51] |
| 13 | Tb(III) | H3L1 | None | N/A g | [Tb(L1)(H2O)3]·H2O | [31] |
| 14 | Gd(III) | H3L1 | None | N/A | [Gd(L1)(H2O)3]·H2O | [31] |
| 15 | Nd(III) | H3L1 | None | N/A | [Nd(L1)(H2O)3]·H2O | [31] |
| 16 | La(III) | H3L1 | None | N/A | [La(L1)(EtOH)(H2O)2]·2.5H2O | [31] |
| 17 | CaCl2 | H3L1 | py | rt/H2O/MeOH (1:1) | [Ca(HL1)(H2O)2] | [52] |
| 18 | CaCl2 | H3L1 | py | rt/H2O/MeOH (1:1) | [Ca6(L1)4(H2O)14](H2O)3 | [52] |
| 19 | Ca(NO3)2·3H2O | H3L1 | NaOAc | rt/H2O/EtOH (1:1) | [Ca2(HL1)2(μ-H2O)(H2O)5]·3H2O | [23] |
| 20 | Sr(NO3)2 | H3L1 | NaOAc | rt/H2O/EtOH (1:1) | [Sr2(HL1)2(H2O)7]·H2O | [23] |
| 21 | Cu(NO3)2·3H2O | H3L2 | KOH/am h | 80 °C/48 h/MeOH/DMF (10:1) | [Cu4(HL2)2(H2O)4(MeO)4] | [53] |
| 22 | CuCl2·2H2O | H3L2 | KOH | rt/EtOH/DMF (4:1) | [Cu12(L2)8(H2O)12]·8EtOH·40H2O | [54] |
| 23 | Zn(NO3)2·6H2O | H3L2 | bpy/KOH | rt/H2O/MeOH (3:8) | [Zn3(L2)2(bpy)4]·24H2O | [55] |
| 24 | Ni(NO3)2·2H2O | H3L2 | bpy/KOH | 95 °C/48 h/H2O/EtOH (1:1) | [Ni3(L2)2(bpy)4]·2EtOH·14H2O | [56] |
| 25 | Co(NO3)2·2H2O | H3L2 | bpy/KOH | rt/H2O/MeOH (3:8) | [Co3(L2)2(bpy)4]·28H2O | [56] |
| 26 | Cd(NO3)2·4H2O | H3L2 | bpy/teda i | 100 °C/72 h/H2O/DMF (1:1) | [Cd8(L2)6(bpy)3(H2O)4](H3O)2 | [18] |
| 27 | Ni(NO3)2·2H2O | H3L3 | bpy/KOH | 95 °C/48 h/H2O/EtOH (1:1) | [Ni3(L3)2(bpy)4]·2EtOH·14H2O | [56] |
| 28 | Co(NO3)2·2H2O | H3L3 | bpy/KOH | rt/H2O/MeOH (3:8) | [Co3(L3)2(bpy)4]·28H2O | [56] |
| 29 | Cd(NO3)2·4H2O | H3L3 | bpy/teda | 100 °C/72 h/H2O/DMF (1:1) | [Cd8(L3)6(bpy)3(H2O)4](H3O)2 | [18] |
| Entry | Aldehyde | Amine | Alkyne | Yield a (%) |
|---|---|---|---|---|
| 1 | cyclohexane carboxaldehyde | pyrrolidine | phenylacetylene | 99 |
| 2 | cyclohexane carboxaldehyde | piperidine | phenylacetylene | 99 |
| 3 | cyclohexane carboxaldehyde | azepane | phenylacetylene | 94 |
| 4 | cyclohexane carboxaldehyde | morpholine | phenylacetylene | 99 |
| 5 | cyclohexane carboxaldehyde | diethylamine | phenylacetylene | 77 |
| 6 | cyclohexane carboxaldehyde | N-methylaniline | phenylacetylene | 58 |
| 7 | benzaldehyde | pyrrolidine | phenylacetylene | 67 |
| 8 | 4-methyl benzaldehyde | pyrrolidine | phenylacetylene | 61 |
| 9 | 4-methoxy benzaldehyde | pyrrolidine | phenylacetylene | 36 |
| 10 | 4-chloro benzaldehyde | pyrrolidine | phenylacetylene | 64 |
| 11 | cyclohexane carboxaldehyde | pyrrolidine | 1-hexyne | 95 |
| 12 | cyclohexane carboxaldehyde | pyrrolidine | phenylacetylene | 96 b |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loukopoulos, E.; Michail, A.; Kostakis, G.E. A 12-Fold ThSi2 Interpenetrated Network Utilizing a Glycine-Based Pseudopeptidic Ligand. Crystals 2018, 8, 47. https://doi.org/10.3390/cryst8010047
Loukopoulos E, Michail A, Kostakis GE. A 12-Fold ThSi2 Interpenetrated Network Utilizing a Glycine-Based Pseudopeptidic Ligand. Crystals. 2018; 8(1):47. https://doi.org/10.3390/cryst8010047
Chicago/Turabian StyleLoukopoulos, Edward, Alexandra Michail, and George E. Kostakis. 2018. "A 12-Fold ThSi2 Interpenetrated Network Utilizing a Glycine-Based Pseudopeptidic Ligand" Crystals 8, no. 1: 47. https://doi.org/10.3390/cryst8010047
APA StyleLoukopoulos, E., Michail, A., & Kostakis, G. E. (2018). A 12-Fold ThSi2 Interpenetrated Network Utilizing a Glycine-Based Pseudopeptidic Ligand. Crystals, 8(1), 47. https://doi.org/10.3390/cryst8010047