Abstract
Silicon remains the most important material for electronic technology. Presently, some efforts are focused on the use of Si nanoparticles—not only for saving material, but also for improving the efficiency of optical and electronic devices, for instance, in the case of solar cells coated with a film of Si nanoparticles. The synthesis by a bottom-up approach based on condensation from low temperature plasma is a promising technique for the massive production of such nanoparticles, but the knowledge of the basic processes occurring at the atomistic level is still very limited. In this perspective, numerical simulations can provide fundamental information of the nucleation and growth mechanisms ruling the bottom-up formation of Si nanoclusters. We propose to model the low temperature plasma by classical molecular dynamics by using the reactive force field (ReaxFF) proposed by van Duin, which can properly describe bond forming and breaking. In our approach, first-principles quantum calculations are used on a set of small Si clusters in order to collect all the necessary energetic and structural information to optimize the parameters of the reactive force-field for the present application. We describe in detail the procedure used for the determination of the force field and the following molecular dynamics simulations of model systems of Si gas at temperatures in the range 2000–3000 K. The results of the dynamics provide valuable information on nucleation rate, nanoparticle size distribution, and growth rate that are the basic quantities for developing a following mesoscale model.
1. Introduction
The synthesis of nanoparticles in the gas phase by a bottom-up process is emerging as a promising technology compared to traditional production techniques. This is because, even though the wet phase synthesis has a better control of the nanoparticle morphology, it requires solvents and cannot be easily used for industrial production. In contrast, the milling techniques are relatively simple and can be adapted to large scale productions, but do not provide a good control of size and morphology of the nanoparticles. Furthermore, some bottom-up processes do not use solvents and are therefore more eco-friendly. In the case of hard materials that are stable at high temperatures, the synthesis from gaseous phase in plasma reactors [1], flame reactors [2], and hot-wall reactors [3] is especially interesting. The operating conditions—in terms of density and high temperature (2000–3000 K) of the gas—in these types of reactors make the environment in which the synthesis occurs extremely complex, difficult to monitor and investigate with experimental techniques. This is due to the non-equilibrium thermodynamic processes, the kinetics that may develop in the range of ns or even of ps, and the chains of chemical reactions between the precursors and any contaminant.
For such complex systems, computational modelling might in principle provide valuable insights into the basic mechanisms that affect the synthesis process. These are, for example, nucleation, growth of the embryo-particles, and their surface chemical activity. Of course, numerical modelling has its objective difficulties too, due to the size of the models and the time scale of the simulations required to obtain results comparable to experiments. The basic approach in this case is to define—when possible—different modeling regions of spatial and temporal dimensions that are contiguous and partially superimposed. In the specific case of the plasma synthesis, the modelling range extends from the atomic size of the primary species up to the macroscopic continuous reactor and from the femtoseconds of molecular dynamics up to the milliseconds of diffusion and turbulence. Chemical engineers have developed and already employ numerical models on the macroscopic scale of the reactors, based on empirical parameters which, however, are not easily measurable (as in the case of the reactors here considered). This has led to the development of meso-scale modelling for a better understanding of the systems at a microscopic level. However, such models also depend on a set of basic parameters derived from thermodynamical data obtained on bulk material that can generally have properties very different from that of a nanoparticle. Atomistic modelling can provide insight about the basic processes that occur on the scale of nanometers and picoseconds, and then drive the subsequent models on larger scales to a better description of the system.
Atomistic modelling is the subject of the present report concerning the bottom-up synthesis of nanoparticles from Si low temperature plasma. The main goal is to estimate—by means of first-principle and molecular mechanics calculations with force fields (FF)–the morphology of embryo-particles (small size clusters) that form through homogeneous nucleation of the monoatomic gas and their growth during the first steps of the nucleation process. The numerical procedures described here can be easily extended to other hard materials. They are based on density functional theory (DFT), basin hopping [4] (DFT-BH) calculations, and molecular dynamics simulations based on reactive force fields. These force fields can describe bond breaking and formation during the evolution of the systems, which is fundamental in the cases examined in this study.
After a brief overview of the features of the reactive force field [5] and of the procedure to optimize its parameters for the case of Si nanoparticles, the results of the dynamics in terms of inception time of the nanoparticles as a function of their size and of the environmental conditions (temperature and density of the primary gas) will be presented. These results will be critically discussed and compared to the predictions provided by the Classical Nucleation Theory (CNT) [6], which, in different forms, is largely used to describe homogeneous nucleation.
2. Model and Computational Details
The atomistic model adopted to simulate the early stages of the gas phase condensation of Si nanoparticles consists of classical molecular dynamics simulations based on a reactive force field (ReaxFF) originally developed by van Duin [5], where the parameters were recently optimized [7] for Si-containing systems. ReaxFF is flexible, computationally efficient, and has been successfully applied to a large variety of material modelling problems [8,9,10,11,12,13,14,15]. For these reasons, it has been selected to model the early stages of the plasma-assisted growth of Si nanostructures.
For the simulation of this kind of process in Si-containing materials, two formally simpler potentials—namely, Tersoff and Stillinger–Weber potentials—have been frequently employed. Among the most recent applications there are the computational studies for the simulation of the condensation, growth, and crystallization of Si nanoclusters obtained by magnetron sputtering [16,17,18]. In this technique, the fragments of Si (mainly embryo clusters but also isolated atoms) produced by sputtering at very high temperatures (even 6000 K) pass through a condensation region occupied by an inert gas (Ar) at a much lower temperature. The sample is obviously very far from equilibrium, and has been simulated by molecular dynamics for an ensemble with constant number of atoms, volume and total energy (NVE) with a time gradient of the temperature that is taken as large as 30 K per ns. In our case we intend to refer to environmental conditions much closer to equilibrium and then better represented by simulations for an ensemble with constant number of atoms, volume and average temperature (NVT) with large fluctuations induced by the Berendsen’s thermostat adopted. These conditions correspond locally to those that are present in the thermal plasma reactors of industrial type employed for large-scale production of nanoparticles.
ReaxFF is expressed by an extended set of analytical functions that reflect the complex picture of the atomic interactions and depend on a number of different parameters. These parameters have been derived, in the present case, from quantum chemistry calculations on model systems representative of the species present in the Si plasma. The complex analytic form of the force field is, anyway, much less computationally expensive than the Hamiltonian formulation of the quantum molecular dynamics (QMD) methods, and as a consequence, classical simulations are the only practical choice for describing large aggregates containing up to one million atoms. In comparison to most of the force fields available in the literature, which essentially provide a physical picture of the system, ReaxFF allows the simulation of bond breaking and formation. This aspect is very relevant in order to properly simulate not only interactions, but also reactions, and then dynamically characterize the various processes that determine and regulate the early formation of nanomaterial aggregates from the plasma. The potential energy in ReaxFF is calculated as the sum of a number of energy terms depending on the bond distance/bond order and bond order/bond energy relationships. The algorithm appropriately describes under- and over-coordination and dismisses the energy terms depending on bond distance, bond, and dihedral angles upon bond breaking. Non-bonded interactions (Coulomb and van der Waals forces) are calculated between atom pairs with a shielding factor [19] to dampen close-range interactions; atomic charges are calculated using the electronegativity equalization method (EEM) [20] and long-range Coulomb interactions are calculated by means of a taper function with an outer cut-off radius.
2.1. Optimization of the ReaxFF Parameters
Starting from a parametrization of the FF for Si based materials (VD) available in the literature [7] and designed to describe the bulk structure of SiC and SiO, we re-optimized the FF for the specific systems under study in order to consider surface effects of small Si clusters. These effects can be fundamental in the nucleation and growth processes, leading to nanoparticles with maximum diameter of about 1 nm. The FF parameters have been optimized in order to reproduce a training-set (t-set) of properties (i.e., energies and geometries) derived from quantum chemical (QC) calculations on selected model systems. Few experimental measurements of dissociation/ionization energies and mobilities of the ionized structures are available in the literature [21,22,23,24], whereas geometries and electronic structures of small silicon clusters have been studied extensively by QC approaches over the last few years [25,26,27,28,29,30,31,32,33,34,35,36,37,38].
The first-principle quantum method we have adopted is based on DFT [39], which is widely used for studying medium-large molecular systems. Calculations have been performed by the ESPRESSO package [40] using ultrasoft pseudo potentials [41] for an approximate description of the core electrons that do not directly participate in the formation of molecular bonds, whereas the electronic structure of the valence electrons (4 for Si) is described by the GGA-PBE [42] functional. Energy spin-restricted optimizations were performed in a cubic box with side between 10 an 20 Å (depending on the size of the cluster) and periodic boundary conditions in order to effectively project the wave function on a set of plane waves with cut-offs on the wave-function/electronic density of about 140/1100 eV, respectively. Other details of the DFT calculations are: gaussian smearing for the occupation of the Kohn–Sham single-particle states of 0.03 eV; Brillouin zone explored at the Gamma point only; single local minimization with a threshold on energy/forces of / au.
A small Si cluster may adopt various conformations corresponding to slightly different energy values; at the plasma temperatures (>2000 K), a great number of such conformations is accessible. For the optimization of the force field, we selected the most populated minimum energy configurations of the clusters that have been identified by a specific algorithm based on geometry optimization and Basin-Hopping [4]. This is one of the several global optimization methods that are used to find the minimum of a multi-variable Potential Energy Surface (PES). As any other similar method, this approach does not guarantee a successful identification of the absolute energy minimum in a finite time, but provides an efficient algorithm for exploring the PES and accumulating a voluminous structural database. In our approach, for each cluster size, we performed a search for the minimum by running several BH calculations (from three to five), composed of 500 steps each. At each step, the cluster geometry is slightly modified randomly and a DFT geometry optimization is performed; the step is accepted or refused according to the Monte Carlo/Metropolis criterion [43] with a kT value in the range 0.5–1.0 eV [44]. The lowest-energy structures resulting from this search are summarized in Figure 1 for Si clusters composed of an even number of atoms with a size in the range 6 to 36. These structures were considered as model systems for computing the benchmark data forming the training set.
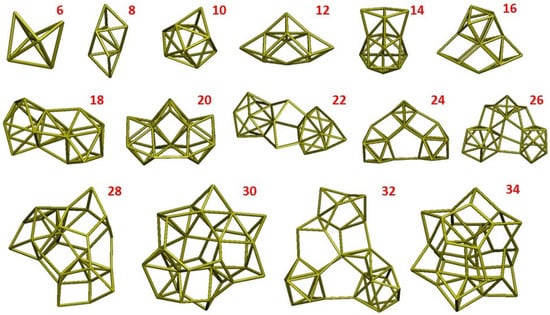
Figure 1.
Lowest-energy structures of Si clusters with an even number of atoms and size in the range 6–34.
To refine the VD FF, we adopted the ReaxFF standalone code (version 2.0). The t-set adopted was given by geometries and energies of a number of model systems selected among the structures optimized by the DFT-BH searches, namely: Si, Si, Si, Si, Si, Si, Si, Si (two structures), see Figure 2, to which the basic unit of silicon diamond cubic crystal was added. Starting from the original parametrization (VD) [7], we optimized only the relevant parameters responsible for the flexibility of the Si clusters at high temperature against our t-set: (i) Si-Si bond (14 parameters); (ii) Si-Si-Si valence angle (5 parameters); (iii) Si-Si-Si-Si torsion angle (5 parameters). These latest parameters were not present in the original parametrization, but were added in this new force field to get a better agreement with the t-set data. Starting values of the parameters were taken from carbon in VD.
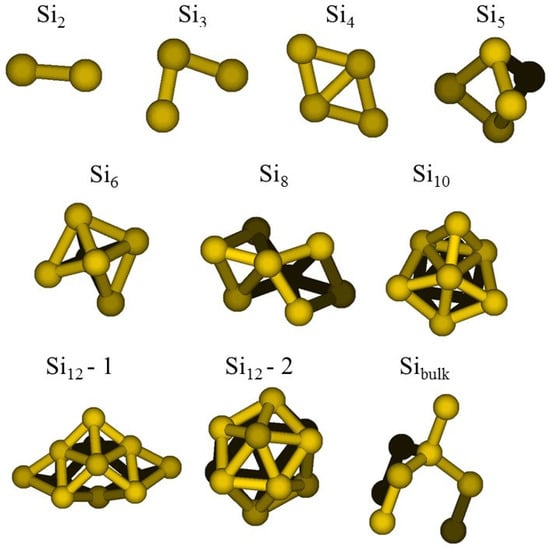
Figure 2.
Si clusters included in the training set used in the FF optimization.
The ReaxFF code adopts a sequential optimization procedure of the parameters by the minimization of an error function (EF) which is calculated in the following way:
where and are, respectively, the running FF and the benchmark QC values of the i-th entry of the t-set and is its weight. The sum is extended to all the geometrical/energetic entries of the t-set. The ReaxFF optimization procedure works in the following way: at each step, a single parameter is considered and its starting value is varied by adding and subtracting a given quantity; for these three values of the considered parameter, the total EF is estimated and a parabolic extrapolation is achieved by finding (in correspondence of the minimum of the parabolic expression) the optimal value of the parameter within the chosen window; if this new predicted value corresponds to a lower value of the EF, the change is accepted and the algorithm moves to the successive parameter. As the parameters are strongly entangled, several sequential optimization cycles have to be repeated in order to achieve a real reduction of the EF against the t-set. It should be added that at each step the QC geometries optimized by the DFT-BH procedure are compared to those optimized by using the running FF; this is done in view of using the FF for an effective and realistic search of the different stable conformations of the clusters.
From the point of view of this performance it is convenient to analyze the predicted lowest-energy structures by using the two synthetic structural descriptors: R and θ. The first one refers to an average value of the interatomic distances:
where N is the number of atoms in the cluster, each atom has first-neighbours, and is the distance between atoms i and j; the second one refers to an average value of the molecular angles:
2.2. Reactive Molecular Dynamics
Classical Molecular Dynamics simulations of the Si + Ar plasma have been carried out by using the LAMMPS code [45] with the presently optimized reactive force field (in the following named ND) in the NVT ensemble with the Berendsen’s thermostat in order to have temperature fluctuations around a constant average temperature. In our approach, such simulations—performed at different compositions and temperatures—intend to model the environment present in thermal plasma reactors employed for large-scale production of Si nanoparticles. The results are employed to investigate nucleation processes and estimate specific parameters and mathematical expressions that can be used to feed mesoscale models for the simulation of much larger systems.
On the basis of the good performance of the ND FF on the exploration of the PES at 0 K, extensive MD simulations have been carried out by using this FF; nevertheless, a comparison with VD on a specific set of thermodynamic conditions will be discussed in the next section as well, in order to show how the two different parameterizations influence the nucleation and growth processes.
The computational details of the nucleation simulations are the following:
- Simulation box: an appropriate value of the length of the cubic simulation box has been chosen in order to reduce the effect of the periodic boundary conditions. The number of events that occur on the border becomes less and less relevant than the number of total events when the size of the box is increased. In the limit of a box with an infinite size, the biasing constraint of the translational periodicity disappears. According to a series of simulations in which the volume of a cubic unit cell has been progressively increased (from a minimum of nm to a maximum of nm), the volume chosen was nm.
- Temperature: each simulation has been carried out at fixed average temperature. The temperature range between 1800 and 2400 K has been explored by increasing the temperature by steps of 100 K in each simulation.
- Atom density: the number of atoms within the simulation box has been chosen on the basis of particular experimental conditions: total gas pressure within the plasma reactor around 1 atmosphere (standard ambient pressure). On the basis of this latter value, the density of the simulation gas (which is composed of Si and Ar atoms) is about 3.6 × 10 atoms/m, which corresponds—in the chosen cubic box—to an ensemble of about 12,000 atoms.
- Composition of the mixture: the content of the (Si, Ar) mixture has been varied according to the following percentages: (i) 25% Si and 75% Ar; (ii) 50% Si and 50% Ar; and (iii) 75% Si and 25% Ar.
- Time step: The time step of the MD simulations has been set to 2 fs, having verified that a smaller time step of 0.5 fs reproduces the same results at both the qualitative and quantitative levels.
We underline that the adopted models refer to environmental conditions close to equilibrium corresponding, locally, to those that are present in the thermal plasma reactors of industrial type for large-scale production of nanoparticles.
3. Results and Discussion
From the point of view of assessing the performance of the reactive FF in exploring the PES of the small Si clusters, it is interesting to compare the structures predicted by the VD and the presently refined parametrization ND of the reactive FF against DFT results. In Figure 3, the lowest-energy structures predicted at sizes 12, 20, 30, and 36 are reported, together with a structural and energetic analysis of the conformations obtained. It should be noted that the lowest-energy structures shown in Figure 3 have been obtained by adopting the same BH search protocol described above, with energies and forces calculated at the FF level, but the energy of such structures has been then computed at the DFT level for a correct comparison with the DFT-BH results.
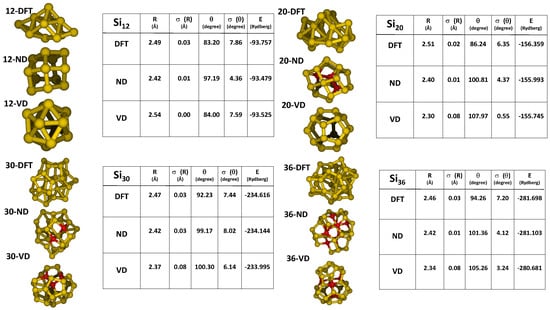
Figure 3.
Structural and energetic results obtained by using different parametrizations of ReaxFF for Si clusters of size 12, 20, 30, and 36. The structural data R and θ are defined in Equations (2) and (3), respectively, while σ refers to their root-mean-square dispersion. DFT: density functional theory; ND: present optimization of ReaxFF; VD: parametrization for Si-based materials available in the literature [7].
From the structural point of view, we can observe two main features that characterize the geometries of small Si clusters:
- (i)
- a compact arrangement of atoms with coordination numbers and coordination angles much different from bulk silicon, where the coordination number is strictly four and the angles are perfectly tetrahedral; in the DFT-BH optimized structures, instead, the number of nearest neighbours can be as large as 6 and even 7, and the bond angles are remarkably different from tetrahedral;
- (ii)
- cluster surfaces are not smooth, but sharp with the presence of convex re-entrances, leaving the surface atoms with a very low coordination of one, or maximum two, neighbours.
By inspecting the results of the two FFs, we can see that the presently optimized parametrization ND using the t-set shown in Figure 1 better reproduces both the structural and energetic DFT results. Indeed, the two structural features discussed above can be found in the geometries predicted by the ND FF, and the relative FF energies are closer—compared to the VD values—to the benchmark DFT ones. The limitations exhibited in the present application by the VD FF could be due to its apparent tendency to favor Si atoms with “bulk-character”. As a consequence, in the structures predicted for the small clusters, the average coordination number is about four, as evidenced by the high number of red atoms in Figure 3, which are tetrahedrally shaped, and by very smooth surfaces; moreover, the DFT energy of the VD FF optimized structures is higher than that predicted by the ND FF [44].
A direct comparison of our present MD results with those available in the literature [16,17,18]—using the Tersoff or Stillinger–Weber potentials in order to evaluate the performance of different force fields—would be biased by the large differences in the model systems adopted and in the characteristics of the MDs employed. An accurate comparison would require calculations in which the features of the simulations are more homogeneous, which is outside the scope of the present work.
Two typical results of the nucleation MD simulations at different temperatures are shown in Figure 4. On a short time scale, the dynamics is dominated by the formation of smaller aggregates (mainly Si dimers, which are quickly formed); the concentration of the smaller clusters reaches a plateau and then starts decreasing due to the formation of the larger aggregates. As can be noted in Figure 4a, a further peak is found at size 8, which corresponds to a remarkably stable structure of the Si system. As the simulation proceeds, larger aggregates start to form. At lower temperature (Figure 4a), several Si clusters can be found at the end of the run (about 300 ns), whereas at higher temperature (Figure 4b), the particles growth is slowed down due to the larger thermal agitation and only few aggregates of size around 30 can be obtained at the end of the simulation. This picture is different from that provided by the classical nucleation theory, in which, out of nothing, a nanoparticle of critical mass is produced without a previous history. In our opinion, this phenomenological model does not apply to the cases where—as for Si—the interaction between atomic components leads to the formation of covalent bonds with bond energy much higher than the available average thermal energy.
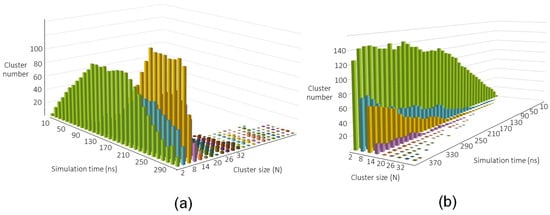
Figure 4.
Nucleation and growth picture at (a) 1800 K and (b) 2400 K.
In Table 1, the characteristic inception times for particles of size 20, 30, and 40 are reported as a function of the temperature for the three mixture compositions specified above. The inception time of a particle composed of N atoms is estimated as the time when the first particle of that size appears in the simulation box. For a specific nuclearity, the inception time increases when increasing the temperature due to the higher thermal agitation that slows down the aggregation process. As could be expected, the inception time decreases at increasing Si content when comparing the same particle size and temperature. The Ar gas present in industrial thermal plasma reactors provides a more effective coupling with the electromagnetic field applied for the formation and maintenance of the plasma; the case of a pure Si gas is practically not realized. Our simulations (not included in this study) of such a system show that the processes of condensation and growth of Si nanoparticles remain substantially unchanged; the presence of Ar is not necessary for their implementation.

Table 1.
Inception times (ns) for Si particles of 20, 30, and 40 atoms.
When using the force field VD, the results of the nucleation and growth processes are completely different: at T = 2000 K with a plasma made of 50% Si and 50% Ar (within a time scale of around 160 ns), only a very low percentage of atoms form aggregates, as it can be evinced by inspecting Figure 5a; moreover, only dimers are formed (about 50 during the simulation, see Figure 5b) with the sporadic appearance of trimers, which soon dissociates again in dimers and single atoms (see Figure 5c). This behaviour could be due to an instability of the trimer that originates a bottleneck in the growth process [44], as this nuclearity is a mandatory step in the growth of larger aggregates, due to the fact that the formation of a tetramer due to the aggreagtion of two dimers is a very unlikely event in the present conditions.
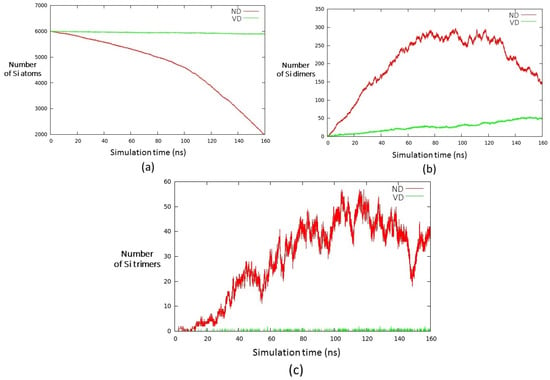
Figure 5.
Number of (a) unreacted Si atoms; (b) dimers; and (c) trimers as a function of time at T = 2000 K. The results from the ND and VD FFs are shown in red and green, respectively.
To conclude, we compare the results of the present MD simulations with the ND FF and the only—to our knowledge—work in the literature reporting the values of partial pressures of small Si clusters (N = 1–6) in the vapour phase in equilibrium with the Si liquid phase calculated on the basis of classical thermodynamics [46]. In order to compare with the reported data, in Figure 6 we have plotted the ratio between the numbers of dimers and single atoms vs. the ratio between the number of trimers and dimers as a function of the simulation time. In this case, an NVT dynamics calculation has been performed at a total pressure of 1.22 × MPa (1 atm = 0.1 MPa) and a temperature of T = 3000 K, matching one of the conditions considered in the experiment. The choice to operate at a higher temperature (with respect to the previous simulations) has been done to work with a higher value of Si pressure, and hence collect more statistical information about the investigated system. The simulation box contained 2860 atoms, and time-step has been set to 2.0 fs; the Berendsen’s thermostat has been employed, in agreement with the methodological procedure applied for the estimation of the nucleation rates. According to the results reported in Figure 6, it can be seen that the dimers/atoms ratio is quite stable and reaches a plateau around the value 0.05; on the other side, the trimers/dimers ratio is less regular and is characterized by remarkable oscillations, mainly due to statistical reasons (lower absolute number of trimers in the simulation box during the dynamics); nevertheless, we can estimate that the latter ratio oscillates around the value 0.10 during the simulation. The obtained values of the ratios are in qualitative agreement with the data of Reference [46], which reports a dimers/atoms ratio of about 0.11 and a trimers/dimers ratio of about 0.25 at the considered values of temperature and pressure.
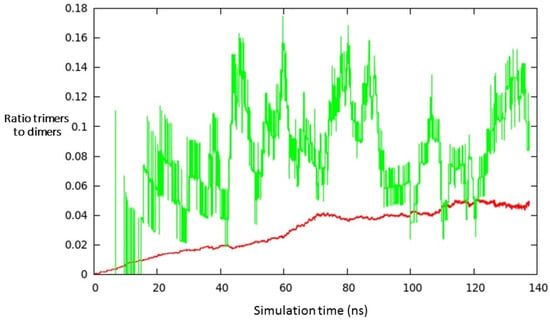
Figure 6.
Ratio between the number of dimers and unreacted atoms (in red) vs. ratio between trimers and dimers (in green) for a set of thermodynamic variables specified in the main text.
4. Conclusions
We have proposed an theoretical approach to the difficult problem of modelling the nucleation and growth of embryo- particles in a low temperature plasma, with application to the Si gas. The approach is based on reactive molecular dynamics with a FF specifically optimized using DFT calculations as benchmark for a set of small Si clusters. The presently proposed (ND) FF—optimized starting from the one (VD) available in the literature—shows appreciable improvements in the description of Si nanoparticles. The results obtained by exploring the PES at 0 K evidenced the tendency of VD FF to over-estimate the bulk behavior in Si clusters. On the contrary, ND FF was capable of more confidently reproducing small clusters and favoring the formation of compact aggregates. The results of the molecular dynamics simulations performed with the ND FF are in reasonable agreement with the very few experimental data available for this kind of system, although the experimental environment corresponding to Si vapor in equilibrium with the liquid phase is only roughly represented by our simulation model. Unfortunately, an extension of the model including a representation of the liquid phase would lead to too-CPU-demanding calculations. Our results can be viewed as describing a state of quasi-equilibrium only in the sense that the obtained ratios are quite stable in a wide time window; consequently, the agreement of theory and experiment can be expected only at a qualitative level. The results obtained show that the proposed theoretical model can be satisfactorily applied to systems where the experimental studies are very difficult to perform and an accurate description of the basic processes at the atomistic level is presently missing.
Acknowledgments
This work has been developed as part of the European project Nanodome that has received funding form the European Union’s Horizon 2020 Research and Innovation Programme, under Grant Agreement number 646121.
Author Contributions
Susanna Monti, Vincenzo Carravetta and Giovanni Barcaro conceived and designed the theoretical approach; Giovanni Barcaro and Luca Sementa performed the calculations; Giovanni Barcaro, Vincenzo Carravetta and Luca Sementa analysed the results; Vincenzo Carravetta, Susanna Monti, Giovanni Barcaro and Luca Sementa wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shigeta, M.; Murphys, A.B. Thermal plasmas for nanofabrication. J. Phys. D Appl. Phys. 2011, 44, 174025–174041. [Google Scholar] [CrossRef]
- Koirala, R.; Pratsinisa, S.E.; Baiker, A. Synthesis of catalytic materials in flames: Opportunities and challenges. Chem. Soc. Rev. 2016, 45, 3053–3068. [Google Scholar] [CrossRef] [PubMed]
- Schwade, B.; Roth, P. Simulation of nano-particle formation in a wall-heated aerosol reactor including coalescence. J. Aerosol Sci. 2003, 34, 339–357. [Google Scholar] [CrossRef]
- Wales, D.; Doye, P.K. Global Optimization by Basin-Hopping and the Lowest Energy Structures of Lennard-Jones Clusters Containing up to 110 Atoms. J. Phys. Chem. A 1997, 101, 5111–5116. [Google Scholar] [CrossRef]
- Van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A Reactive Force Field for Hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- Kashchiev, D. Nucleation: Basic Theory with Applications; Butterworth-Heinemann: Oxford, UK, 2000. [Google Scholar]
- Newsome, D.A.; Sengupta, D.; Foroutan, H.; Russo, M.F.; van Duin, A.C. Oxidation of Silicon Carbide by O2 and H2O: A ReaxFF Reactive Molecular Dynamics Study, Part I. J. Phys. Chem. C 2012, 116, 16111–16121. [Google Scholar] [CrossRef]
- Han, Y.; Jiang, D.; Zhang, J.; Li, W.; Gan, Z.; Gu, J. Development, applications and challenges of ReaxFF reactive force field in molecular simulations. Front. Chem. Sci. Eng. 2016, 10, 16–38. [Google Scholar] [CrossRef]
- Li, C.; Monti, S.; Carravetta, V. Journey toward the Surface: How Glycine Adsorbs on Titania in Water Solution. J. Phys. Chem. C 2012, 116, 18318–18326. [Google Scholar] [CrossRef]
- Monti, S.; Li, C.; Carravetta, V. Reactive dynamics simulation of monolayer and multilayer adsorption of glycine on Cu(110). J. Phys. Chem. C 2013, 117, 5221–5228. [Google Scholar] [CrossRef]
- Carravetta, V.; Monti, S.; Li, C.; Ågren, H. Theoretical simulations of structure and X-ray photoelectron spectra of glycine and diglycine adsorbed on Cu(110). Langmuir 2013, 29, 10194–10204. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Monti, S.; Ågren, H.; Carravetta, V. Cysteine on TiO2(110): A theoretical study by reactive dynamics and photoemission spectra simulation. Langmuir 2014, 30, 8819–8828. [Google Scholar] [CrossRef] [PubMed]
- Monti, S.; Li, C.; Ågren, H.; Carravetta, V. Dropping a Droplet of Cysteine Molecules on a Rutile (110) Interface: Reactive versus Nonreactive Classical Molecular Dynamics Simulations. J. Phys. Chem. C 2015, 119, 6703–6712. [Google Scholar] [CrossRef]
- Monti, S.; Carravetta, V.; Ågren, H. Simulation of Gold Functionalization with Cysteine by Reactive Molecular Dynamics. J. Phys. Chem. Lett. 2016, 7, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, Á.M.; Mocellin, A.; Monti, S.; Li, C.; Marinho, R.R.T.; Medina, A.; Agren, H.; Carravetta, V.; de Brito, A.N. Surface-Altered Protonation Studied by Photoelectron Spectroscopy and Reactive Dynamics Simulations. J. Phys. Chem. Lett. 2015, 6, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Singh, V.; Grammatikopoulos, P.; Cassidy, C.; Kengo, A.; Sowwan, M.; Nordlund, K.; Djurabekova, F. Crystallization of silicon nanoclusters with inert gas temperature control. Phys. Rev. B 2015, 91, 35419–35431. [Google Scholar] [CrossRef]
- Zhao, J.; Baibuz, E.; Vernieres, J.; Grammatikopoulos, P.; Jansson, V.; Nagel, M.; Steinhauer, S.; Sowwan, M.; Kuronen, A.; Nordlund, K.; et al. Formation Mechanism of Fe Nanocubes by Magnetron Sputtering Inert Gas Condensation. ACS Nano 2016, 10, 4684–4694. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Cassidy, C.; Grammatikopoulos, P.; Djurabekova, F.; Nordlund, K.; Sowwan, M. Heterogeneous Gas-Phase Synthesis and Molecular Dynamics Modeling of Janus and Core-Satellite Si-Ag Nanoparticles. J. Phys. Chem. C 2014, 118, 13869–13875. [Google Scholar] [CrossRef]
- Janssens, G.O.A.; Baekelandt, B.G.; Toufar, H.; Mortier, W.J.; Schoonheydt, R.A. Comparison of Cluster and Infinite Crystal Calculations on Zeolites with the Electronegativity Equalization Method (EEM). J. Phys. Chem. 1995, 99, 3251–3258. [Google Scholar] [CrossRef]
- Mortier, W.J.; Ghosh, S.K.; Shankar, S. Electronegativity-equalization method for the calculation of atomic charges in molecules. J. Am. Chem. Soc. 1986, 108, 4315–4320. [Google Scholar] [CrossRef]
- Liu, B.; Lu, Z.Y.; Pan, B.; Wang, C.Z.; Ho, K.M.; Shvartsburg, A.A.; Jarrold, M.F. Ionization of medium-sized silicon clusters and the geometries of the cations. J. Chem. Phys. 1998, 109, 9401–9409. [Google Scholar] [CrossRef]
- Hudgins, R.R.; Imai, M.; Jarrold, M.F.; Dugourd, P. High-resolution ion mobility measurements for silicon cluster anions and cations. J. Chem. Phys. 1999, 111, 7865. [Google Scholar] [CrossRef]
- Bachels, T.; Schäfer, R. Binding energies of neutral silicon clusters. Chem. Phys. Lett. 2000, 324, 365–372. [Google Scholar] [CrossRef]
- Muller, J.; Liu, B.; Shvartsburg, A.A.; Ogut, S.; Chelikowsky, J.R.; Siu, K.W.M.; Ho, K.M.; Gantefor, G. Spectroscopic evidence for the tricapped trigonal prism structure of semiconductor clusters. Phys. Rev. Lett. 2000, 85, 1666–1669. [Google Scholar] [CrossRef] [PubMed]
- Kaxiras, E.; Jackson, K. Shape of Small Silicon Clusters. Phys. Rev. Lett. 1993, 71, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.M.; Shvartsburg, A.A.; Pan, B.; Lu, Z.Y.; Wang, C.Z.; Wacker, J.G.; Fye, J.L.; Jarrold, M.F. Structures of medium-sized silicon clusters. Nature 1998, 392, 582–585. [Google Scholar]
- Mitas, L.; Grossman, J.; Stich, I.; Tobik, J. Silicon Clusters of Intermediate Size: Energetics, Dynamics, and Thermal Effects. Phys. Rev. Lett. 2000, 84, 1479–1482. [Google Scholar] [CrossRef] [PubMed]
- Rata, I.; Shvartsburg, A.A.; Horoi, M.; Frauenheim, T.; Siu, K.W.M.; Jackson, K.A. Single-parent evolution algorithm and the optimization of Si clusters. Phys. Rev. Lett. 2000, 85, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, G.; Ding, F.; Lee, H.; Shen, W.; Zhao, J. Structural transition of Si clusters and their thermodynamics. Chem. Phys. Lett. 2001, 341, 529–534. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, Q.; Jena, P.; Rao, B.K.; Kawazoe, Y. Stabilization of Si60 Cage Structure. Phys. Rev. Lett. 2003, 90, 135503. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Zeng, X.C. Global geometry optimization of silicon clusters described by three empirical potentials. J. Chem. Phys. 2003, 119, 1442–1450. [Google Scholar] [CrossRef]
- Jackson, K.A.; Horoi, M.; Chaudhuri, I.; Frauenheim, T.; Shvartsburg, A.A. Unraveling the shape transformation in silicon clusters. Phys. Rev. Lett. 2004, 93, 013401. [Google Scholar] [CrossRef]
- Li, B.X. Stability of medium-sized neutral and charged silicon clusters. Phys. Rev. B Condens. Matter Mater. Phys. 2005, 71, 235311. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, X.; Wang, G.; Zhao, J. Optimally stuffed fullerene structures of silicon nanoclusters. Phys. Rev. B Condens. Matter Mater. Phys. 2005, 71, 113412. [Google Scholar] [CrossRef]
- Ona, O.; Bazterra, V.E.; Caputo, M.C.; Facelli, J.C.; Fuentealba, P.; Ferraro, M.B. Modified genetic algorithms to model cluster structures in medium-sized silicon clusters: Si18–Si60. Phys. Rev. A 2006, 73, 1–11. [Google Scholar] [CrossRef]
- Tereshchuk, P.L.; Khakimov, Z.M.; Umarova, F.T.; Swihart, M.T. Energetically competitive growth patterns of silicon clusters: Quasi-one-dimensional clusters versus diamond-like clusters. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 76, 125418. [Google Scholar] [CrossRef]
- Zhou, R.L.; Pan, B.C. Low-lying isomers of Si and Si (n = 31–50) clusters. J. Chem. Phys. 2008, 128, 234302. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Shao, N.; Zeng, X.C. Structures and relative stability of medium- and large-sized silicon clusters. VI. Fullerene cage motifs for low-lying clusters Si39, Si40, Si50, Si60, Si70, and Si80. J. Chem. Phys. 2008, 128, 104316. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density Functional Theory of Electronic Structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Metropolis, N.; Ulam, S. The Monte Carlo method. J. Am. Stat. Assoc. 1949, 44, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Barcaro, G.; Carravetta, V.; Monti, S.; Sementa, L. Force Field Parameters for Reactive Molecular Dynamics Simulation of Si Nanoparticles. J. Chem. Theory Comput. 2017. in preparation. [Google Scholar]
- LAMMPS Molecular Dynamics Simulator. Available online: http://lammps.sandia.gov (accessed on 12 February 2017).
- Sevast’yanov, V.G.; Nosatenko, P.Y.; Gorskii, V.V.; Ezhov, Y.S.; Sevast’yanov, D.V.; Simonenko, E.P.; Kuznetsov, N.T. Experimental and theoretical determination of the saturation vapor pressure of silicon in a wide range of temperatures. Russ. J. Inorg. Chem. 2010, 55, 2073–2088. [Google Scholar] [CrossRef]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).