Abstract
The conformational preferences of benznidazole were examined through the application of DFT, PCM and QTAIM calculations, whose results were compared with crystallography data. The geometries were fully optimized with minimum potential energy surface by means of the Relaxed Potential Energy Surface Scan (RPESS) at AM1, followed by the B3LYP/6-311++G(d,p) theoretical level. As a result, the s-cis conformation (1C) was shown to be more stable (4.78 kcal∙mol−1) than s-trans (1T). The Quantum Theory Atoms in Molecules (QTAIM) was applied in order to characterize the (N–H∙∙∙O=N) and (C–H∙∙∙=N) intramolecular hydrogen bonds. The simulation of solvent effect performed by means of the implicit Polarized Continuum Model (PCM) revealed great results, such as, for instance, that the conformation 1W is more stable (23.17 kcal∙mol−1) in comparison to 1C. Our main goal was stressed in the topological description of intramolecular hydrogen bonds in light of the QTAIM approach, as well as in the solvent simulation to accurately obtain an important conformation of benznidazole.
1. Introduction
In medicinal chemistry, the studies of new compounds with high biological activities and less toxic effects have attracted the attention of many researchers around the world [1]. This scientific interest concerns the potential of several compounds in treating serious diseases, ranging from “neglected” diseases such as Schistosoma mansoni [2] to disorders such as cancer [3]. In the context of neglected diseases, for which big pharmaceutical companies are not investing in the search for therapeutic alternatives, Chagas disease, also called American trypanosomiasis, is considered the most important parasitic infection of Latin America [4]. Chagas disease is manifested in humans and domestic animals living in extreme poverty and rural areas [5]. In practice, the infection is caused by the Trypanosoma cruzi protozoan although it is transmitted by the Triatoma infestans insect. In the actual treatment, basically two drugs have been widely used: Benznidazole (Rochagan®, Basel, Switzerland) and Nifurtimox (Lampit®, Leverkusen, Germany). Due to the possibility to obtain new compounds to be used in the treatment against Chagas disease, a lot of studies have been conducted in order to improve the potentiality of benznidazole.
Some time ago, a theoretical conformational study of benznidazole was performed [6]. The results point to the existence of local minima in the potential energy surface, whose structures were compared with some active compounds against T. cruzi: derivatives of tetrahydro-β-carboline. This study was carried out in gas phase by using the AM1 semi empirical Hamiltonian, whose application was conditioned to the modeling of the Potential Energy Surface Scan (PESS). Only then, the optimized geometry in each point of minimum has been determined through the B3LYP functional jointly with the 6-311+G(d) basis set. From an experimental viewpoint, Soares Sobrinho et al. [7] have published a crystallography study about the benznidazole structure based on X-ray diffraction, whose results were very different from those in gas phase, including the formation of intermolecular hydrogen bonds on the crystal packaging. In molecular modeling [8], it is well established that the identification of hydrogen bonds is one of the most important criteria in analysis of biological compounds [9]. These studies may be performed by virtual screening [10] or quantum chemical calculations [11,12] in the pursuit of achieving structure-activity relationships. Historically, the formation of intermolecular or intramolecular hydrogen bonds has been discussed by taking into account the van der Waals radii of the electron donor-acceptor. If we consider this statement [5], the distance values for typical hydrogen bonds should be exactly the same as or shorter than 2.6 Å, of course regarding the F, O and N atoms, or even the π cloud as proton-acceptor centers [13,14,15,16]. In fact, the characterization of hydrogen bonds is not just dependent on structural parameters, actually, the application of quantum mechanical criteria is feasible in this regard.
The Quantum Theory of Atoms in Molecules (QTAIM) [17] represents a useful tool in studies of molecular stability and strength of chemical bonds [18], but is also widely applied in investigations of hydrogen bonds through the analysis of electronic density and its topological parameters [19]. According to some publications [20], the QTAIM approach has been also useful in studies of biological systems [21], mainly to characterize the intramolecular hydrogen bonds of compounds with biological activity [22]. In this sense, we are convinced that QTAIM can be decisive in our investigation not only regarding the intramolecular hydrogen bonds [23,24,25,26,27,28,29,30,31], but also to unveil the most stable conformation of benznidazole. Another interesting viewpoint is that the solvent effect may be responsible for drastic changes in several molecular properties, e.g., geometrical deformations; increase in the reaction rate; control of products along the reaction paths, for instance [32,33]. If we consider that intramolecular hydrogen bonds can be formed in the minimum structure of benznidazole, the specialized literature informs us that the application of the Polarized Continuum Model (PCM) [34] to evaluate the solvent effect is recommended [35,36,37,38,39]. By taking into account the importance of drug action in human organisms, that are predominantly aqueous, this article also presents a theoretical investigation of the solvent effect on the benznidazole structure through the application of PCM protocol, demonstrating the importance of solvation in the conformational study of this bioactive molecule [40]. In the context of molecular stabilization, a topological description of intramolecular hydrogen bonds using the QTAIM approach is another important contribution of this work.
2. Computational Procedure
The procedure of Relaxed Potential Energy Surface Scan (RPESS) was performed by taking particular care with the dihedral angles (θ1, θ2, θ3, and θ4), which are illustrated in Figure 1. These calculations were executed by using the AM1 semiempirical level with angle variations from 0° to 360°, in intervals of 10°. After that, the minimum conformation was fully optimized by the B3LYP/6-311++G(d,p) level of calculation without any symmetry constraint and with all geometries modeled at a minimum of potential energy because no imaginary frequencies were found. As a result, the conformation recognized as 1C was generated.
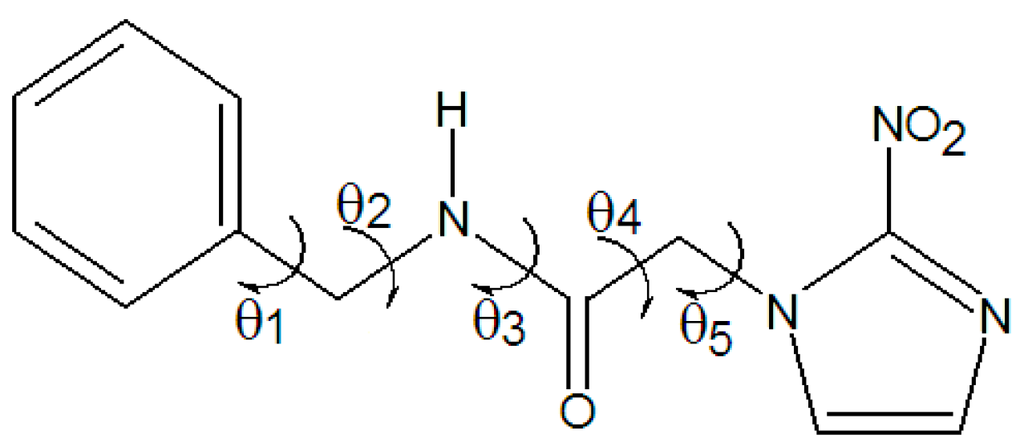
Figure 1.
Structure of benznidazole and dihedral angles examined in RPESS procedure.
The crystal structure (1K) was optimized using the same level of calculation presented above. The conformation 1T was obtained from an arbitrary variation in the geometry of 1C, corresponding to a previously reported geometry [6], which remains as a minimum conformation (less stable than 1C) after B3LYP/6-311++G(d,p) optimization. The solvent effect was evaluated on both 1K and 1C through the arguments of the Polarized Continuum Model (PCM) both providing 1W. All these calculations were carried out by GAUSSIAN 03W quantum suite of codes [41]. Both the nature of electronic density and the characterization of intramolecular hydrogen bonds were investigated through the QTAIM formalism, whose calculations were performed by the AIM 2000 1.0 software package [42].
3. Results and Discussion
3.1. Minimum Structure, Bond Lenghts and Vibration Modes
Particularly, the study of dihedral θ3 (Figure 1) is important due to the high energetic barrier of amide group (HN–C=O) [43]. In line with this, it is worthy to assume that two conformations (s-trans and s-cis) must coexist (Figure 2) when the energetic interconversion between them is about 20–22 kcal∙mol−1. The terminology of s-cis and s-trans (s means to conformation into N–C single bond) was used to describe the relative positioning between the R (RNH) and C=O groups when they are in the same direction (s-cis) or even in an opposite alignment (s-trans), respectively. From RPESS protocol, the lowest energy conformation was selected, and after undergoing an optimization procedure at the B3LYP/6-311++G(d,p) level of theory, the conformation symbolized as 1C was obtained (Figure 3). As such, 1C shows both RN and C=O groups in s-cis position. Moreover, the conformation 1T (Figure 4) was also obtained from the geometry described previously [6] through the optimization at B3LYP/6-311++G(d,p) level of theory. Note that the groups (R–N and C=O) are in s-trans position in 1T.
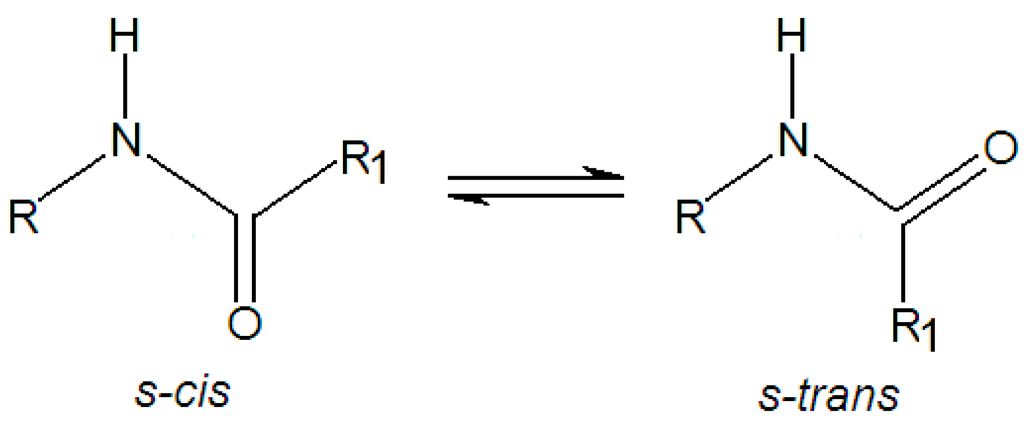
Figure 2.
Conformations s-cis and s-trans.
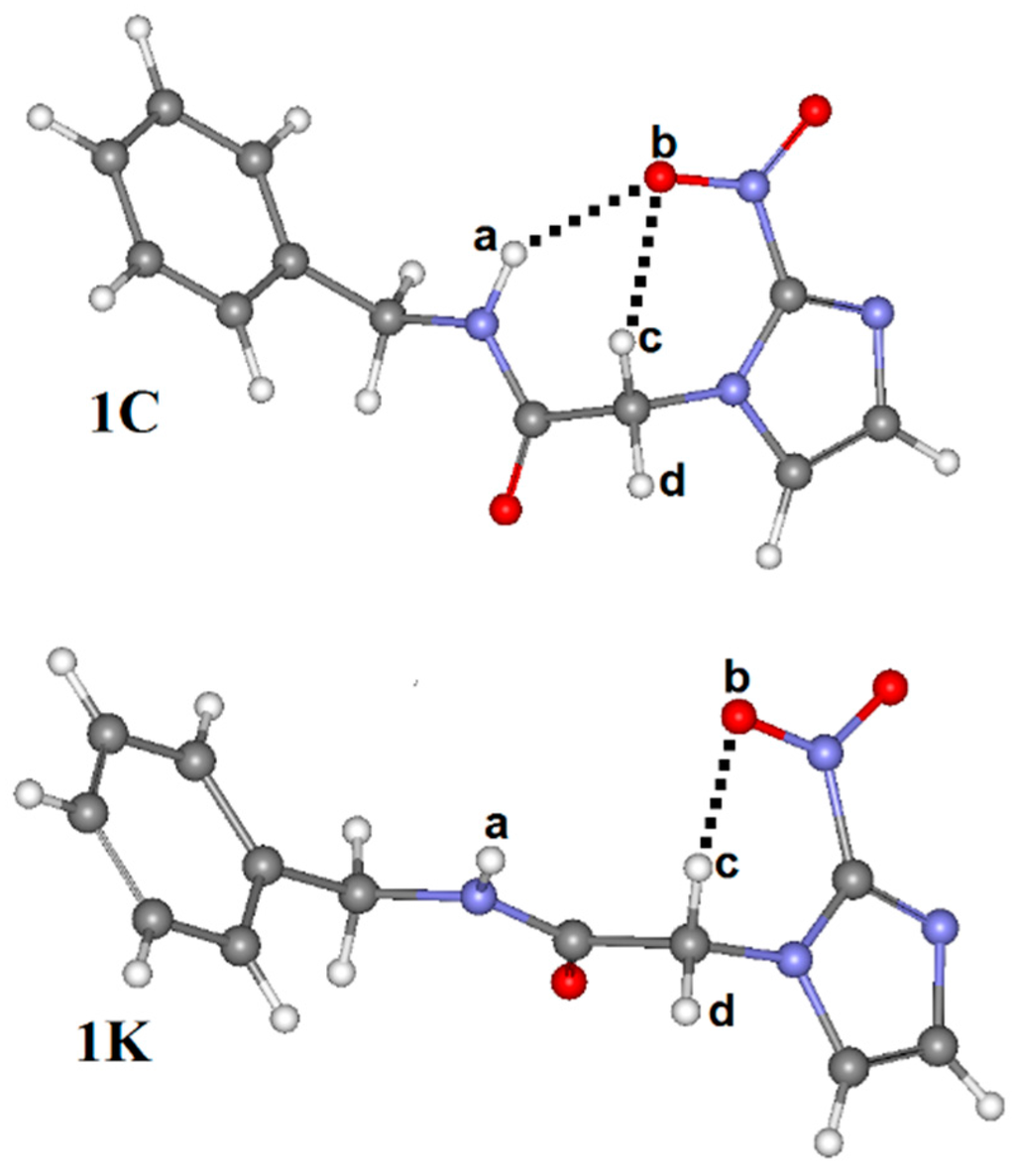
Figure 3.
Conformations (1C and 1K) of benznidazole.
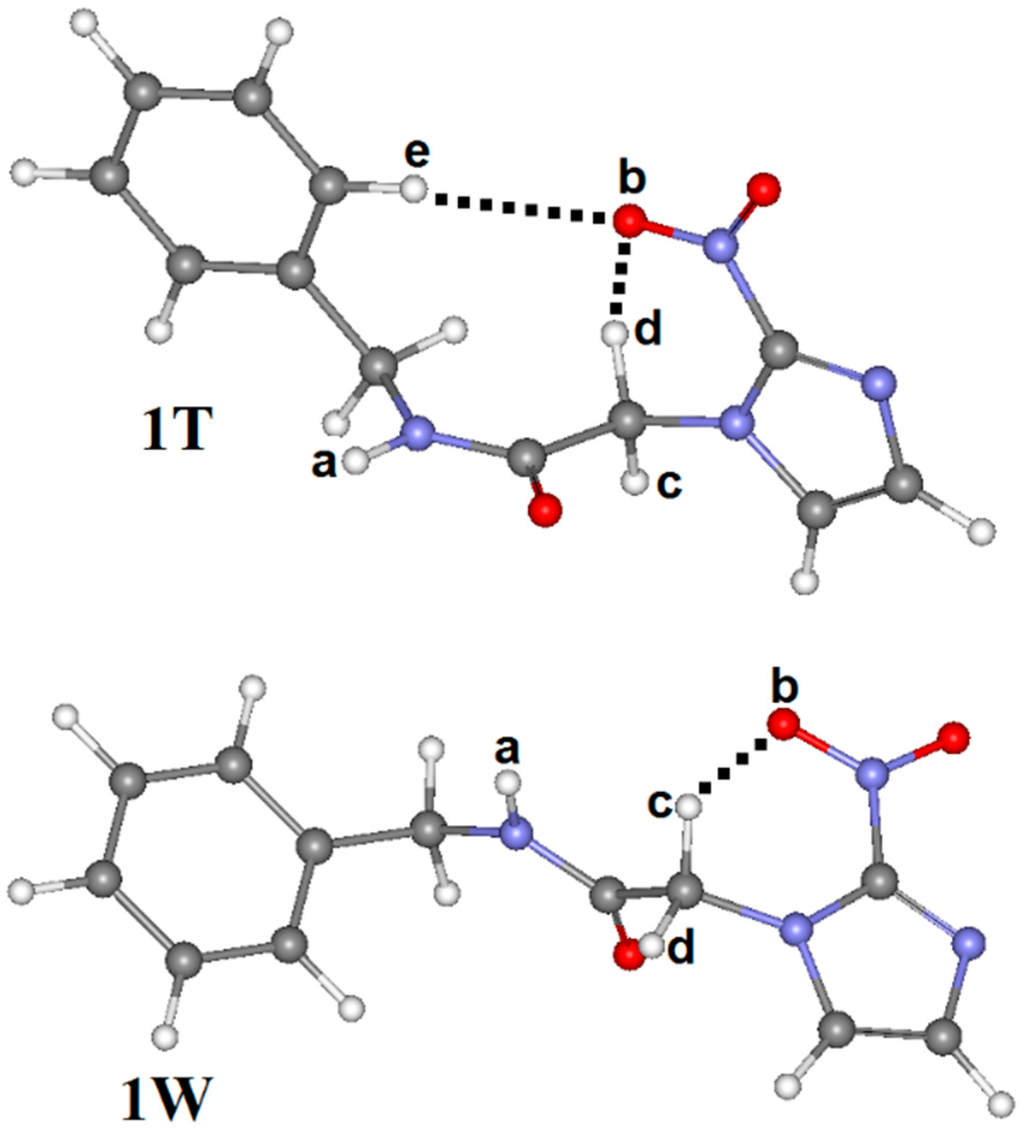
Figure 4.
Conformations (1T and 1W) of benznidazole.
By analyzing Figure 3, it can be seen that 1C leads to the formation of two intramolecular hydrogen bonds (N–Ha∙∙∙Ob=N) and (C–Hc∙∙∙Ob=N) with respective distance values of 2.125 Å and 2.287 Å, which contributes to a substantial stabilization of this conformation. In 1T, however, the C–Hd∙∙∙Ob=N and C–He∙∙∙Ob=N hydrogen bonds were considered, but their lengths of 2.347 Å and 2.562 Å are longer than those results of 1C. From a structural viewpoint, this discovery provides solid evidence about the preferential conformation of 1C, s-cis, as has already been demonstrated in many similar works [21,44,45,46,47]. Regarding 1K and 1W (see Figure 3 and Figure 4), B3LYP/6-311++G(d,p) calculations revealed that 1K presents the same intramolecular C–Hc∙∙∙Ob=N hydrogen bond of 1C, once the distance values of 2.340 Å (1K) differ slightly from the value of 2.287 Å (1C). At last, 1W presents only one hydrogen bond, C–Hc∙∙∙Ob=N, whose distance value of 2.468 Å is much longer than C–Hd∙∙∙Ob=N and N=Ob∙∙∙C–He. Although it should be expected that the most stable conformation must be the solvated structure [48,49], geometrically the strength of the intramolecular N=Ob∙∙∙Hc–C hydrogen bond is not sufficient evidence for that. The strongest statement regarding the hydrogen bonds’ formation, either intra or intermolecular, actually has its basis in the deformations of the bond lengths that compose it.
Besides the intramolecular hydrogen bonds, Table 1 also enumerates the bond length results of the donors X–H (N–Ha, C–Hc, C–He and C–Hd) and acceptors Y (Ob=N) of protons. Although a precise measurement of the bond length variation is not allowed because the monomer configuration of the proton donors cannot be attained, it can be seen that the HBond distances are not in agreement with the bond length variations of X–H as well as of Y, whose values are quite similar. Regarding the characterization of the infrared vibration modes as one of the preconditions to recognize the most stable conformation, the values of stretch frequencies and absorption intensities are invariable, even though a slight difference of 97.3 (1C) and 107.6 km∙mol−1 (1K) for IN–Ha has been observed. Nevertheless, the new vibration modes could not be unveiled, although by taking into account the long values of the HBond distances beyond 2.000 Ǻ, their stretch frequencies and absorption intensities must be active in an infrared region lower than 50 cm−1.

Table 1.
Values of HBond distances and bond lengths of donors and acceptors of protons in the 1C, 1T, 1K and 1W conformations of benznidazole.
3.2. PCM Calculations
The stabilization of 1C, 1T and 1W can be discussed on the basis of thermodynamic properties, whose results organized in Table 2 represent the sum of electronic and zero-point energies (ε0 + ZPE), sum of electronic and thermal free energies (ε0 + Gcorr) and ΔG, obtained from the difference between (e0 + Gcorr) for conformation and the corresponding value for 1C. The PCM calculations were performed on 1C, although this procedure has been based on the X-Ray crystal structure (1K), which both culminated with the development of the 1W conformation. Regarding Figure 3, there is less tendency to form an intramolecular hydrogen bond, which can be explained by the solvent effect in the stabilization of benznidazole, although the structural interaction strength recommended by the RC–Hc∙∙∙Ob=N contact points out that 1K is slightly more strongly bonded than 1C. The value of −23.17 kcal∙mol−1 may mean that 1W is the most stable structure. However, this HBond presents a distance of 2.468 Ǻ, which, being one of the longest interactions, which seems to conflict with the stabilization statement presented above. It is important to point out that the solvent effect should reveal electronic energy values that may differ from a hypersurface investigated in the gas phase. Thus, the displayed geometry is consistent with the weakening of the intramolecular interaction favoring the influence of the external environment (aqueous). Really, the solvent effect should be carefully used to explain the additional molecular stabilization, depending on the type of system studied [50], including drug molecules that act in predominantly aqueous environments.
Table 2.
Thermochemical parameters calculated at B3LYP/6-311++G(d,p) level (298.15 K and 1 atm) obtained from gas phase and PCM solvent model a.
3.3. QTAIM Parameters and Intramolecular Hydrogen Bonds
Bond pathways, Bond Critical Points (BCP), and values of ρ followed by ∇2ρ were characterized in terms of the QTAIM topological integrations of the electronic density. We can see, in Figure 5 and Figure 6, the presence of a Bond Critical Point (BCP) between (N–Ha∙∙∙Ob=N), indicating the formation of intramolecular hydrogen bonds, corroborating therefore the formulated hypothesis in this work. The same conformation shows a BCP between (C–Hc∙∙∙Ob=N) groups, forming a ring of six members [9]. The participation of the C–H group in intramolecular hydrogen bonds has been established [51]. In our current case, the simultaneous positions of the α-carbonyl and α-nitrogen-hydrogen can contribute to a peculiar acidity of C–H and also lead to the formation of an intramolecular hydrogen bond. The values of ρ and ∇2ρ that characterize these interactions can be visualized in Table 3. By taking into account the virial theorem [17], G and U idealize the kinetic (always positive) and potential (always negative) energy density functions. Then, positive values of ∇2ρ indicate a depletion of electronic potential energy density at BCP in favor of the kinetic energy because it outweighs the potential one, showing a concentration of electronic charge in separated nuclei [51,52,53].
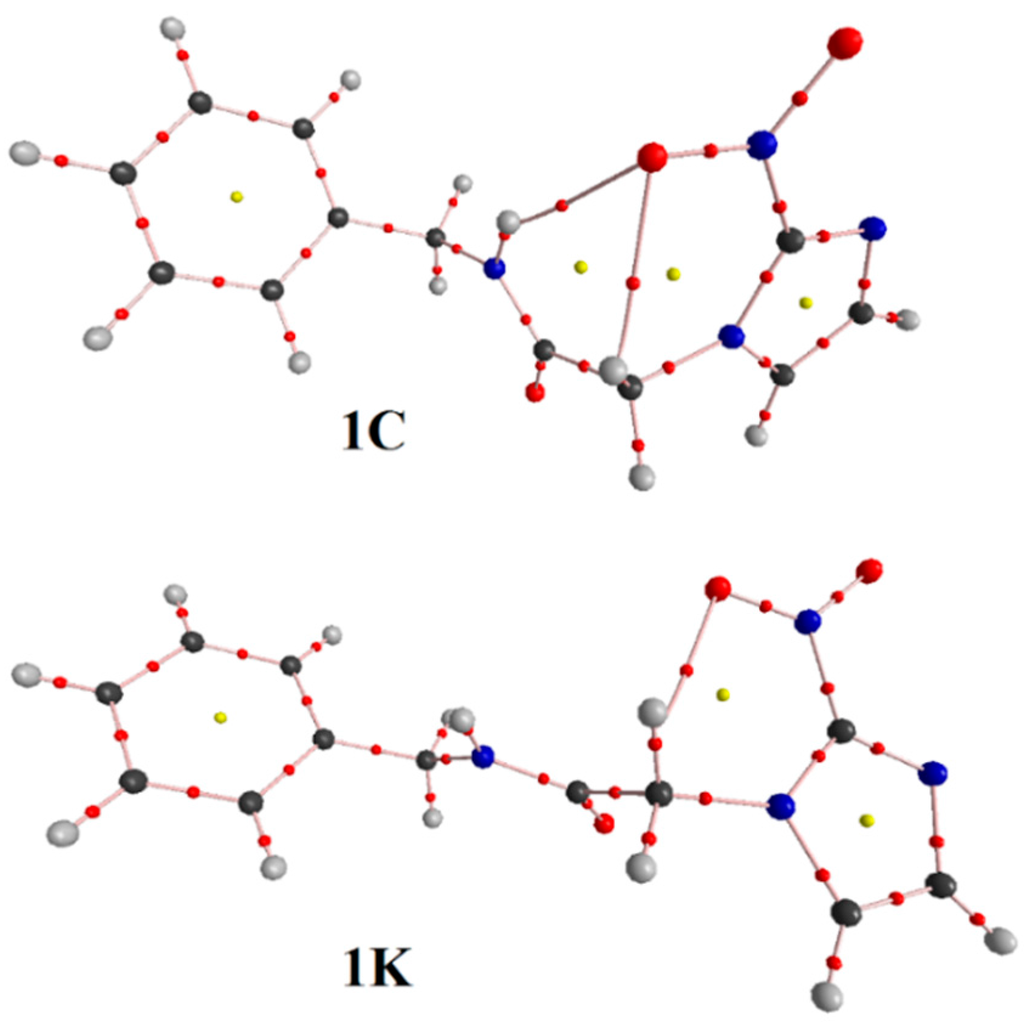
Figure 5.
BP and BCP for conformations (1C and 1K) of benznidazole.
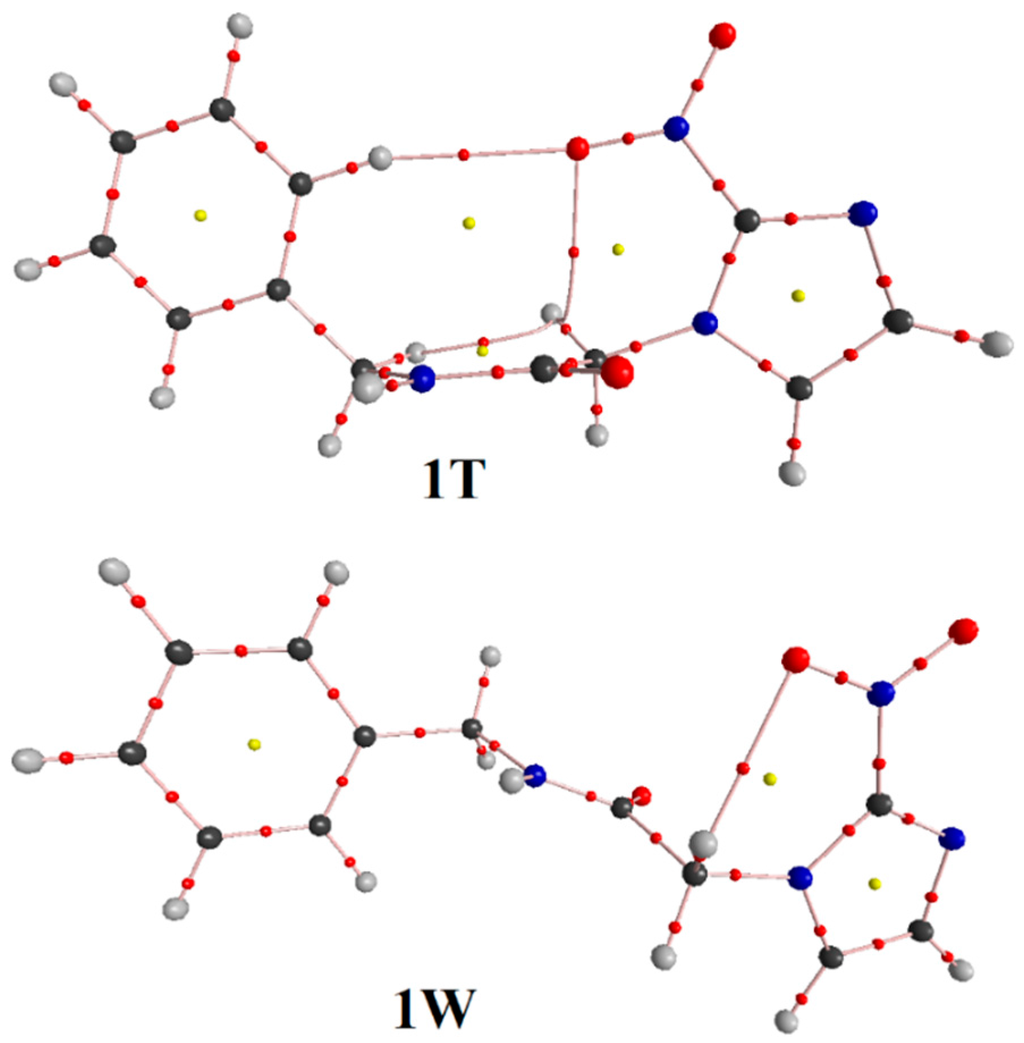
Figure 6.
BP and BCP for conformations (1T and 1W) of benznidazole.
Table 3.
Topological parameters obtained from QTAIM calculations.
These values also indicate intramolecular hydrogen bonds with a significant covalent character, as argued by Gilli et al. [54] and Siskos et al. [55]. All these considerations can also be visualized in terms of the Resonance Assisted Hydrogen Bond phenomenon (RAHB) [56,57], in which the π electrons of the pirazole system are in resonance with the NO2 group, increasing the electron density at the BCP and decreasing the bond lengths, that are considerably smaller than sum of van der Walls radii. On the other hand, it is important to be highlighted that the values of ∇2ρ, ρ presented here are much smaller than the ones obtained at BCP for common covalent interactions.
Regarding the interaction strength, the −G/U ratio is a qualitative manner to express the appearing of covalent effect [32]. According to values of 0.733 and 0.738 gathered in Table 3, the C–Hc···Ob=N intramolecular hydrogen bonds behave with a trend of covalence in 1K and 1W, although the electronic density values are not the largest, and the bond lengths are not the shortedones, as demonstrated in Figure 7. This can be stated once these results are smaller than 1.0 [32,58]. In regards to the remaining hydrogen bonds, all of them are non-covalent. However, besides the hydrogen bonds, 1T also reveals the formation of a dihydrogen bond [59,60,61], namely C–Hd···Hf–C, wherein by means of the values of ρ and ∇2ρ as well as −G/U, this interaction is also non-covalent.
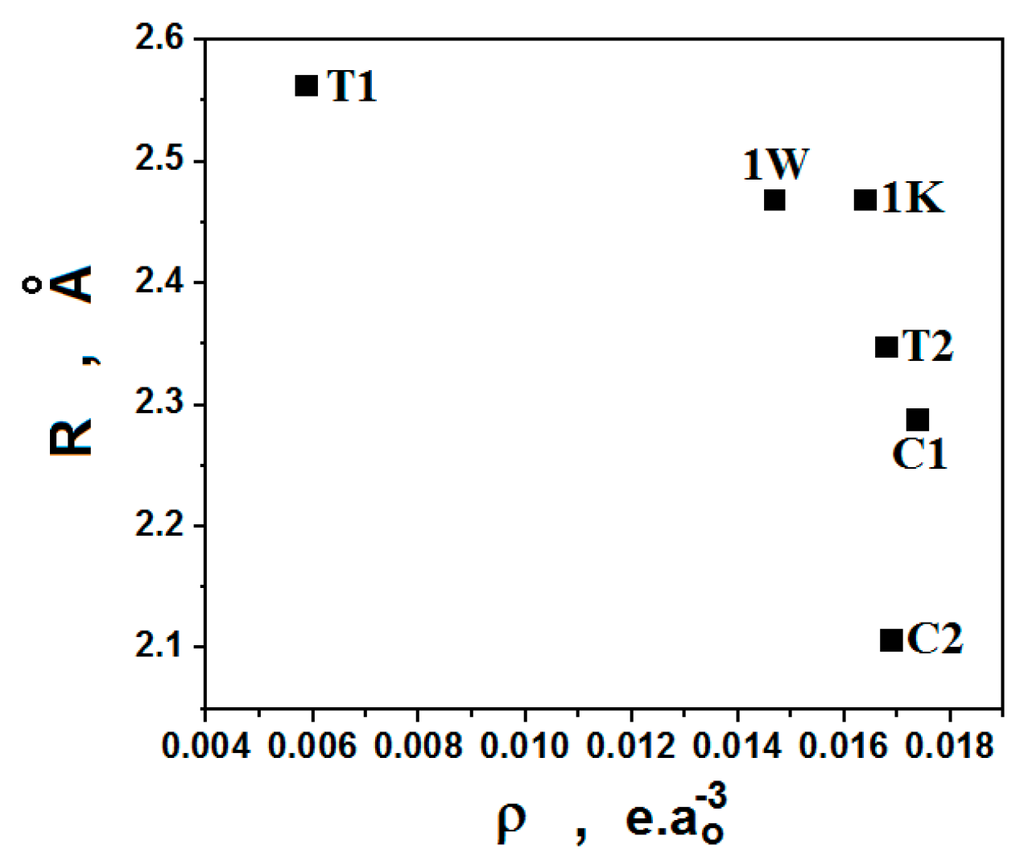
Figure 7.
Relationship between the distance values of intramolecular hydrogen bonds and QTAIM electronic density amounts.
4. Conclusions
In this work, we observed that intramolecular hydrogen bonds can control the conformations of benznidazole. The structural parameters embodied as bond lengths and intermolecular distances revealed unsystematic tendencies in comparison with the stabilization criteria. Also, the new vibration modes of the intramolecular hydrogen bonds could not be unveiled, and in addition, the difficulties in identifying the frequency shifts of the proton donors prevent us from correlating this parameter with the stabilization criteria. The QTAIM calculations were able to characterize classical and non-classical intramolecular hydrogen bonds by means of the values of electronic density and Laplacian. Also, the appearance of a partial covalent character was observed in the C–Hc∙∙∙Ob=N links of 1K and 1W. Approaches considering solvent or gas phase effect can lead to deviations of the crystal structure, but they are all s-cys and not s-trans. This must be taken into account in studies of the structure-activity relationships of benznidazole.
Acknowledgments
The authors thank “Coordenação de Aperfeiçoamento de Pessoal de Nível Superior” (CAPES) and “Conselho Nacional de Desenvolvimento Científico e Tecnológico” (CNPq) for financial support.
Author Contributions
Mário L. A. A. Vasconcellos and Edilson B. Alencar Filho conceived and designed the experiments; Boaz G. Oliveira, Mário L. A. A. Vasconcellos and Edilson B. Alencar Filho performed the experiments; Boaz G. Oliveira, Mário L. A. A. Vasconcellos and Edilson B. Alencar Filho analyzed the data; Boaz G. Oliveira, Mário L. A. A. Vasconcellos and Edilson B. Alencar Filho contributed reagents/materials/analysis tools; Boaz G. Oliveira, Mário L. A. A. Vasconcellos and Edilson B. Alencar Filho wrote the paper. Authorship must be limited to those who have contributed substantially to the work reported.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Thomas, G. Medicinal Chemistry: An Introduction; John Wiley and Sons: Chichester, UK, 2000. [Google Scholar]
- Pitta, M.G.R.; Silva, A.C.A.; Neves, J.K.A.L.; Silva, P.G.; Irmão, J.I.; Malagueño, E.; Santana, J.V.; Lima, M.C.A.; Galdino, S.L.; Pitta, I.R.; et al. New imidazolidinic bioisosters: Potencial candidates for antischistosomal drugs. Mem. Inst. Oswaldo Cruz 2006, 101, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Goodell, J.R.; Ougolkov, A.V.; Hiasa, H.; Kaur, H.; Remmel, R.; Billadeau, D.D.; Ferguson, D.M. Acridine-based agents with Topoisomerase II activity inhibit pancreatic cancer cell proliferation and induce apoptosis. J. Med. Chem. 2008, 51, 179–182. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: http://www.who.int/whr/2004/en/ (accessed on 14 September 2009).
- Tarleton, R.L.; Reithinger, R.; Urbina, J.A.; Kitron, U.; Gurtler, R.E. The challenges of Chagas disease—Grim outlook or glimmer of hope? PLoS Med. 2007, 4, 1852–1957. [Google Scholar] [CrossRef] [PubMed]
- Tonin, L.T.D.; Barbosa, V.A.; Bocca, C.C.; Ramos, E.R.F.; Nakamura, C.V.; Costa, W.F.; Basso, E.A.; Nakamura, T.U.; Sarragiotto, M.H. Comparative study of the trypanocidal activity of the methyl 1-nitrophenyl-1,2,3,4-9H-tetrahydro-β-carboline-3-carboxylate derivatives and benznidazole using theoretical calculations and cyclic voltammetry. Eur. J. Med. Chem. 2008, 44, 1745–1750. [Google Scholar] [CrossRef] [PubMed]
- Soares-Sobrinho, J.L.; Cunha-Filho, M.S.S.; Rolim-Neto, P.J.; Torres-Labandeira, J.J.; Dacunha-Marinho, B. Benznidazole. Acta Crystallogr. E 2008, 64, o634. [Google Scholar] [CrossRef] [PubMed]
- Höltje, H.-H.; Sippl, W.; Rognan, D.; Folkers, G. Molecular Modeling: Basic Principles and Applications; Wiley-VHC: Weinhelm, Germany, 2003. [Google Scholar]
- Filho, E.B.A.; Ventura, E.; do Monte, A.S.; Oliveira, B.G.; Junior, C.G.L.; Rocha, G.B.; Vasconcellos, M.L.A.A. Synthesis and conformational study of a new class of highly bioactive compounds. Chem. Phys. Lett. 2007, 449, 336–340. [Google Scholar] [CrossRef]
- Santos Filho, J.M.; Leite, A.C.L.; Oliveira, B.G.; Moreira, D.R.M.; Lima, M.S.; Soares, M.B.P.; Leite, L.F.C.C. Design, synthesis and cruzain docking of 3-(4-substituted-aryl)-1,2,4-oxadiazole-N-acylhydrazones as anti-Trypanosoma cruzi agents. Bioorg. Med. Chem. 2009, 17, 6682–6691. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.G.; Araujo, R.C.M.U.; Carvalho, A.B.; Ramos, M.N. DFT calculations on the cyclic ethers hydrogen-bonded complexes: Molecular parameters and the non-linearity of the hydrogen bond. Spectrochim. Acta A 2007, 68, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.G.; Leite, L.F.C.C. A quantum chemical study of red-shift and blue-shift hydrogen bonds in bimolecular and trimolecular methylhydrazine-hydrate complexes. J. Mol. Struct. THEOCHEM 2009, 915, 38–42. [Google Scholar] [CrossRef]
- Oliveira, B.G.; Araujo, R.C.M.U.; Carvalho, A.B.; Ramos, M.N. A chemometrical study of intermolecular properties of hydrogen-bonded complexes formed by C2H4O∙∙∙HX and C2H5N∙∙∙HX with X = F, CN, NC, and CCH. J. Mol. Model. 2009, 15, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.G.; Araujo, R.C.M.U. Theoretical aspects of binary and ternary complexes of aziridine⋯ammonia ruled by hydrogen bond strength. J. Mol. Model. 2012, 18, 2845–2854. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.G.; Araújo, R.C.M.U.; Ramos, M.N. Evidence for blue-shifting and red-shifting effects in the C2H4⋯HCF3, C2H3(CH3)⋯HCF3 and C2H2(CH3)2∙∙∙HCF3 complexes: π and improper-π hydrogen bonds. J. Mol. Struct. THEOCHEM 2010, 944, 168–172. [Google Scholar] [CrossRef]
- Oliveira, B.G.; Zabardasti, A.; Goudarziafshar, H.; Salehnassaj, M. The electronic mechanism ruling the dihydrogen bonds and halogen bonds in weakly bound systems of H3SiH∙∙∙HOX and H3SiH∙∙∙XOH (X = F, Cl, and Br). J. Mol. Model. 2015, 21, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Cortés-Guzmán, F.; Bader, R.F.W. Complementarity of QTAIM and MO theory in the study of bonding in donor-acceptor complexes. Coord. Chem. Rev. 2005, 249, 633–662. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Šafář, P.; Žúžiová, J.; Marchalín, Š.; Prónayová, N.; Švorc, Ľ.; Vrábel, V.; Šesták, S.; Rendić, D.; Tognetti, V.; Joubert, L.; et al. Combined chemical, biological and theoretical DFT-QTAIM study of potent glycosidase inhibitors based on quaternary indolizinium salts. Eur. J. Org. Chem. 2012, 2012, 5498–5514. [Google Scholar] [CrossRef]
- LaPointe, S.M.; Farrag, S.; Bohrquez, H.J.; Boyd, R.J. QTAIM study of an alpha-helix hydrogen bond network. J. Phys. Chem. B 2009, 113, 10957–10964. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.G.; Lima, M.C.A.; Pitta, I.R.; Galdino, S.L.; Hernandes, M.Z. A theoretical study of red-shifting and blue-shifting hydrogen bonds occurring between imidazolidine derivatives and PEG/PVP polymers. J. Mol. Model. 2010, 16, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Rozas, I.; Alkorta, I.; Elguero, J. Intramolecular hydrogen bonds in ortho-substituted hydroxybenzenes and in 8-susbtituted 1-hydroxynaphthalenes: Can a methyl group be an acceptor of hydrogen bonds? J. Phys. Chem. A 2001, 105, 10462–10467. [Google Scholar] [CrossRef]
- Zevallos, J.; Toro-Labbé, A.; Mó, O.; Yáñez, M. The role of intramolecular hydrogen bonds versus other weak interactions on the conformation of hyponitrous acid and its mono- and dithio-derivatives. Struct. Chem. 2005, 16, 295–303. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Małecka, M. Intramolecular H-Bonds: DFT and QTAIM studies on 3-(aaminomethylene)pyran-2,4-dione and its derivatives. J. Phys. Chem. A 2006, 110, 11847–11854. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, M.M.; Gadre, S.R.; Bartolotti, L.J. Estimation of intramolecular hydrogen bond energy via molecular tailoring approach. J. Phys. Chem. A 2006, 110, 12519–12523. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, M.M.; Suresh, C.H.; Gadre, S.R. Intramolecular hydrogen bond energy in polyhydroxy systems: A critical comparison of molecular tailoring and isodesmic approaches. J. Phys. Chem. A 2007, 111, 6472–6480. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, J.; Clausen, H.F.; Poulsen, R.D.; Overgaard, J.; Schiøtt, B. Short strong hydrogen bonds in 2-acetyl-1,8-dihydroxy-3,6-dimethylnaphthalene: An outlier to current hydrogen bonding theory? J. Phys. Chem. A 2007, 111, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, M.M.; Gadre, S.R. Estimation of N−H∙··O=C intramolecular hydrogen bond energy in polypeptides. J. Phys. Chem. A 2009, 113, 7927–7932. [Google Scholar] [CrossRef] [PubMed]
- Fuster, F.; Grabowski, S.J. Intramolecular hydrogen bonds: The QTAIM and ELF characteristics. J. Phys. Chem. A 2011, 115, 10078–10086. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, M.M.; Bartolotti, L.J.; Gadre, S.R. Intramolecular hydrogen bond energy and cooperative interactions in α-, β-, and γ-cyclodextrin conformers. J. Comput. Chem. 2011, 32, 2996–3004. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.G. Structure, energy, vibrational spectrum, and Bader’s analysis of π∙∙∙H hydrogen bonds and H−δ∙∙∙H+δ dihydrogen bonds. Phys. Chem. Chem. Phys. 2013, 15, 37–79. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, C. Solvents and Solvent Effects in Organic Chemistry; Wiley-VCH: Weinheim, Germany, 1988. [Google Scholar]
- Tomasi, J.; Persico, M. Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Yasuda, T.; Ikawa, S.-I. On the dielectric continuum solvent model for theoretical estimates of the conformational equilibrium of molecules with an intramolecular hydrogen bond. Chem. Phys. 1998, 238, 173–178. [Google Scholar] [CrossRef]
- Abkowicz-Bieńko, A.; Biczysko, M.; Latajka, Z. Solvent effect on hydrogen bonded ammonia-hydrogen halide complexes: Continuum medium versus cluster models. Comput. Chem. 2000, 24, 303–309. [Google Scholar] [CrossRef]
- Oliveira, B.G.; Araújo, R.C.M.U.; Carvalho, A.B.; Ramos, M.N.; Hernandes, M.Z.; Cavalcante, K.R. A theoretical study of the solvent effects in ethylene oxide: Hydrofluoric acid complex using continuum and new discrete models. J. Mol. Struct. THEOCHEM 2007, 802, 91–97. [Google Scholar] [CrossRef]
- López-Vallejo, F.; Medina-Franco, J.L.; Hernández-Campos, A.; Rodríguez-Morales, S.; Yépez, L.; Cedillo, R.; Castillo, R. Molecular modeling of some 1H-benzimidazole derivatives with biological activity against Entamoeba histolytica: A comparative molecular field analysis study. Bioorg. Med. Chem. 2007, 15, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1933, 98, 1372–1377. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03 Revision D.02; Gaussian Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Biegler-König, F. AIM 2000 1.0; University of Applied Sciences: Bielefeld, Germany, 2000. [Google Scholar]
- Wang, W.; Pu, X.; Zheng, W.; Wong, N.-B.; Tian, A. Hyperconjugation versus intramolecular hydrogen bond: Origin of the conformational preference of gaseous glycine. Chem. Phys. Lett. 2003, 370, 147–153. [Google Scholar] [CrossRef]
- Shagidullin, R.R.; Chernova, A.V.; Shagidullin, R.R. Intramolecular hydrogen bonds and conformations of the 1,4-butanediol molecule. Rus. Chem. Bull. 1993, 42, 1505–1510. [Google Scholar] [CrossRef]
- Lee, H.-J.; Jung, H.-J.; Kim, J.K.; Park, H.-M.; Lee, K.-B. Conformational preference of azaglycine-containing dipeptides studied by PCM and IPCM methods. Chem. Phys. 2003, 294, 201–210. [Google Scholar] [CrossRef]
- Hopkins, W.S.; Hasan, M.; Burt, M.; Marta, R.A.; Fillion, E.; McMahon, T.B. Persistent intramolecular C–H···X (X = O or S) hydrogen-bonding in benzyl meldrum’s acid derivatives. J. Phys. Chem. A 2014, 118, 3795–3803. [Google Scholar] [CrossRef] [PubMed]
- Alcântara, A.F.C.; Teixeira, A.F.; Silva, I.F.; Almeida, W.B.; Piló-Veloso, D. NMR Investigation and theoretical calculations of the effect of solvent on the conformational analysis of 4’,7-di-hydroxy-8-prenylflavan. Quim. Nova 2004, 27, 371–377. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Honda, K.; Uchimaru, T.; Mikami, M.; Tanabe, K. The magnitude of the CH/π interaction between benzene and some model hydrocarbons. J. Am. Chem. Soc. 2000, 122, 3746–3753. [Google Scholar] [CrossRef]
- Takahashi, O.; Kohno, Y.; Gondoh, Y.; Saito, K.; Nishio, M. General preference for alkyl/phenyl folded conformations. Relevance of the CH/pi and CH/O interactions to stereochemistry as evidenced by ab Initio MO calculations. Bull. Chem. Soc. Jpn. 2003, 76, 369–374. [Google Scholar] [CrossRef]
- Lithoxoidou, A.T.; Bakalbassis, E.G. PCM study of the solvent and substituent effects on the conformers, intramolecular hydrogen bonds and bond dissociation enthalpies of 2-substituted phenols. J. Phys. Chem. A 2005, 109, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.G.; Araújo, R.C.M.U. Bonding topology, hydrogen bond strength, and vibrational chemical shifts on hetero-ring hydrogen-bonded complexes—Theoretical insights revisited. Can. J. Chem. 2012, 90, 368–375. [Google Scholar] [CrossRef]
- Oliveira, B.G.; Araújo, R.C.M.U.; Carvalho, A.B.; Ramos, M.N. The molecular properties of heterocyclic and homocyclic hydrogen-bonded complexes evaluated by DFT calculations and AIM densities. J. Mol. Model. 2009, 15, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.G.; Araújo, R.C.M.U.; Chagas, F.F.; Carvalho, A.B.; Ramos, M.N. The electronic structure of the C2H4O∙∙∙2HF tri-molecular heterocyclic hydrogen-bonded complex: A theoretical study. J. Mol. Model. 2008, 14, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Gilli, P.; Bertolasi, V.; Ferretti, V.; Gilli, G. Evidence for resonance-assisted hydrogen bonding. 4. Covalent nature of the strong homonuclear hydrogen bond. Study of the O–H--O system by crystal structure correlation methods. J. Am. Chem. Soc. 1994, 116, 909–915. [Google Scholar] [CrossRef]
- Małecka, M. Intramolecular N–H∙∙∙O resonance-assisted hydrogen bonds in crystal structures of oxaphosphinanes and chromones—DFT calculations and AIM analysis. J. Mol. Struct. 2007, 831, 135–143. [Google Scholar] [CrossRef]
- Sanz, P.; Mó, O.; Yáñez, M.; Elguero, J. Bonding in tropolone, 2-aminotropone, and aminotroponimine: No evidence of resonance-assisted hydrogen-bond effects. Chemistry 2008, 14, 4225–4232. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, C.; Sánchez-Sanz, G.; Alkorta, I.; Elguero, J.; Mó, O.; Yáñez, M. Resonance assisted hydrogen bonds in open-chain and cyclic structures of malonaldehyde enol: A theoretical study. J. Mol. Struct. 2013, 1048, 138–151. [Google Scholar] [CrossRef]
- Grabowski, S.J. What is the covalency of hydrogen bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.G.; Araújo, R.C.M.U.; Silva, J.J.; Ramos, M.N. A theoretical study of three and four proton donors on linear HX∙∙∙BeH2∙∙∙HX and bifurcate BeH2∙∙∙2HX trimolecular dihydrogen-bonded complexes with X = CN and NC. Struct. Chem. 2010, 21, 221–228. [Google Scholar] [CrossRef]
- Oliveira, B.G.; Vasconcellos, M.L.A.A.A. B3LYP and QTAIM study of a new proton donor for dihydrogen bonds: The case of the C2H5+∙∙∙nBeH2 complexes (n = 1 or 2). Struct. Chem. 2010, 20, 897–902. [Google Scholar] [CrossRef]
- Oliveira, B.G. Interplay between dihydrogen and alkali–halogen bonds: Is there some covalency upon complexation of ternary systems? Comput. Theor. Chem. 2012, 998, 173–182. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).