3. Experimental Procedures
3.1. General Information for the Synthesis of Compounds (1)–(6)
All reagents were purchased from Sigma Aldrich Co., St Louis, MS, USA. NMR spectra were obtained using a JEOL ECS-400 MHz spectrometer at room temperature unless otherwise stated. Chemical shifts are reported in ppm and referenced via residual solvent resonances to Me
4Si (
1H and
13C). Melting points were taken in open capillary tubes and are uncorrected. Absorption spectra were recorded in acetonitrile (from 6 × 10
−4 to 2 × 10
−5 M at room temperature) on a Cary 300 UV-Vis spectrophotometer. NMR (
Figure S1–S12) and UV-Vis spectra (
Figure S13–S18) for all compounds can be found in the
Supplementary Information.
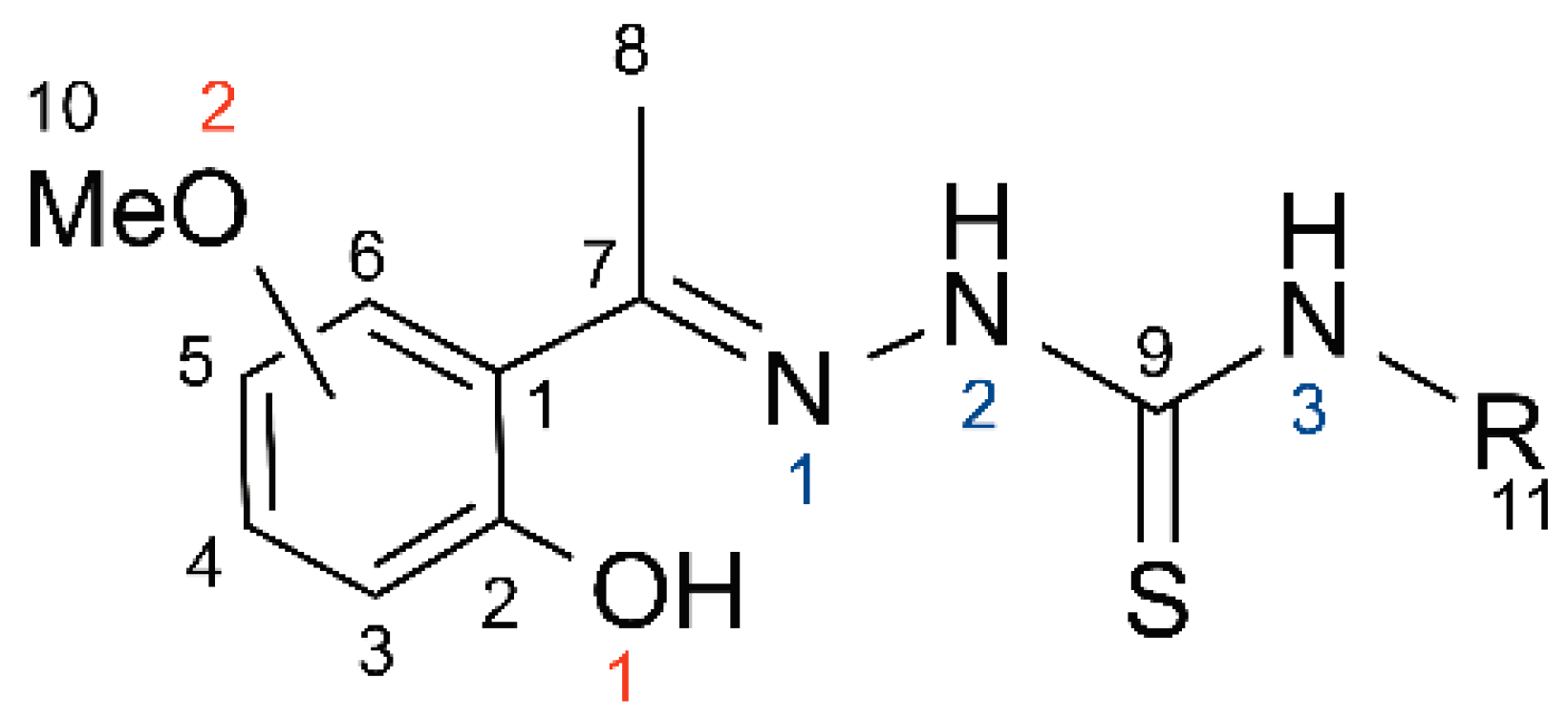
3.2. Synthesis and Spectroscopic Analysis of 2-[1-(2-Hydroxy-5-Methoxyphenyl)Ethylidene]-N-Methyl-Hydrazinecarbothioamide (1)
A 25 mL round bottom flask was charged with 5 mL of 1:1 mixture of ethanol: deionized water solution and then 0.2 g (1 equiv) of starting ketone and 0.17 g (1 equiv) of the thiosemicarbazide were added. The solution was refluxed for 48 h, after dichloromethane (5 mL) and deionized water (5 mL) were added, and the organic layer was separated. The aqueous layer was then extracted with an additional 5 mL of dichloromethane, the organic layers were combined, washed with brine (2 × 5 mL), dried with magnesium sulfate, and the solvent removed in vacuo. The resulting product was crystallized from acetonitrile or dichloromethane layered with hexanes in 58% yield. M.p. 399–402 K. 1H NMR (acetone-d6, δ): 11.7 (br s, 1H, NH), 9.52 (br s, 1H, OH), 7.73 (br s, 1H, NH), 7.5 (d, J = 8.6 Hz, 1H, Ar), 6.47 (d, J = 8.6 Hz, 1H, Ar), 6.42 (d, 1H, Ar), 3.80 (s, 3H, CH3), 3.13 (d, J = 4.7 Hz, 3H, CH3), 2.43 (s, 3H, CH3). 13C {1H} NMR (acetone-d6, δ): 180.4 (C=S), 162.3 (C=N), 160.1 (Ar), 155.1 (Ar), 129.9 (Ar), 113.5 (Ar), 105.9 (Ar), 101.5 (Ar), 54.9 (OMe), 29.6 (Me), 13.6 (Me).
3.3. Synthesis and Spectroscopic Analysis of 2-[1-(2-Hydroxy-5-Methoxyphenyl)Ethylidene]-N-Ethyl-Hydrazinecarbothioamide (2)
A 50 mL round bottom flask was charged with 2′-hydroxy-4′-methoxyacetophenone (0.206 g, 1.24 mmol), and 4-ethyl-3-thiosemicarbazide (0.148 g, 1.24 mmol), 20 mL of methanol and one drop of concentrated sulfuric acid. The colorless solution was then refluxed for 24 h, after which time the resulting yellow solution was transferred to a 125 mL glass separatory funnel and dichloromethane (15 mL) and water (15 mL) were added. The layers were separated and the aqueous layer was extracted with an additional 10 mL of dichloromethane. The organic layer (top) was removed, the aqueous layer was extracted with an additional 10 mL, the organic layers were combined and washed with brine (15 mL), dried with magnesium sulfate, and the solvent was removed in vacuo. The resulting yellow solid was recrystallized from minimal hot acetonitrile and allowed to cool to room temperature yielding yellow crystals (0.260 g, 78%). M.p. 411–416 K. 1H NMR (acetone-d6, δ): 10.6 (br s, 1H, OH), 9.50 (br s, 1H, NH), 7.83 (br s, 1H, NH), 7.05 (d, J = 3 Hz, 1H, Ar), 6.87 (dd, J = 9, 3 Hz, 1H, Ar), 6.80 (d, J = 9 Hz, 1H, Ar), 3.74 (s, 3H, OCH3), 3.69 (m, 2H, CH2CH3), 2.42 (s, 3H, CH3), 1.19 (t, J = 8 Hz, 3H, CH2CH3). 13C {1H} NMR (acetone-d6, δ): 179.5 (C=S), 152.6 (C=N), 152.4 (Ar), 151.6 (Ar), 121.5 (Ar), 117.7 (Ar), 117.0 (Ar), 113.2 (Ar), 55.187 (OMe), 39.2 (Me), 13.9, 13.8.
3.4. Synthesis and Spectroscopic Analysis of 2-[1-(2-Hydroxy-4-Methoxyphenyl)Ethylidene]-N-Ethyl-Hydrazinecarbothioamide (3)
A 50 mL round bottom flask was charged with 2′-hydroxy-4′-methoxyacetophenone (0.206 g, 1.24 mmol), and 4-ethyl-3-thiosemicarbazide (0.148 g, 1.24 mmol), 20 mL of methanol and one drop of concentrated sulfuric acid. The colorless solution was then refluxed for 24 h, after which time the resulting yellow solution was transferred to a 125 mL glass separatory funnel and dichloromethane (15 mL) and water (15 mL) were added. The layers were separated and the aqueous layer was extracted with an additional 10 mL of dichloromethane. The organic layer (top) was removed, the aqueous layer was extracted with an additional 10 mL, the organic layers were combined and washed with brine (15 mL), dried with magnesium sulfate, and the solvent was removed in vacuo. The resulting yellow solid was recrystallized from minimal hot acetonitrile and allowed to cool to room temperature yielding yellow crystals (0.260 g, 78%). M.p. 418–420 K. 1H NMR (CDCl3, δ): 11.20 (s, 1H, NH), 8.54 (s, 1H, OH), 7.35 (d, J = 7 Hz, 1H, Ar), 6.65 (br s, 1H, NH), 6.50-6.46 (m, 2H, Ar), 3.81 (s, 3H, CH3), 3.80–3.68 (m, 2H, CH2), 2.33 (s, 3H, CH3), 1.28 (t, J = 7 Hz, 3H, CH3). 13C {1H} NMR (acetone-d6, δ): 179.5 (C=S), 162.3 (C=N), 160.1 (Ar), 154.9 (Ar), 129.9 (Ar), 113.5 (Ar), 105.9 (Ar), 101.5 (Ar), 54.9 (OMe), 39.3 (Me), 13.8, 13.5.
3.5. Spectroscopic Data for Compounds (4), (5), and (6)
Compound 4: 1H NMR (acetone-d6, δ): 11.7 (br s, 1H, NH), 9.52 (br s, 1H, OH), 7.73 (br s, 1H, NH), 7.5 (d, J = 8.6 Hz, 1H, Ar), 6.47 (d, J = 8.6 Hz, 1H, Ar), 6.42 (d, 1H, Ar), 3.80 (s, 3H, CH3), 3.13 (d, J = 4.7 Hz, 3H, CH3), 2.43 (s, 3H, CH3). 13C {1H} NMR (acetone-d6, δ): 180.4 (C=S), 162.3 (C=N), 160.1 (Ar), 155.1 (Ar), 129.9 (Ar), 113.5 (Ar), 105.9 (Ar), 101.5 (Ar), 54.9 (OMe), 29.6 (Me), 13.6 (Me). M.p. 448–453 K.
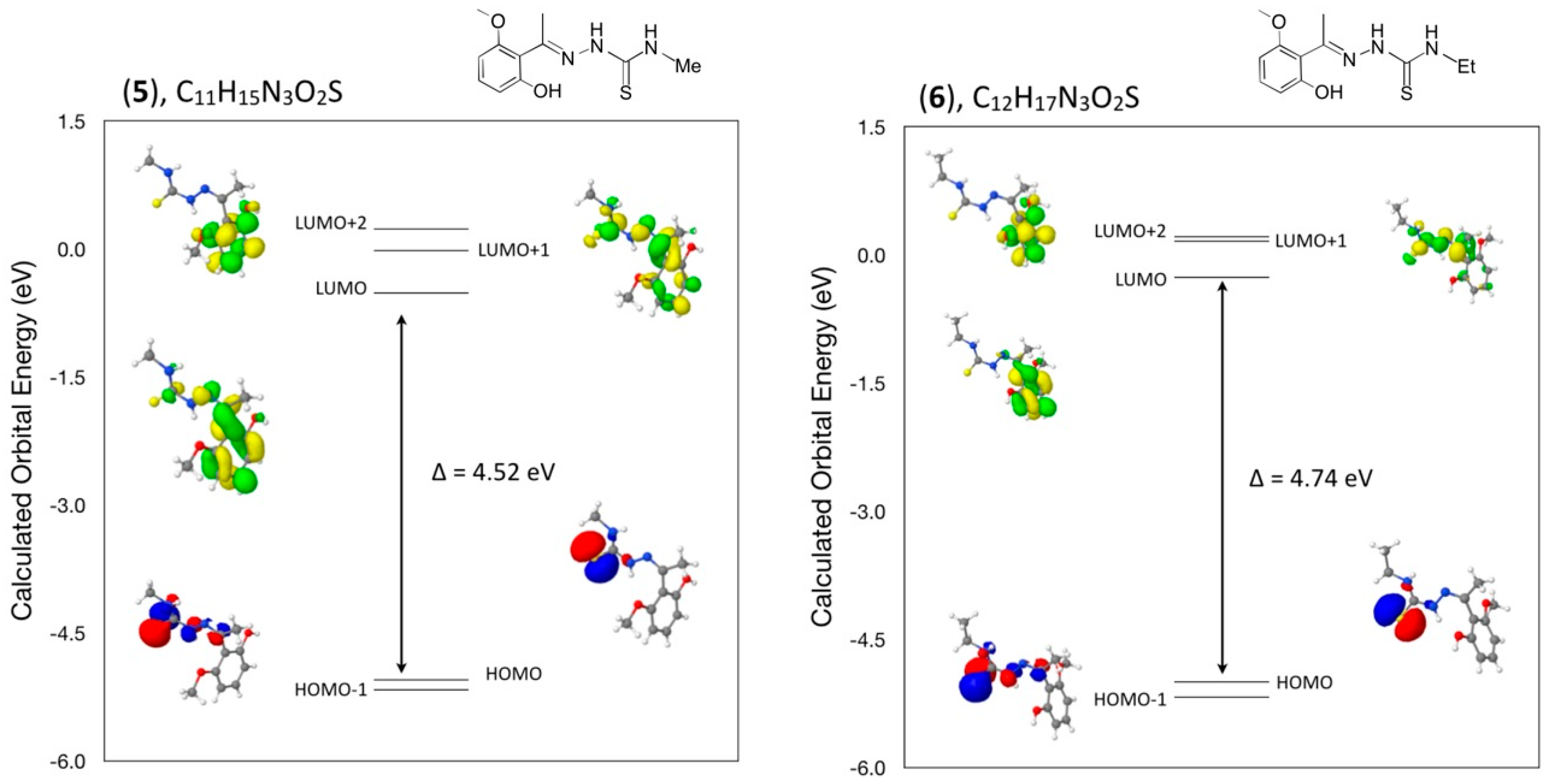
Compound 5: 1H NMR (acetone-d6, δ): 8.9 (br, 1H, NH), 8.23 (br, 1H, OH), 8.03 (br, 1H, NH), 7.25 (t, J = 8 Hz, 1H, Ar), 6.62 (d, J = 8 Hz, 2H, Ar), 3.78 (3H, OMe), 3.08 (d, J = 4 Hz, 3H, NMe), 2.11 (3H, Me). 13C {1H} NMR (acetone-d6, δ): 178.9 (C=S), 157.2 (C=N), 154.5 (Ar), 145.3 (Ar), 131.5 (Ar), 109.4 (Ar), 108.9 (Ar), 102.9 (Ar), 55.4 (OMe), 30.5 (Me), 22.7 (Me). M.p. 415–420 K.
Compound 6: 1H NMR (acetone-d6, δ): 8.97 (br, 1H, NH), 8.18 (br, 1H, OH), 8.04 (br, 1H, NH), 7.25 (t, J = 8 Hz, 1H, Ar), 6.64 (d, J = 8 Hz, 2H, Ar), 3.78 (3H, OMe), 3.67-3.60 (m, 2H, CH2CH3), 2.11 (3H, Me), 1.18 (t, J = 7 Hz, CH2CH3, 3H). 13C {1H} NMR (acetone-d6, δ): 177.8 (C=S), 157.2 (C=N), 154.5 (Ar), 145.4 (Ar), 131.5 (Ar), 109.3 (Ar), 108.8 (Ar), 102.9 (Ar), 55.4 (OMe), 38.8 (Me), 22.6, 14.0. M.p. 458–460 K.
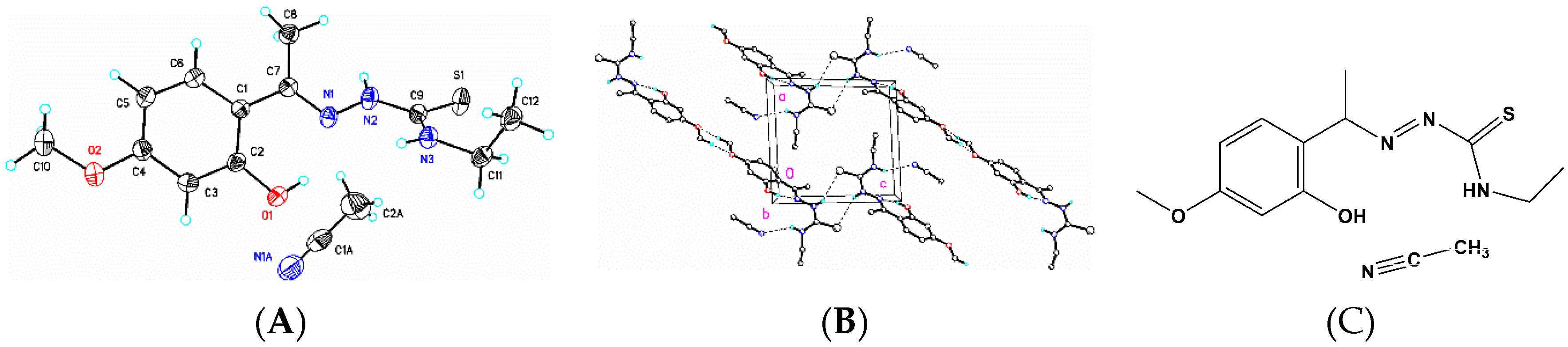
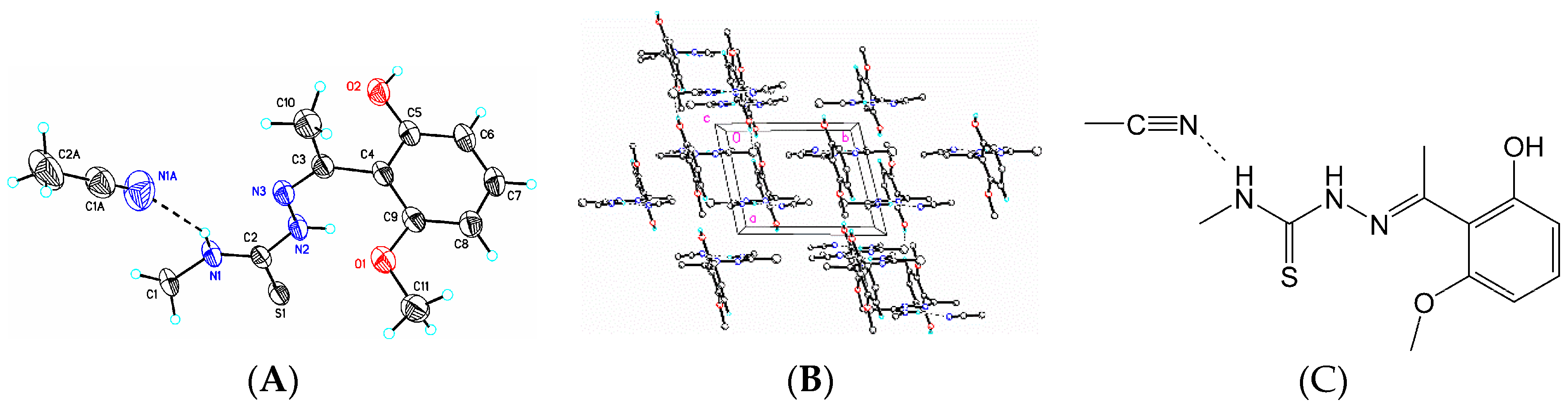
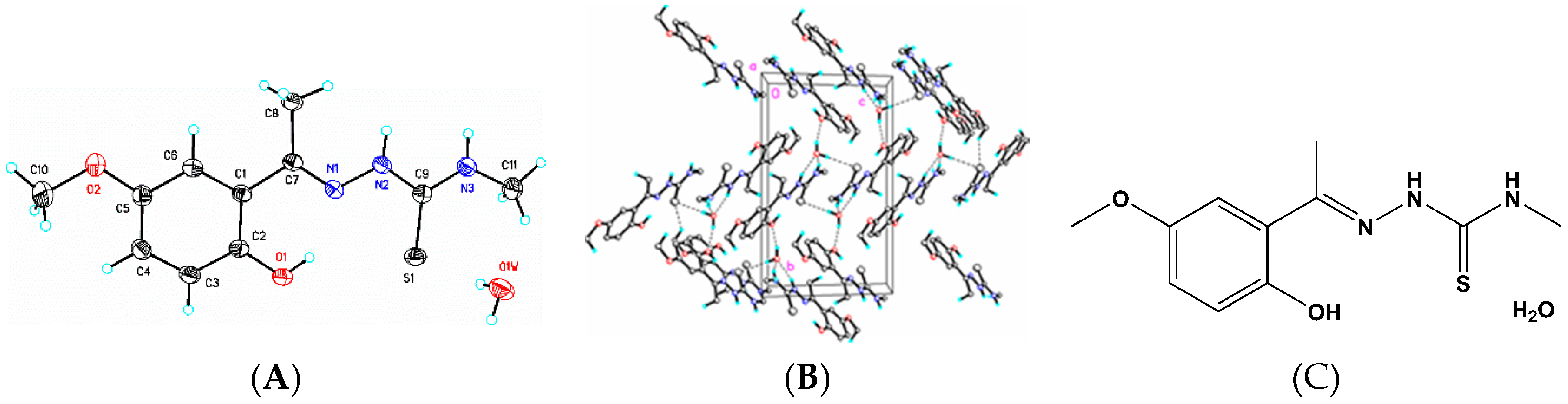
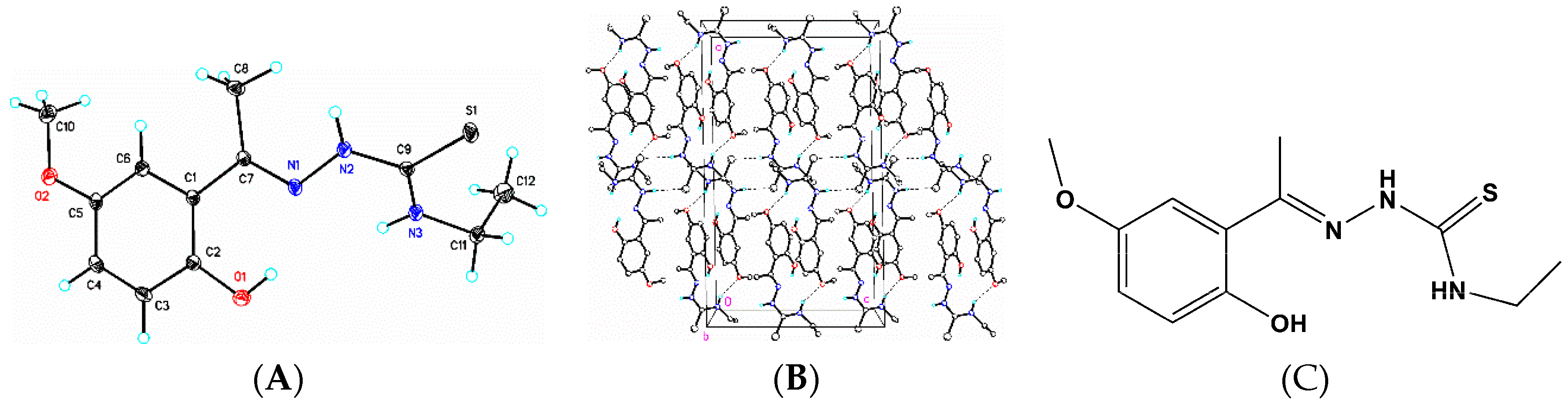
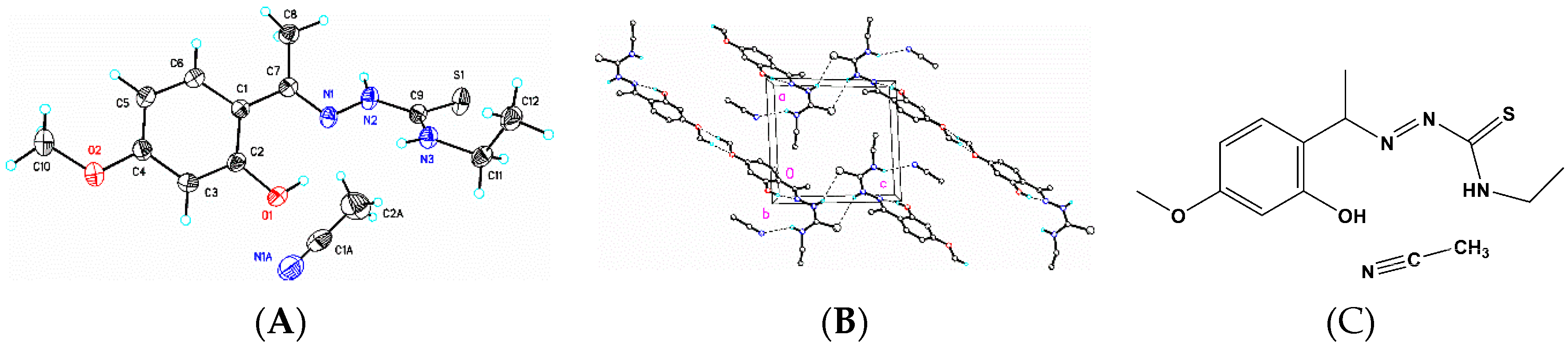
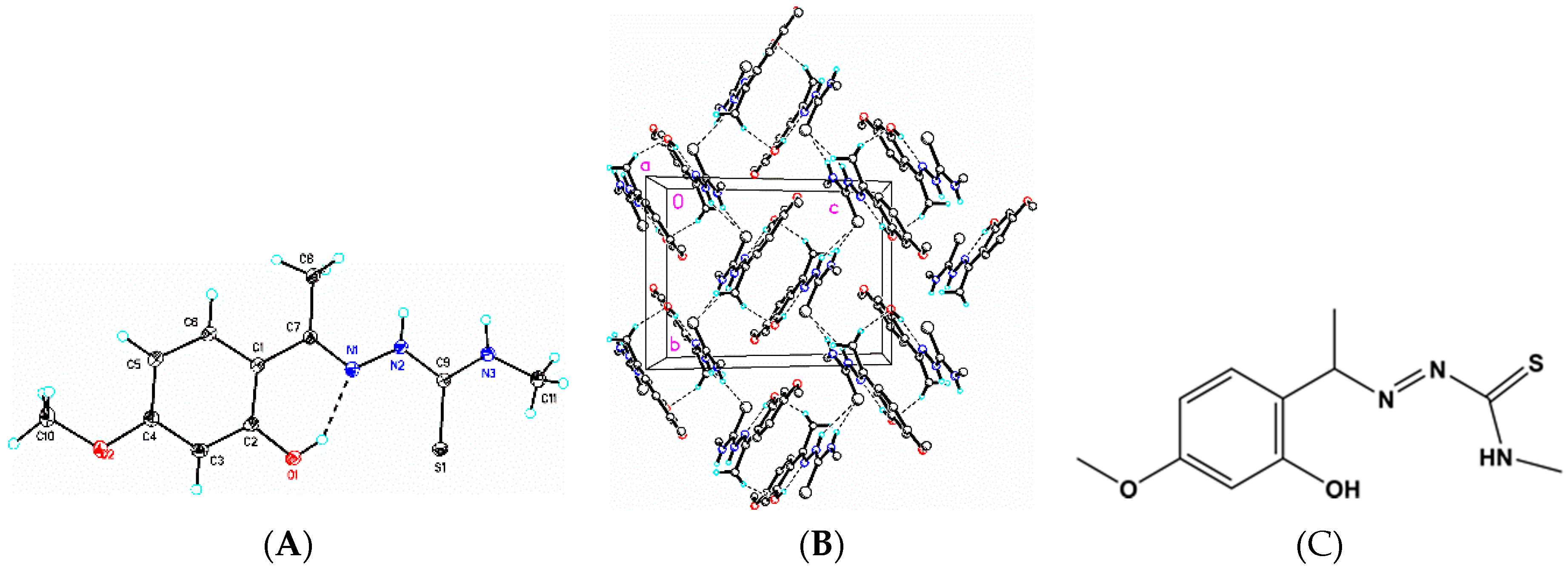
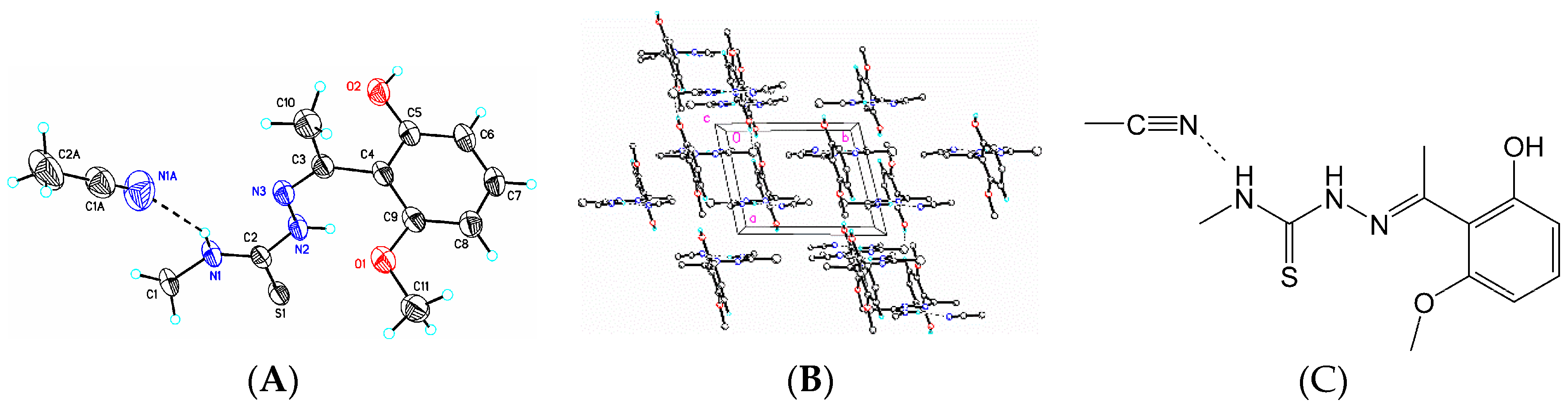
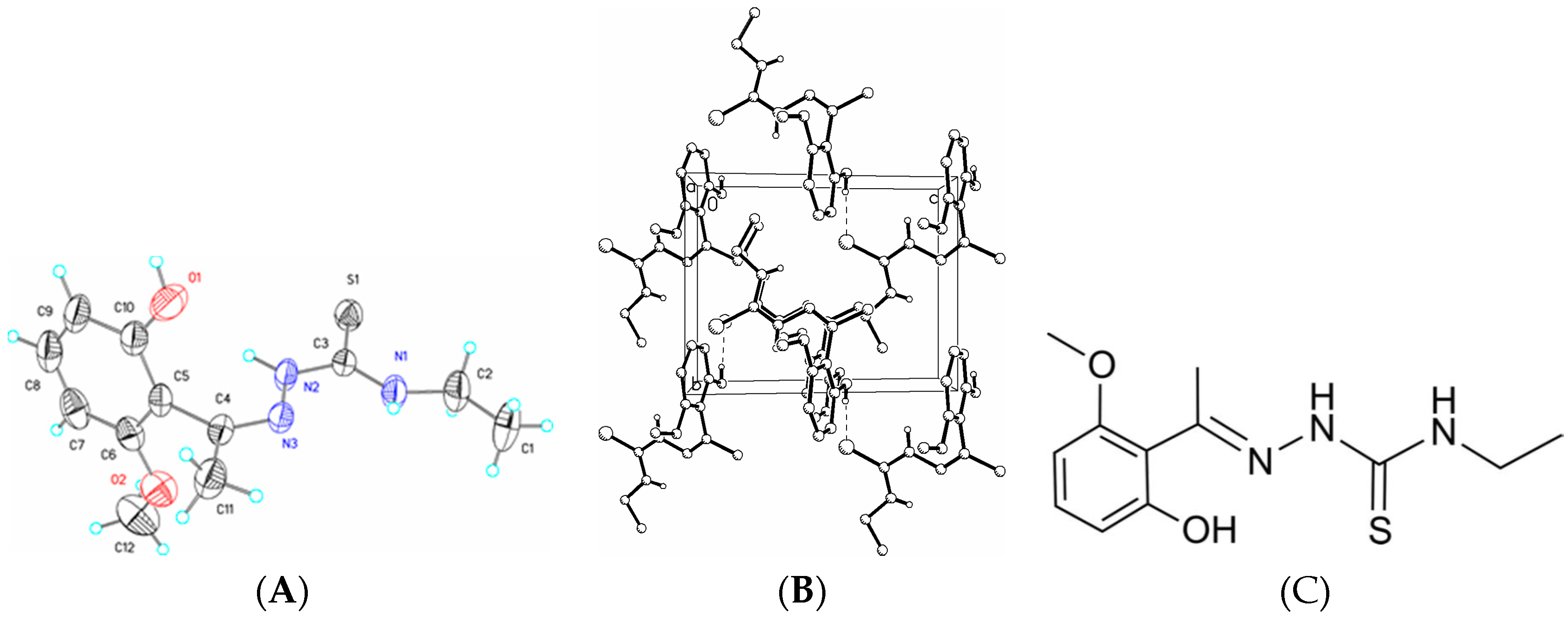
3.6. X-Ray Structure Analysis and Refinement
Individual crystals of compounds (
1), (
2) and (
3) were mounted on a CryoLoop (Hampton Research, 34 Journey, Aliso Viejo, CA, USA) and placed in a −100 °C compressed air stream on an Agilent Gemini-EOS Single Crystal Autodiffractometer at Keene State College (Agilent Technologies, LTD, Yarnton, England, Keene, NH, USA). Crystallographic data were collected using graphite monochromated 0.71073 Å Mo-Kα radiation and integrated and corrected for absorption using the CrysAlisRed (Oxford Diffraction, 2010 software package) [
18]. The structures were solved using direct methods and refined using least-square methods on F-squared [
19]. The hydrogen atoms were placed in their calculated positions and included in the refinement using the riding model. All other pertinent crystallographic details such as h, k, l ranges, 2θ ranges, and R-factors can be found in
Table 10.
Crystallographic Data for (1), (2) and (3)
Table 10.
Crystal and experimental data for (1), (2) and (3).
Table 10.
Crystal and experimental data for (1), (2) and (3).
| Compound | 1 | 2 | 3 |
|---|
| Formula | C11H15N3O2S ·H2O | C12H17N3O2S | C12H17N3O2S.C2H3N |
| Formula weight | 271.33 | 267.34 | 308.40 |
| Crystal color, habit | Colorless, block | Colorless, irregular | Colorless, irregular |
| Crystal size (mm) | 0.46 × 0.22 × 0.14 | 0.36 × 0.32 0.26 | 0.45 × 0.38 × 0.32 |
| Crystal system | Monoclinic | Monoclinic | Triclinic |
| Space Group, Z | P21/c, 4 | C2/c, 8 | P-1, 2 |
| Temperature, K | 173(s) | 173(2) | 173 (2) |
| a (Å) | 8.2304(3) | 23.3083(6) | 8.9384(7) |
| b (Å) | 16.2787(6) | 8.2956(2) | 9.5167(8) |
| c (Å) | 9.9708(4) | 13.5312(3) | 10.0574(8) |
| α (°) | 90 | 90 | 110.773(7) |
| β (°) | 103.355(4) | 91.077(2) | 92.413(6) |
| γ (°) | 90 | 90 | 90.654(7) |
| Volume, Å3 | 1299.77(9) | 2615.87(12) | 798.89(12) |
| F (0,0,0) | 576.0 | 1136.0 | 328.0 |
| m (mm−1) | 0.254 | 0.246 | 0.213 |
| Qcalc (Mg m−3) | 1.387 | 1.358 | 1.282 |
| Radiation | Mo Kα (λ =0.7107) | Mo Kα (λ = 0.7107) | Mo Kα (λ = 0.7107) |
| 2Θ range for data collection | 6.318 to 65.744° | 6.036 to 65.736° | 6.14 to 65.516° |
| Reflections collected | 15566 | 16344 | 9837 |
| Independent Reflections/Rint | 4479/0.0343 | 4463/0.0353 | 5259/0.0379 |
| Data/restraints/parameters | 4479/0/170 | 4463/0/167 | 5259/0/196 |
| Collection range | | | |
| h | −11 to 12 | −33 to 35 | −13 to 13 |
| k | −24 to 24 | −12 to 12 | −13 to 13 |
| l | −15 to 14 | −20 to 17 | −14 to 14 |
| GOF on F2 | 1.073 | 1.060 | 1.047 |
| Final R indexes [I ≥ 2σ (I)] | R1 = 0.0429, wR2 = 0.1038 | R1 = 0.0381, wR2 = 0.0948 | R1 = 0.0539, wR2 = 0.1317 |
| Final R indexes (all data) | R1 = 0.0584, wR2 = 0.1132 | R1 = 0.0498, wR2 = 0.1023 | R1 = 0.0769, wR2 = 0.1516 |
| Largest diff. Peak/hole/eÅ−3 | 0.37/−0.22 | 0.39/−0.31 | 0.40/−0.35 |
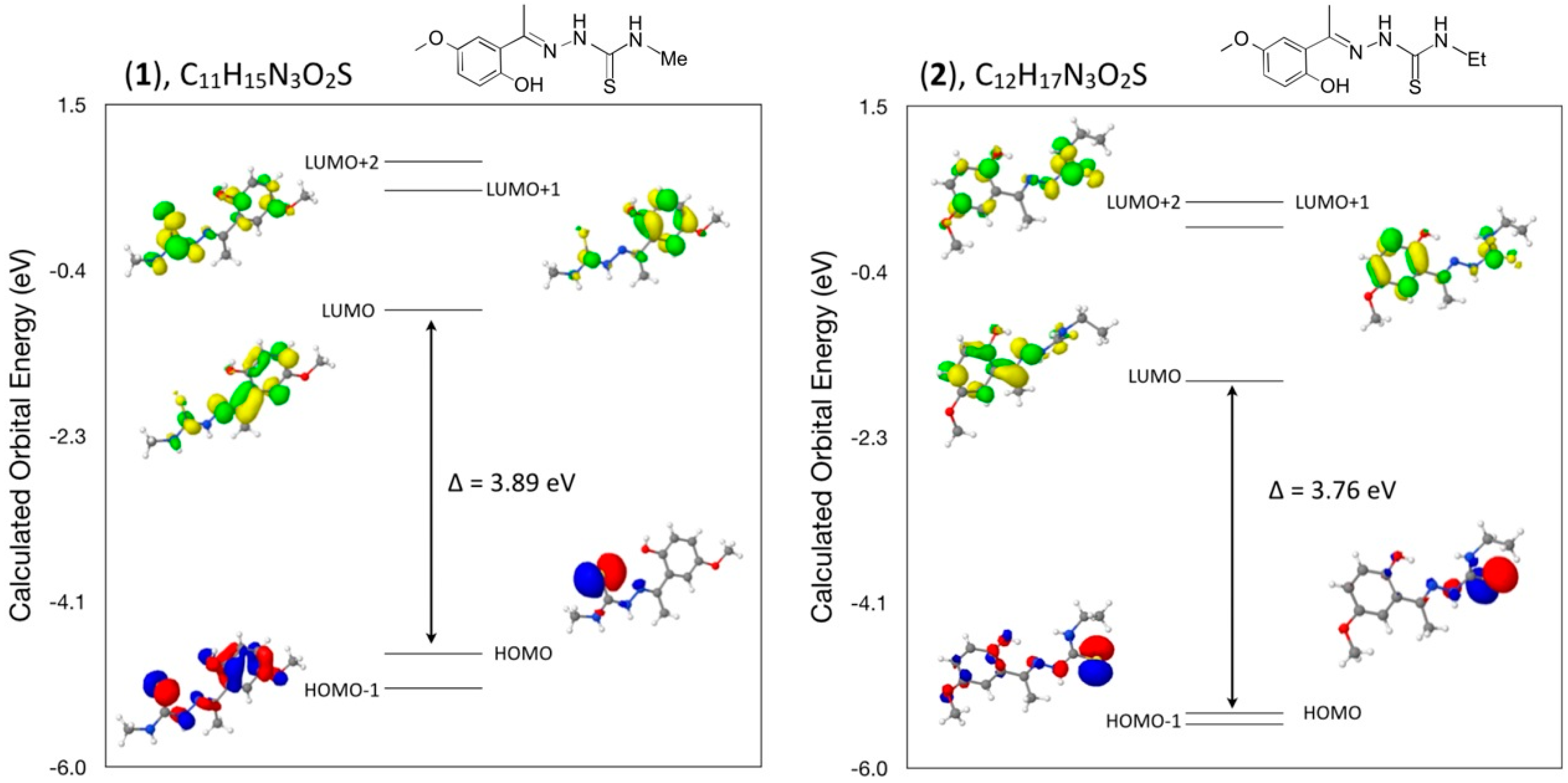
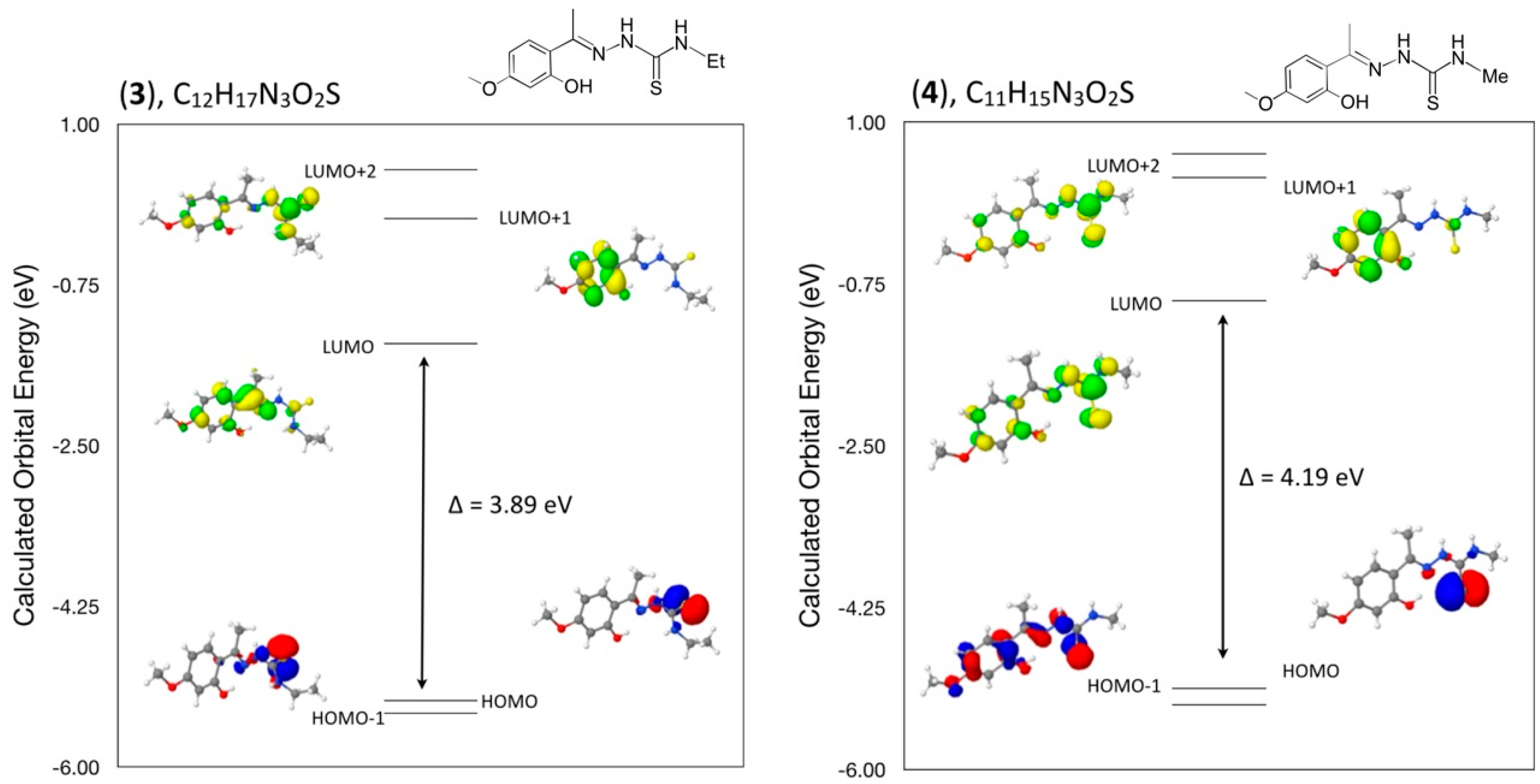
3.7. Computational Details
A density functional theory (DFT) molecular orbital calculation (WebMo Pro [
20]) with the GAUSSIAN-03 program package [
21] employing the B3LYP (Becke three parameter Lee-Yang-Parr exchange correlation functional), which combines the hybrid exchange functional of Becke [
22,
23] with the gradient correlation functional of Lee, Yang and Parr [
21] and the 6–31 G(d) basis set [
24] was performed on each of the six compounds. No solvent corrections were made with these calculations. Starting geometries were taken from X-ray refinement data. The optimized results in the free molecule state are, therefore, compared to those in the crystalline state. Discrepancies between the experimental and calculated band centers and band intensities exist. However, this does not prohibit us from making informed decisions on the observations since it is generally known that DFT often underestimates HOMO-LUMO gaps, thereby having a tendency to give excitations far too low in energy. All calculations were performed on a workstation PC using default convergence criteria.
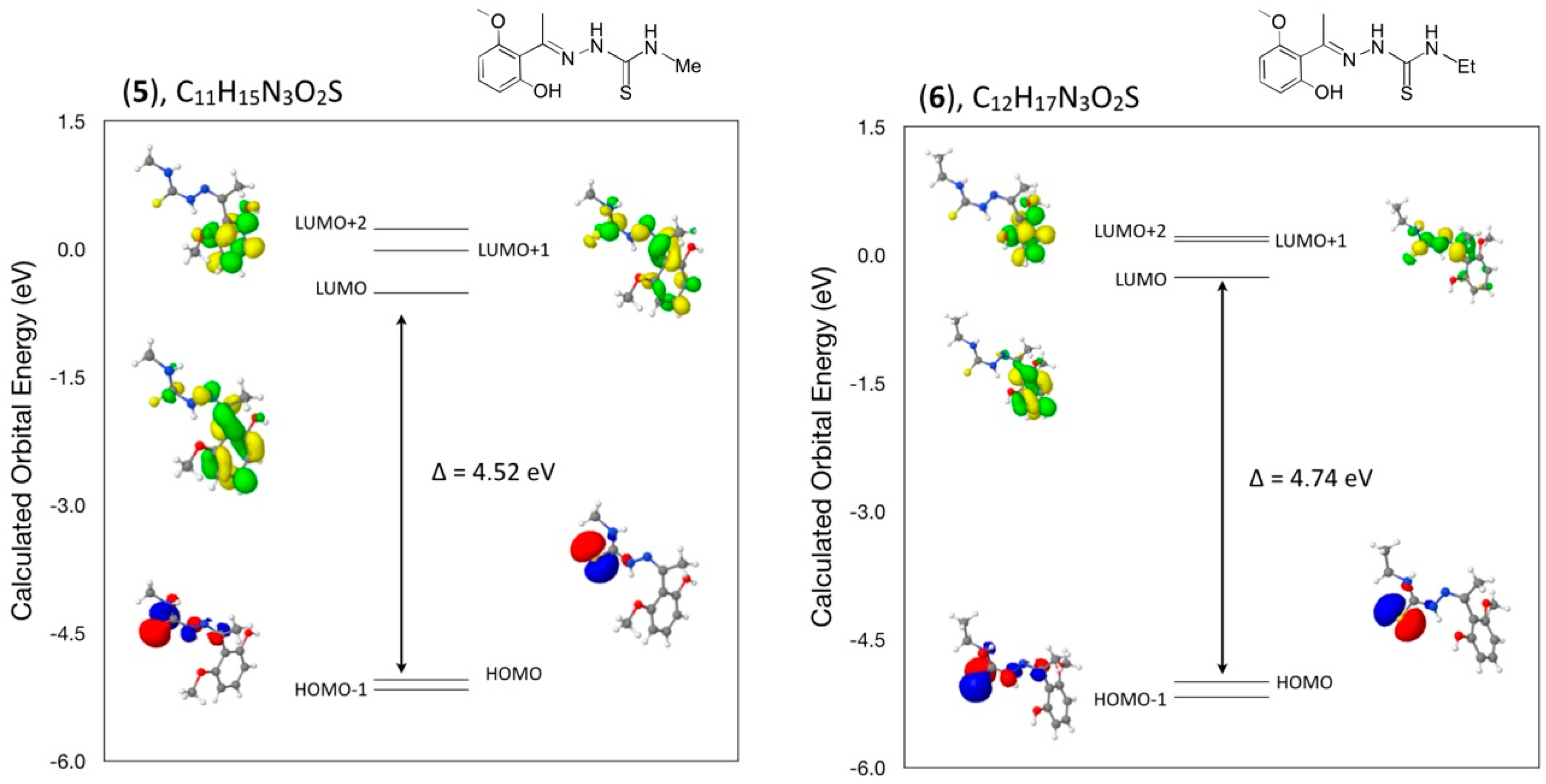
3.8. Density Functional Theory (DFT) Calculations
A comparison of selected bond angles and bond distances in crystals (
1), (
2) and (
3) to that from the geometry optimized DFT calculations at the B3LYP 6–31 G(d) level is given in
Table 1,
Table 3 and
Table 5. The differences between the two values are within normal ranges and generally consistent with bond lengths and angles for similar types of compounds such as in compounds (
4), (
5) and (
6).
In addition, a comparison of the angles between mean planes of the indole and phenylsulfonyl rings in the crystal and with the DFT geometry optimized calculation in concert with strong and weak intermolecular hydrogen bond interactions has been included in a discussion of the structural aspects for each molecule. From a DFT molecular orbital calculation for each compound, surface plots for the two highest occupied molecular orbital (HOMO and HOMO−1) and three lowest unoccupied molecular orbitals (LUMO, LUMO+1, LUMO+2) are displayed to provide visual evidence of the molecular orbitals involved in the spectroscopic electronic energy transitions examined. Based on correlation of the energies of these HOMO-LUMO frontier surfaces to the UV-VIS absorption spectra, electronic excitation transition predications are suggested.
3.9. Electronic Absorption Spectra
Electronic absorption spectra of all compounds were obtained using a Cary-300 UV-Vis spectrophotometer. All samples were dissolved in acetonitrile and spectra were recorded at room temperature. Stock solutions of ~1 × 10
−3 M solution were prepared and dilutions were prepared ranging down to ~2 × 10
−5. All Spectra were scanned from 800 to 190 nm. Deconvolution of the spectra to obtain the λ
max was carried out by the IGOR program [
25].




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}